

Abstract
Mitochondrial DNA mutations have been associated with cardiovascular disease. We report here the clinical, genetic, and molecular characterization of 1 Han Chinese family with suggestively maternally transmitted hypertension. Matrilineal relatives in this family exhibited the variable degree of hypertension at the age at onset of 44 to 55 years old. Sequence analysis of entire mitochondrial DNA in this pedigree identified the known homoplasmic 4435A>G mutation, which is located immediately at the 3 prime end to the anticodon, corresponding with the conventional position 37 of tRNAMet, and 35 other variants belonging to the Asian haplogroup B5a. The adenine (A37) at this position of tRNAMet is extraordinarily conserved from bacteria to human mitochondria. This modified A37 was shown to contribute to the high fidelity of codon recognition, the structural formation, and stabilization of functional tRNAs. In fact, a 40% reduction in the levels of tRNAMet was observed in cells carrying the 4435A>G mutation. As a result, a failure in mitochondrial tRNA metabolism, caused by the 4435A>G mutation, led to ≈30% reduction in the rate of mitochondrial translation. However, the homoplasmic form, mild biochemical defect, and late onset of hypertension in subjects carrying the 4435A>G mutation suggest that the 4435A>G mutation itself is insufficient to produce a clinical phenotype. The other modifier factors, such as nuclear modifier genes, environmental, and personal factors may also contribute to the development of hypertension in the subjects carrying this mutation. Our findings imply that the 4435A>G mutation may act as an inherited risk factor for the development of hypertension in this Chinese pedigree.
Keywords: hypertension, mitochondria, mutation, tRNA metabolism, maternal inheritance, risk factor, Chinese
Cardiovascular disease is the leading cause of death in America and the world. Cardiovascular disease includes high blood pressure, coronary heart disease, heart failure, and stroke. In particular, hypertension affects ≈1 billion individuals worldwide and 130 million in China.1 The etiology of cardiovascular disease is not well understood because of the multifactorial causes. Cardiovascular disease can be caused by single-gene or multifactorial conditions, resulting from interactions between environment and inherited risk factors. Of hereditary factors, the maternal transmissions of cardiovascular disease have been implicated in some pedigrees, suggesting that the mutation(s) in mitochondrial DNA (mtDNA) is one of the molecular bases for this disorder.2–6 Recently, several mtDNA point mutations have been identified to be associated with cardiovascular disease. These mutations included the 1555A>G mutation in the 12S rRNA gene,7 the 3260A>G and 3303C>T mutations in the tRNALeu(UUR) gene,8,9 the 8348A>G and 8363A>G mutations in the tRNALys gene,10,11 and the 4295A>G, 4300A>G, and 4317A>G mutations in the tRNAIle gene.12–14 Most recently, the 4291T>C mutation in the tRNAIle gene has been associated with a cluster of metabolic defects, including essential hypertension, hypercholesterolemia, and hypomagnesemia in a large family.15
However, the molecular pathogenesis of hypertension in the Chinese population remains poorly understood. To understand a role of mitochondrial genomes in the pathogenesis of cardiovascular diseases in the Chinese population, we have initiated a systematic and extended mutational screening of mtDNA in a large cohort of hypertension subjects at the Chinese People’s Liberation Army (PLA) General Hospital Geriatric Cardiology Clinic.16,17 In the present study, we performed the clinical, genetic, and molecular characterizations of another Han Chinese family with suggestive maternally transmitted hypertension. Mutational analysis of a mitochondrial genome in this Chinese family has identified the known tRNAMet 4435A>G mutation, which is localized at the 3 prime end adjacent to the anticodon (position 37) of tRNAMet.18 In fact, the adenine at this position of tRNAMet is extraordinarily conserved from bacteria to human mitochondria. The mitochondrial genome in this Chinese family belonged to the Eastern Asian haplogroup B5a.19 To investigate the pathogenic mechanism of the 4435A>G mutation in this Chinese family, these lymphoblastoid cell lines derived from an affected matrilineal relative carrying the 4435A>G mutation and from a Chinese control individual belonging to the same mitochondrial haplogroup lacking the mutation were assessed for the effects of the mtDNA mutation on the mitochondrial tRNA metabolism, including tRNAMet, tRNAGly, tRNALeu(UUR), and tRNALeu(CUN), and the rate of mitochondrial protein synthesis.
Materials and Methods
Subjects
As a part of a genetic screening program for hypertension, a Han Chinese family (Figure 1) was ascertained at the Institute of Geriatric Cardiology of Chinese PLA General Hospital. Informed consent, blood samples, and clinical evaluations were obtained from all of the participating family members, under protocols approved by the ethics committee of Chinese PLA General Hospital and the Cincinnati Children’s Hospital Medical Center Institute Review Board. Members of this family were interviewed and evaluated to identify both personal or medical histories of hypertension and other clinical abnormalities. The 242 control DNA samples were obtained from a panel of unaffected Han Chinese individuals from the same area.
Figure 1.

The Chinese pedigree with hypertension. Affected individuals are indicated by filled symbols. Arrowhead denotes proband.
Measurements of Blood Pressure
Members of this Chinese family underwent a physical examination, laboratory assessment of cardiovascular disease risk factors, and routine electrocardiography. A physician measured the systolic and diastolic blood pressures of subjects using a mercury column sphygmomanometer and a standard protocol. The first and the fifth Korotkoff sounds were taken as indicative of systolic and diastolic blood pressures, respectively. The average of 3 such systolic and diastolic blood pressure readings was taken as the examination blood pressure. Hypertension was defined according to the recommendation of the Sixth Joint National Committee on the Detection, Evaluation, and Treatment of High Blood Pressure20 and the World Health Organization International Society of Hypertension21 as a systolic blood pressure of ≥140 mm Hg and/or a diastolic blood pressure of ≥90 mm Hg.
Mutational Analysis of Mitochondrial Genome
Genomic DNA was isolated from the whole blood of participants using Puregene DNA Isolation kits (Gentra Systems). The entire mitochondrial genomes of the proband II-1 and an unaffected Han Chinese control subject A25 were PCR amplified in 24 overlapping fragments by use of sets of the light-strand and the heavy-strand oligonucleotide primers, as described elsewhere.22 Each fragment was purified and subsequently analyzed by direct sequencing in an ABI 3700 automated DNA sequencer using the Big Dye Terminator Cycle sequencing reaction kit. The resultant sequence data were compared with the revised consensus Cambridge sequence (GenBank accession No. NC_001807).23
For the quantification of the 4435A>G mutation, the PCR segments (700 bp) were amplified using genomic DNA as a template and oligodeoxynucleotides corresponding with mtDNA at positions 3861 to 4560 and subsequently digested with a restriction enzyme NlaIII. In fact, the 4435A>G mutation creates a novel site for this enzyme.18 Equal amounts of various digested samples were then analyzed by electrophoresis through 7% polyacrylamide gel. The proportions of digested and undigested PCR products were determined by the Image-Quant program after ethidium bromide staining to determine whether the 4435A>G mutation is in the homoplasmy in these subjects.
Mitochondrial tRNA Analysis
Lymphoblastoid cell lines were immortalized by transformation with the Epstein-Barr virus, as described elsewhere.24 Cell lines derived from one of the probed II-1s carrying the 4435A>G mutation and 1 Han Chinese control A25 belonging to the same mtDNA haplogroup lacking this mutation were grown in RPMI 1640 (Invitrogen), supplemented with 10% FBS. Total mitochondrial RNA were obtained using a TOTALLY RNA kit (Ambion) from mitochondria isolated from lymphoblastoid cell lines (≈4.0×108 cells), as described previously.25 Two μg of total mitochondrial RNA were electrophoresed through a 10% polyacrylamide/7 mol/L urea gel in Tris-borate-EDTA buffer (after heating the sample at 65°C for 10 minutes) and then electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis with oligodeoxynucleotide probes. For the detection of tRNAMet, tRNAGly, tRNALeu(UUR), and tRNALeu(CUN), the following nonradioactive digoxigenin (DIG)-labeled oligodeoxynucleotides specific for each RNA were used: 5′-TAGTACGGGAAGGGTATAACC-3′ (tRNAMet); 5′-TGTTAAGAA GAGGAATTGAA-3′ (tRNALeu(UUR)); 5′-TACTCTTTTTTGAATG TTGTC-3′ (tRNAGly); and 5′-TACTTTTATTTGGAGTTGCACC-3′ (tRNALeu(CUN)).23 DIG-labeled oligodeoxynucleotides were generated by using the DIG oligonucleotide Tailing kit (Roche). The hybridization was carried out as detailed elsewhere.26 Quantification of density in each band was made as detailed previously.26–28
Analysis of Mitochondrial Protein Synthesis
Pulse labeling of the cell lines for 30 minutes with [35S]methionine-[35S]cysteine in methionine-free DMEM in the presence of emetine, electrophoretic analysis of the translation products, and quantification of radioactivity in the whole-electrophoretic patterns or in individual well-resolved bands was carried out as detailed previously.29
Results
Clinical Presentation
The proband (II-1) began suffering from hypertension at the age of 44 years old. His blood pressure was 200/100 mm Hg by then. He came to the Chinese PLA General Hospital Geriatric Cardiology Clinic for further clinical evaluations at the age of 64 years old. His blood pressure was 180/100 mm Hg. After the administration of an angiotensin-converting enzyme inhibitor, calcium channel blocker, and diuretic, his blood pressure has ranged from 130/80 to 160/100 mm Hg. Laboratory assessment of cardiovascular disease risk factors showed that he had a normal range of the index of liver and kidney metabolic function, the blood routine, and 24-hour urinary sodium but hypercholesterolemia (9.2 mmol/L). As shown in Table 1, the echocardiogram showed that he had an increase of the interventricular septal and posterior ventricular wall thickness (13 mm) with normal atrial and ventricular dimension. Physical examination showed that he did not have other clinical abnormalities, including diabetes mellitus, vision and hearing impairments, and renal and neurological disorders. Therefore, he exhibited a typical essential hypertension. The family is originated from Beijing in Northern China. As shown in Figure 1, this familial history is suggestive of a maternal inheritance. None of the offspring of the affected father had hypertension. His mother (I-1) was diagnosed as having hypertension at 55 years old, with a blood pressure of ≤160/100 mm Hg, and his younger brother (II-3) suffered from hypertension at the age of 54 years old, with a blood pressure of 150/98 mm Hg. However, other members of this family had normal blood pressure. Comprehensive family medical histories of these individuals showed no other clinical abnormalities, including diabetes mellitus, vision and hearing impairments, and renal and neurological disorders.
Table 1.
Summary of Echocardiogram of the Proband at the Various Ages
| Variables | Age at Examination, y |
||||
|---|---|---|---|---|---|
| 64 | 66 | 70 | 72 | Chinese Reference | |
| IVST, mm | 13 | 15 | 14 | 17 | 8 to 11 |
| LVPW, mm | 10 | 11 | 14 | 8 to 11 | |
| AID, mm | 32 | 34 | 34 | 38 | 26±3 |
| LAID, mm | 34 | 37 | 43 | 43 | <40 |
| LEDID, mm | 53 | 49 | 58 | 50 | 46±4 |
| LESID, mm | 32 | 33 | 36 | 33 | 30±4 |
| EF, % | 69 | 61 | 67 | 62 | 50 to 70 |
IVST indicates interventricular septal thickness; LVPW, left ventricular posterior wall; AID, atrial inner dimension; LAID, left atrial inner dimension; LEDID, left ventricular end-systolic inner dimension; LESID, left ventricular end-diastolic inner dimension; EF, ejection fraction.
Mitochondrial DNA Analysis
The suggestively maternal transmission of hypertension in this family implied the mitochondrial involvement and led us to analyze the mitochondrial genome of matrilineal relatives. For this purpose, the DNA fragments spanning the entire mtDNA of the proband II-1 were PCR amplified, and each fragment was purified and subsequently analyzed by direct sequence. As shown in Table 2, the comparison of the resultant sequences with the revised Cambridge consensus sequence23 identified 36 nucleoside changes, belonging to the Eastern Asian haplogroup B5a.19 Furthermore, the sequences of entire mtDNA in a Han Chinese control subject A25 belonging to the haplogroup B5a were determined. Of these nucleoside changes in mtDNA of the proband II-1, there were 14 polymorphisms in the D-loop region, 4 variants in the 12S rRNA gene, 1 variant in the 16S rRNA gene, the 4435A>G mutation in the tRNAMet gene, the previously identified CO2/tRNALys intergenic 9-bp deletion corresponding with mtDNA at positions 8271 to 8279,30 and the 8 known silent mutations and 7 missense mutations in protein encoding genes (http//www.mitomap.org or http://www.genpat.uu.se/mtDB).31 These missense mutations are the 8141G>A (186A>T) in the CO2 gene, the 8584G>A (20A>T) and 8860A>G (112T>A) in A6 gene, the 10398A>G (114T>A) in the ND3 gene, and the 14766C>T (7T>I), 15236A>G (164I>V), and 15326A>G (194T>A) in the Cytb gene. These variants in rRNAs and polypeptides were further evaluated by phylogenetic analyses of these variants and sequences from other organisms, including the mouse,32 bovine,33 and Xenopus laevis.34 None of the variants in the polypeptides and rRNAs were highly evolutionarily conserved and implicated to have a significantly functional consequence. In addition, there were 32 variants in the mitochondrial genome in a Chinese control subject, A25. As shown in Table 2, both proband II-1 and control subject A25 shared 25 known mtDNA variants belonging to the haplo-group B5a.19 On the hand, the control subject A25 lacked the 4435A>G mutation in the tRNAMet gene, the 1393G>A variant in the 12S rRNA, and the 8141G>A variant in the CO2 gene.
Table 2.
mtDNA Variants in 1 Han Chinese Subject (II-1) With Hypertension and 1 Han Chinese Control Subject
| Gene | Position | Replacement | Conservation (H/M/B/X)* | rCRS† | II-1 | A25 | Previously Reported‡ |
|---|---|---|---|---|---|---|---|
| D-loop | 73 | A to G | A | G | G | Yes | |
| 210§ | A to G | A | G | G | Yes | ||
| 235 | A to G | A | G | Yes | |||
| 263 | A to G | A | G | G | Yes | ||
| 294 | T to C | T | C | Yes | |||
| 310 | T to CTC | T | CTC | CTC | Yes | ||
| 16093 | T to C | T | C | Yes | |||
| 16140§ | T to C | T | C | C | Yes | ||
| 16183 | A to C | A | C | C | Yes | ||
| 16189 | T to C | T | C | C | Yes | ||
| 16190 | C to CC | C | CC | Yes | |||
| 16260 | C to T | C | T | Yes | |||
| 16266§ | C to A | C | A | A | Yes | ||
| 16291 | C to T | C | T | Yes | |||
| 16319 | G to A | G | A | Yes | |||
| 16519 | T to C | T | C | C | Yes | ||
| 12S rRNA | 709§ | G to A | G/A/A/A | G | A | A | Yes |
| 750 | A to G | A/G/A/A | A | G | G | Yes | |
| 1393 | G to A | G/T/A/A | G | A | Yes | ||
| 1438 | A to G | A/A/A/G | A | G | G | Yes | |
| 16S rRNA | 2706 | A to G | A/A/G/A | A | G | G | Yes |
| ND1 | 3537§ | A to G | A | G | G | Yes | |
| tRNAMet | 4435 | A to G | A/A/A/A | A | G | Yes | |
| ND2 | 4769 | A to G | A | G | G | Yes | |
| CO1 | 6970 | C to T | C | T | Yes | ||
| 7001 | A to G | A | G | Yes | |||
| 7028 | C to T | C | T | T | Yes | ||
| CO2 | 7852 | G to A | G | A | Yes | ||
| 8065 | G to A | G | A | No | |||
| CO2 | 8141 | G to A (Ala to Thr) | A/S/S/A | G | A | No | |
| NC7 | 8271 to 79§ | 9 bp del | 9bp del | 9bp del | Yes | ||
| A6 | 8584§ | G to A (Ala to Thr) | A/V/V/I | G | A | A | Yes |
| 8860 | A to G (Thr to Ala) | T/A/A/T | A | G | G | Yes | |
| CO3 | 9950§ | T to C | T | C | C | Yes | |
| ND3 | 10398§ | A to G (Thr to Ala) | T/T/T/A | A | G | G | Yes |
| Nd4L | 10754 | A to G | A | G | Yes | ||
| ND4 | 11719 | G to A | G | A | A | Yes | |
| Cytb | 14766 | C to T (Thr to Ile) | T/I/S/S | C | T | T | Yes |
| 14989 | C to T | C | T | Yes | |||
| 15229 | T to C | T | C | Yes | |||
| 15235 | A to G | A | G | G | Yes | ||
| 15236 | A to G (Ile to Val) | I/I/I/S | A | G | Yes | ||
| 15326 | A to G (Thr to Ala) | T/I/M/I | A | G | G | Yes |
Data show the conservation of amino acid for polypeptides or nucleotide for rRNAs, in human (H), mouse (M), bovine (B), and Xenopus laevis (X).
rCRS indicates revised Cambridge reference sequence.23
See http//www.mitomap.org and http://www.genpat.uu.se/mtDB/ for more information.
Sequence variations used to establish the haplogroup affiliation of each mtDNA are shown.
The known 4435A>G mutation in the tRNAMet gene, as shown in Figure 2, is located immediately at the 3 prime end to the anticodon, corresponding with conventional position 37 of the tRNAMet.35 In fact, an adenine at this position is an extraordinarily conserved base in every sequenced methionine tRNA from bacteria to human mitochondria.35,36 Interestingly, the nucleotide at position 37 is more prone to modification that those at other places of tRNA.37 The nucleotide modification at this position has been shown to play a pivotal role in the stabilization of tertiary structure and the biochemical function of tRNA.37 To determine whether the 4435A>G mutation is present in homoplasmy, the fragments spanning the tRNAMet gene were PCR amplified and subsequently digested with NlaIII. There was no detectable wild-type DNA in 3 matrilineal relatives (data now shown), indicating that the 4435A>G mutation was present in homoplasmy in these matrilineal relatives. In addition, this mutation was absent in 242 Chinese controls.
Figure 2.

Identification of the 4435A>G mutation in the mitochondrial tRNAMet gene. A, Partial sequence chromatograms of the tRNAMet gene from affected individual II-1 and a married-in-control II-2. Arrow indicates the location of the base changes at position 4435. B, The location of the 4435A>G mutation in the mitochondrial tRNAMet. Cloverleaf structure of human mitochondrial tRNAMet is derived from Florentz et al.35 Arrow indicates the position of the 4435A>G mutation.
Mitochondrial tRNA Analysis
To further examine whether the 4435A>G mutation alters the tRNAMet metabolism, the steady-state level of the tRNAMet was determined by isolating total mitochondrial RNA from lymphoblastoid cell lines, separating them by a 10% polyacrylamide/7 mol/L urea gel, electroblotting and hybridizing with a nonradioactive DIG-labeled oligode-oxynucleotide probe specific for tRNAMet. After stripping the blots, the DIG-labeled probes, including tRNALeu(UUR), tRNAGly, and tRNALeu(CUN), were hybridized with the same blots for normalization purposes.
As shown in Figure 3A, the amounts of tRNAMet in the mutant cell line derived from the proband II-1 carrying the 4435A>G mutations were markedly decreased, as compared with those in the cell line derived from a Chinese control A25 belonging to the same haplogroup but lacking those mtDNA mutations. For comparison, the average levels of tRNAMet in the control or mutant cell lines were normalized to the average levels in the same cell line for the tRNALeu(UUR), tRNAGly, and tRNALeu(CUN), respectively. As shown in Figure 3B, the average levels of tRNAMet in the mutant cell line derived from II-1 ranged among ≈55% of the control after normalization to tRNALeu(UUR), ≈67% of controls after normalization to tRNAGly, and ≈57% of controls after normalization to tRNALeu(CUN).
Figure 3.

Northern blot analysis of mitochondrial tRNA. A, Equal amounts (2 μg) of total mitochondrial RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAMet. The blots were then stripped and rehybridized with DIG-labeled tRNALur(UUR), tRNAGly, and tRNALeu(CUN), respectively. B, Quantification of mitochondrial tRNA levels. Average relative tRNAMet content per cell, normalized to the average content per cell of tRNAGly, tRNALeu(UUR), or tRNALeu(CUN) in control and mutant cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on 3 independent determinations of tRNAMet content in each cell line and 3 determinations of the content of each reference RNA marker in each cell line. Error bars indicate 2 SEMs.
Mitochondrial Protein Synthesis Defect
To examine whether a defect in mitochondrial translation occurred in lymphoblastoid cell lines carrying the 4435A>G mutation, cells from 1 lymphoblastoid cell line derived from the proband carrying the 4435A>G mutation and a Chinese control A25 were labeled for 30 minutes with [35S]methionine-[35S]cysteine in methionine-free regular DMEM in the presence of 100 μg/mL of emetine to inhibit cytosolic protein synthesis.29 Figure 4A shows typical electrophoretic patterns of the mitochondrial translation products of the mutant and control cell lines. Patterns of the mtDNA-encoded polypeptides of the cells carrying the 4435A>G mutation were qualitatively identical, in terms of electrophoretic mobility of the various polypeptides, to those of the control cells and of 143B.TK− cells. However, the cell line carrying the 4435A>G mutation showed a clear tendency toward a decrease in the total rate of labeling of the mitochondrial translation products, relative to those of control cell line. Figure 4B shows a quantification of the results of a large number of labeling experiments and electrophoretic runs, which were carried out by densitometric analysis of appropriate exposures of the fluorograms and normalization to data obtained for the 143B.TK− sample. In fact, the overall rate of labeling of the mitochondrial translation products in the cell line derived from the proband carrying the 4435A>G mutation was decreased ≈30% relative to the mean value measured in the control cell line.
Figure 4.
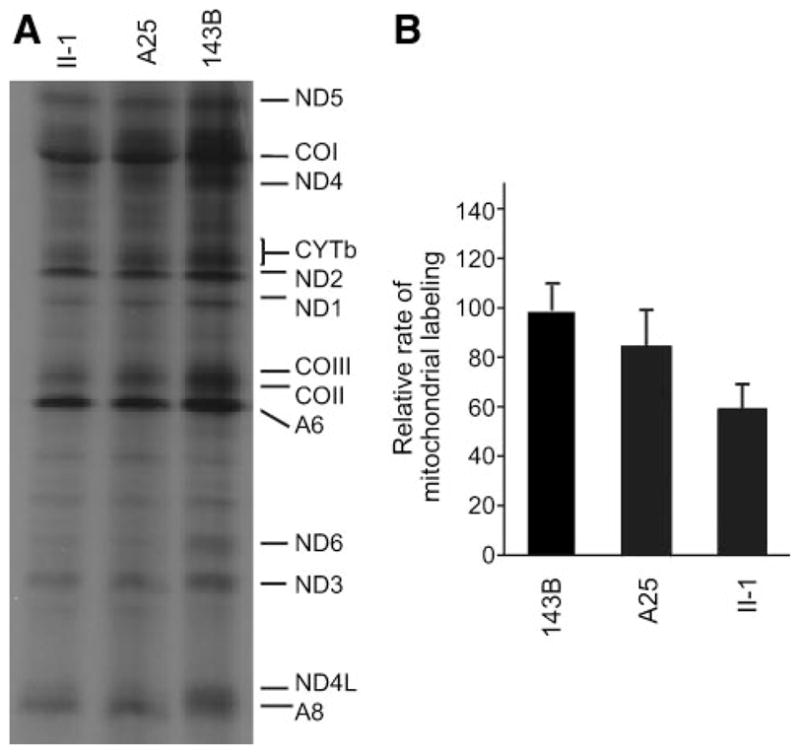
Electrophoretic patterns of the mitochondrial translation products of lymphoblastoid cell lines and of 143B.TK− cells labeled for 30 minutes with [35S]methionine in the presence of 100 μg/mL of emetine. Samples containing equal amounts of protein (30 μg) were run in SDS/polyacrylamide gradient gels. COI, COII, and COIII, subunits I, II, and III of cytochrome c oxidase; ND1, ND2, ND3, ND4, ND4L, ND5, and ND6, subunits 1, 2, 3, 4, 4L, 5, and 6 of the respiratory chain reduced nicotinamide-adenine dinucleotide dehydrogenase; A6 and A8, subunits 6 and 8 of the H+-ATPase; and CYTb, apocytochrome b. B, Quantification of the rates of labeling of the mitochondrial translation products, after a 30-minute [35S]methionine pulse, in lymphoblastoid cell lines. The rates of mitochondrial protein labeling, determined as detailed in Materials and Methods, are expressed as percentages of the value for 143B.TK− in each gel, with error bars representing 2 SEMs. A total of 3 independent labeling experiments and 3 electrophoretic analyses of each labeled preparation were carried out on lymphoblastoid cell lines. The vertical arrows refer to 2 SEs.
Discussion
In the present study, we have performed the clinical, genetic, and molecular characterizations of a Han Chinese family with essential hypertension. The hypertension as a sole clinical phenotype was only present in all of the matrilineal relatives of this 4-generation pedigree. Clinical and genetic evaluations revealed the variable severity and age at onset in hypertension. In particular, the age at onset was 44.0, 54.0, and 55.0 years old in 3 affected matrilineal relatives, with an average of 51 years old. The suggestively maternal transmission of hypertension in this family suggested that the mtDNA mutation(s) is the molecular basis for this disorder. Mutational analysis of the mitochondrial genome in this family identified the tRNAMet 4435A>G mutation and other 35 variants belonging to the Eastern Asian haplogroup B5a.19 On other hand, the 4435A>G mutation was also identified in the haplogroup D5 of a Chinese family18 and a Japanese subject,38 in the haplogroup D of an Asian individual,39 and in the haplogroup M7a2 of 2 Japanese subjects,38 as well as in the haplogroup J of an European subject.39 This suggested that the 4435A>G mutations occurred sporadically and multiplied through evolution of the mtDNA. This mutation was present only in matrilineal relatives of this family in the homoplasmic form but not in other members of this family or in 242 Han Chinese controls. Indeed, our previous study indicated that the 4435A>G mutation may modulate the phenotypic manifestation of Leber’s hereditary optic neuropathy–associated ND4 11778G>A mutation in a Chinese family,18 whereas the 4435A>G mutation was also associated with type 2 diabetes mellitus in 3 Japanese patients.38
The 4435A>G mutation is located at the immediate 3 prime end to the anticodon, corresponding with conventional position 37 of the tRNAMet.35 In fact, the adenine at the 37 position of tRNAMet is extraordinarily conserved among 150 different species (http://w3appli.u-strasbg.fr/mamit-trna/tables.asp?amino acid = 19).36 Almost all of the A37 in tRNAs are modified, eg, thiolation and methylation.37 Indeed, this modified nucleotide contributes to the high fidelity of codon recognition, as well as the structural formation and stabilization of functional tRNAs.40 In Escherichia coli, nucleotide modifications at positions 37 and 34 are responsible for the stabilization of the canonical loop structure in the anticodon domain of tRNALys.41 Also, it has been shown that the modification of A37 stabilizes the 3 prime stacking features of the anticodon, thereby improving its interaction with the codon.42 The deficient modification of A37 decreased the activity of the corresponding tRNA43 and increased +1 frameshifts for tRNAPhe,44 whereas the A-to-G substitution at position 37 led to a 10-fold reduction in the section of tRNAs at the aminoacyl-tRNA binding site.45 Furthermore, the 4295A>G mutation at the 37 position of tRNAIle has been associated with hypertrophic cardiomyopathy in white pedigrees12,46 and hypertension in a Chinese pedigree.16 Most recently, the 4291T>C mutation at the anticodon region of mitochondrial tRNAIle has been associated with hypertension, hypercholesterolemia, and hypomagnesemia.15
In the current study, compared with a control cell lacking the mutation, ≈40% reduction in the levels of tRNAMet was observed in cells carrying the 4435A>G mutation. The lower levels of tRNAMet in cells carrying the 4435A>G mutation most probably result from a defect in nucleotide modification at position 37 of tRNAMet. As a result, a shortage of the tRNAMet is responsible for the reduced rate of mitochondrial protein synthesis. Subsequently, these defects led to an impairment of the function of the mitochondrial respiration chain, reduction of ATP production, and increase of reactive oxygen species production. These mitochondrial dysfunctions likely contribute to the development of hypertension.47,48 However, the levels of total tRNAMet in mutant cells are above a proposed threshold, which is 30% of the control level of tRNA, to support a normal rate of mitochondrial translation,26,27 indicating that the 4435A>G mutation itself is insufficient to produce a clinical phenotype, as in the cases of deafness-associated 12S rRNA 1555A>G mutation49 and Leber’s hereditary optic neuropathy-associated ND4 11778G>A mutation.50 The other modifier factors, eg, nuclear modifier genes, environmental factors, and personal lifestyles, also contribute to the development of hypertension in these subjects carrying the 4435A>G mutation. Therefore, the 4435A>G mutation may act as an inherited risk factor for the development of hypertension in this Chinese pedigree.
Perspectives
The genetic and biochemical evidence of the present study indicate that the mitochondrial tRNAMet 4435A>G mutation is likely associated with essential hypertension. The tissue specificity of this pathogenic mtDNA mutation is likely attributed to the tissue-specific posttranscriptional modification or the contribution of nuclear modifier genes. The 4435A>G mutation should be added to the list of inherited risk factors for future molecular diagnosis. Thus, our finding provides new insights into the molecular mechanism, management, and treatment of maternally inherited hypertension. Future research should further explore the emerging link among hypertension, mitochondrial dysfunction, and their causative-effect relationship.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants RO1DC05230 and RO1DC07696 from the National Institute on Deafness and Other Communication Disorders (to M.-X.G.) and National Key Basic Research and Development Project 973 Fund 2007CB07403 (to S.W.).
Footnotes
Disclosures
None.
References
- 1.Gu D, Reynolds K, Wu X, Chen J, Duan X, Muntner P, Huang G, Reynolds RF, Su S, Whelton PK, He J. Prevalence, awareness, treatment, and control of hypertension in China. Hypertension. 2002;40:920–927. doi: 10.1161/01.hyp.0000040263.94619.d5. [DOI] [PubMed] [Google Scholar]
- 2.Brandao AP, Brandao AA, Araujo EM, Oliveira RC. Familial aggregation of arterial blood pressure and possible genetic influence. Hypertension. 1992;19:II214–II217. doi: 10.1161/01.hyp.19.2_suppl.ii214. [DOI] [PubMed] [Google Scholar]
- 3.Wallace DC. Mitochondrial defects in cardiomyopathy and neuro-muscular disease. Am Heart J. 2000;139:S70–S85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 4.Watson B, Jr, Khan MA, Desmond RA, Bergman S. Mitochondrial DNA mutations in black Americans with hypertension-associated end-stage renal disease. Am J Kidney Dis. 2001;38:529–536. doi: 10.1053/ajkd.2001.26848. [DOI] [PubMed] [Google Scholar]
- 5.Hirano M, Davidson M, DiMauro S. Mitochondria and the heart. Curr Opin Cardiol. 2001;16:201–210. doi: 10.1097/00001573-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz F, Duka A, Sun F, Cui J, Manolis A, Gavras H. Mitochondrial genome mutations in hypertensive individuals. Am J Hypertens. 2004;17:629–635. doi: 10.1016/j.amjhyper.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Santorelli FM, Tanji K, Manta P, Casali C, Krishna S, Hays AP, Mancini DM, DiMauro S, Hirano M. Maternally inherited cardiomyopathy: an atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am J Hum Genet. 1999;64:295–300. doi: 10.1086/302188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeviani M, Gellera C, Antozzi C, Rimoldi M, Morandi L, Villani F, Tiranti V, DiDonato S. Maternally inherited myopathy and cardiomyopathy: association with mutation in mitochondrial DNA tRNALeu(UUR) Lancet. 1991;338:143–147. doi: 10.1016/0140-6736(91)90136-d. [DOI] [PubMed] [Google Scholar]
- 9.Silvestri G, Santorelli FM, Shanske S, Whitley CB, Schimmenti LA, Smith SA, DiMauro S. A new mtDNA mutation in the tRNALeu(UUR) gene associated with maternally inherited cardiomyopathy. Hum Mut. 1994;3:37–43. doi: 10.1002/humu.1380030107. [DOI] [PubMed] [Google Scholar]
- 10.Santorelli FM, Mak SC, El-Schahawi M, Casali C, Shanske S, Baram TZ, Madrid RE, DiMauro S. Maternally inherited cardiomyopathy and hearing loss associated with a novel mutation in the mitochondrial tRNALys gene (G8363A) Am J Hum Genet. 1996;58:933–939. [PMC free article] [PubMed] [Google Scholar]
- 11.Terasaki F, Tanaka M, Kawamura K, Kanzaki Y, Okabe M, Hayashi T, Shimomura H, Ito T, Suwa M, Gong JS, Zhang J, Kitaura Y. A case of cardiomyopathy showing progression from the hypertrophic to the dilated form: association of Mt8348A–>G mutation in the mitochondrial tRNALys gene with severe ultrastructural alterations of mitochondria in cardiomyocytes. Jpn Circ J. 2001;65:691–694. doi: 10.1253/jcj.65.691. [DOI] [PubMed] [Google Scholar]
- 12.Merante F, Myint T, Tein I, Benson L, Robinson BH. An additional mitochondrial tRNAIle point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Hum Mut. 1996;8:216–222. doi: 10.1002/(SICI)1098-1004(1996)8:3<216::AID-HUMU4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Taylor RW, Giordano C, Davidson MM, d’Amati G, Bain H, Hayes CM, Leonard H, Barron MJ, Casali C, Santorelli FM, Hirano M, Lightowlers RN, DiMauro S, Turnbull DM. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003;41:1786–1796. doi: 10.1016/s0735-1097(03)00300-0. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka M, Ino H, Ohno K, Hattori K, Sato W, Ozawa T, Tanaka T, Itoyama S. Mitochondrial mutation in fatal infantile cardiomyopathy. Lancet. 1990;336:1452. doi: 10.1016/0140-6736(90)93162-i. [DOI] [PubMed] [Google Scholar]
- 15.Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Liu Y, Yang L, Wang S, Guan MX. Maternally inherited hypertension is associated with the mitochondrial tRNAIle 4295A>G mutation in a Chinese family. Biochem Biophys Res Commun. 2008;367:906–911. doi: 10.1016/j.bbrc.2007.12.150. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Li Z, Yang L, Wang S, Guan MX. The mitochondrial ND1 T3308C mutation in a Chinese family with the secondary hypertension. Biochem Biophys Res Commun. 2008;368:18–22. doi: 10.1016/j.bbrc.2007.12.193. [DOI] [PubMed] [Google Scholar]
- 18.Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y, Hu Y, Mo JQ, West CE, Guan MX. The novel 4435A>G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci. 2006;47:475–483. doi: 10.1167/iovs.05-0665. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, Shinoda K, Umetsu K, Yamada Y, Oshida Y, Sato Y, Hattori N, Mizuno Y, Arai Y, Hirose N, Ohta S, Ogawa O, Tanaka Y, Kawamori R, Shamoto-Nagai M, Maruyama W, Shimokata H, Suzuki R, Shimodaira H. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;84:1832–1850. doi: 10.1101/gr.2286304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guidelines Subcommittee. 1999 World Health Organization-International Society of Hypertension guidelines for the management of hypertension. J Hypertens. 1999;17:151–183. [PubMed] [Google Scholar]
- 21.Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. The sixth report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. Arch Intern Med. 1997;157:2413–2446. doi: 10.1001/archinte.157.21.2413. [DOI] [PubMed] [Google Scholar]
- 22.Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 24.Miller G, Lipman M. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc Natl Acad Sci U S A. 1973;70:190–194. doi: 10.1073/pnas.70.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King MP, Attardi G. Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J Biol Chem. 1993;1268:10228–10237. [PubMed] [Google Scholar]
- 26.Li X, Fischel-Ghodsian N, Schwartz F, Yan Q, Friedman RA, Guan MX. Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 2004;32:867–877. doi: 10.1093/nar/gkh226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guan MX, Enriquez JA, Fischel-Ghodsian N, Puranam R, Lin CP, Marion MA, Attardi G. The deafness-associated mtDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase ND6 subunit gene expression. Mol Cell Biol. 1998;18:5868–5879. doi: 10.1128/mcb.18.10.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan MX, Yan Q, Li X, Bykhovskaya Y, Gallo-Teran J, Hajek P, Umeda N, Zhao H, Garrido G, Mengesha E, Suzuki T, del Castillo I, Peters JL, Li R, Qian Y, Wang X, Ballana E, Shohat M, Lu J, Estivill X, Watanabe K, Fischel-Ghodsian N. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Lu J, Zhu Y, Yang A, Yang L, Li R, Chen B, Qian Y, Tang X, Wang J, Zhang X, Guan MX. Mitochondrial tRNAThr G15927A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA A1555G mutation in four Chinese families. Pharmacogenet Genomics. 2008;18:1059–1070. doi: 10.1097/FPC.0b013e3283131661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP: a human mitochondrial genome database–2004 update. Nucleic Acids Res. 2005;33:D611–D613. doi: 10.1093/nar/gki079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 33.Gadaleta G, Pepe G, De Candia G, Quagliariello C, Sbisa E, Saccone C. The complete nucleotide sequence of the Rattus norvegicus mitochondrial genome: cryptic signals revealed by comparative analysis between vertebrates. J Mol Evol. 1989;28:497–516. doi: 10.1007/BF02602930. [DOI] [PubMed] [Google Scholar]
- 34.Roe A, Ma DP, Wilson RK, Wong JF. The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J Biol Chem. 1985;260:9759–9774. [PubMed] [Google Scholar]
- 35.Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M. Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci. 2003;60:1356–1375. doi: 10.1007/s00018-003-2343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Björk GR. Biosynthesis and function of modified nucleotides. In: Söll D, RajBhandary UL, editors. tRNA: Structure, Biosynthesis and Function. Washington, DC: ASM Press; 1995. pp. 165–206. [Google Scholar]
- 38.Guo LJ, Oshida Y, Fuku N, Takeyasu T, Fujita Y, Kurata M, Sato Y, Ito M, Tanaka M. Mitochondrial genome polymorphisms associated with type-2 diabetes or obesity. Mitochondrion. 2005;5:15–33. doi: 10.1016/j.mito.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N. Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am J Hum Genet. 2002;70:1152–1171. doi: 10.1086/339933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Björk GR. Stable RNA modification. In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low BK, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia Coli and Salmonella: Cellular and Molecular Biology. Washington, DC: American Society for Microbiology; 1996. pp. 861–886. [Google Scholar]
- 41.Sundaram M, Durant PC, Davis DR. Hypermodified nucleosides in the anticodon of tRNA(Lys) stabilize a canonical U-turn structure. Biochemistry. 2000;39:12575–12584. doi: 10.1021/bi005120y. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Esberg B, Curran JF, Bjork GR. Three modified nucleosides present in the anticodon stem and loop influence the in vivo aa-tRNA selection in a tRNA-dependent manner. J Mol Biol. 1997;271:209–221. doi: 10.1006/jmbi.1997.1176. [DOI] [PubMed] [Google Scholar]
- 43.Buck M, Griffiths E. Iron mediated methylthiolation of tRNA as a regulator of operon expression in Escherichia coli. Nucleic Acids Res. 1982;10:2609–2024. doi: 10.1093/nar/10.8.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001;20:4863–4873. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yarus M, Cline SW, Wier P, Breeden L, Thompson RC. Actions of the anticodon arm in translation on the phenotypes of RNA mutants. J Mol Biol. 1986;192:235–255. doi: 10.1016/0022-2836(86)90362-1. [DOI] [PubMed] [Google Scholar]
- 46.Finnila S, Hassinen I, Majamaa K. Phylogenetic analysis of mitochondrial DNA in patients with an occipital stroke: evaluation of mutations by using sequence data on the entire coding region. Mut Res. 2001;458:31–39. doi: 10.1016/s1383-5726(01)00012-7. [DOI] [PubMed] [Google Scholar]
- 47.Postnov YV, Orlov SN, Budnikov YY, Doroschuk AD, Postnov AY. Mitochondrial energy conversion disturbance with decrease in ATP production as a source of systemic arterial hypertension. Pathophysiology. 2007;14:195–204. doi: 10.1016/j.pathophys.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Lopez-Campistrous A, Hao L, Xiang W, Ton D, Semchuk P, Sander J, Ellison MJ, Fernandez-Patron C. Mitochondrial dysfunction in the hypertensive rat brain: respiratory complexes exhibit assembly defects in hypertension. Hypertension. 2008;51:412–4129. doi: 10.1161/HYPERTENSIONAHA.107.102285. [DOI] [PubMed] [Google Scholar]
- 49.Guan MX, Fischel-Ghodsian N, Attardi G. Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum Mol Genet. 2001;10:573–580. doi: 10.1093/hmg/10.6.573. [DOI] [PubMed] [Google Scholar]
- 50.Qu J, Zhou X, Zhang J, Zhao F, Sun YH, Yong Y, Wei QP, Cai W, West CE, Guan MX. Extremely low penetrance of Leber’s hereditary optic neuropathy (LHON) in eight Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology. 2009;116:558–564. doi: 10.1016/j.ophtha.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]