

Abstract
FeII/α-ketoglutarate-dependent hydroxylases uniformly possess a double-stranded β-helix fold with two conserved histidines and one carboxylate coordinating their mononuclear ferrous ions. Oxidative decomposition of the α-keto acid is proposed to generate a ferryl-oxo intermediate capable of hydroxylating unactivated carbon atoms in a myriad of substrates. This perspective focuses on a subgroup of these enzymes that are involved in pyrimidine salvage, purine decomposition, nucleoside and nucleotide hydroxylation, DNA/RNA repair, and chromatin modification. The varied reaction schemes are presented, and selected structural and kinetic information is summarized.
1.0 Introduction
Ferrous ion and α-ketoglutarate (αKG)-dependent dioxygenases comprise an enzyme superfamily with members present in animals, plants, protists, fungi, bacteria and even viruses. Most representatives of this large group of enzymes couple the oxidative decarboxylation of αKG to various hydroxylation reactions (Fig. 1), but others are capable of catalyzing desaturation, epoxidation, halogenation, ring formation, or ring expansion reactions, which allows them to fill a wide variety of biological roles including antibiotic biosynthesis, hypoxic sensing, DNA repair, and various types of metabolite transformations.1–3 Not surprisingly, the primary substrates also are diverse and include proteins, lipids, nucleic acids, and a myriad of small molecules that can be used for synthesis or undergo degradation.
Fig. 1.

General reaction of FeII/αKG hydroxylases
The sequences of FeII/αKG dioxygenases show little overall identity, yet the diverse reactions all are catalyzed by proteins with the same core fold and use similar chemistries.4 The hallmark of these enzymes is their use of FeII to activate molecular oxygen according to a mechanism (Scheme 1) first proposed by Hanauske-Abel and Günzler in 1982 for prolyl 4-hydroxylase.5 (A) In the absence of substrates, a “facial triad” (usually two His plus one Asp or Glu occurring in a His-X-Asp/Glu-Xn-His motif) weakly bind one face of the hexacoordinate metal.6,7 (B) The αKG co-substrate displaces two waters and coordinates FeII in a bidentate fashion with its C1 carboxyl and C2 keto oxygens. The binding of αKG is stabilized by interaction of its C5 carboxylate with other amino acids, often including a conserved Arg located about a dozen residues beyond the second His of the motif.1 (C) The primary substrate does not directly interact with the metal ion, but its binding leads to displacement of the remaining water molecule and creation of an oxygen-binding site. (D) Reaction with oxygen most likely forms an FeIII-superoxo species. (E) Oxidative decomposition of αKG creates an FeIV-oxo intermediate capable of abstracting a hydrogen atom from a non-activated carbon on the primary substrate. (F) The hydroxyl group rebounds as a radical, resulting in substrate hydroxylation and restoring the FeII form of the enzyme. The FeIV-oxo intermediate has been directly observed for taurine/αKG dioxygenase (TauD),8–10 prolyl 4-hydroxylase,11 and a related halogenase,12 and is believed to form in most if not all members of this family. Recent reviews provide greater details regarding the general characteristics of this enzyme family.1,2
Scheme 1.

General mechanism of FeII/αKG hydroxylases
This perspective focuses on the modification of nucleobases, nucleosides, nucleotides, and chromatin by the FeII and αKG dependent hydroxylases (Table 1). We discuss the degradation and salvage of free bases by the fungal enzymes thymine 7-hydroxylase and xanthine hydroxylase. Next, nucleoside hydroxylases are briefly described. We then examine the developmental modification of DNA in kinetoplastid protozoans and summarize evidence that the JBP1 and JBP2 proteins possess thymidine hydroxylase activity. The oxidative repair of DNA by Escherichia coli AlkB is discussed, and the properties of mammalian homologues are summarized. Finally, we present emerging evidence pointing to the existence of FeII/αKG hydroxylases in other nucleotide and chromatin modifying enzymes.
Table 1.
Metal binding and αKG stabilizing residues and primary substrates of selected FeII/αKG dependent hydroxylases
| Enzyme | Metal Binding | αKG C5 stabilization | Primary Substrate | ||
|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | |||
| R. glutinis thymine-7-hydroxylase | His 211 | Asp 213 | His 268 | Arg 284 | thymine, hydroxymethyluracil, 5-formyluracil |
| A. nidulans xanthine hydroxylasea | His 149 | Asp 151 | His 340 | Arg 352 | xanthine |
| pyrimidine deoxyribonucleoside 2′-hydroxylase | NDb | ND | ND | ND | deoxythymidine, deoxyuridine |
| (deoxy)uridine 1′-hydroxylase | ND | ND | ND | ND | deoxyuridine, uridine |
| T. brucei JBP1c | His 213 | Asp 215 | His 263 | Arg 279 | thymidine in telomeric chromatin |
| E. coli AlkB | His 131 | Asp 133 | His 187 | Arg 204 | 1meA, 3meC in ss-DNA or RNA |
| H. sapien ABH2 | His 171 | Asp 173 | His 236 | Arg 248 | 1meA, 3meC in ds-DNA |
| H. sapien ABH3 | His 191 | Asp 193 | His 257 | Arg 269 | 1meA, 3meC in ss-DNA or RNA |
| H. sapien JMJD2A | His 188 | Glu 190 | His 276 | Tyr 132 Lys 206 | di- and tri-methyl Lys at positions 9 or 36 of histone H3 |
No crystal structure is available, but these residues were confirmed by mutagenesis experiments.
ND, Not Determined.
These residues are derived from sequence alignments.
2.0 Pyrimidine and purine hydroxylases
2.1 Thymine 7-hydroxylase
One of the first FeII/αKG-dependent dioxygenases to be discovered was thymine 7-hydroxylase (T7H), an enzyme that catalyzes three consecutive oxidations of thymine to form 5-hydroxymethyl uracil (HMU), 5-formyl uracil (FU), and uracil-carboxylic acid (Scheme 2).13–16 Each of these reactions requires FeII, αKG, and O2 and produces stoichiometric amounts of the respective primary product, CO2, and succinate. One atom of molecular oxygen is incorporated into succinate and the other into the primary products.15–17 The Rhodotorula glutinis T7H sequence includes His 211, Asp 213, and His 268 as putative metal-binding ligands and Arg 284 as a likely αKG stabilizing residue, but no structural studies have been carried out to confirm these assignments.
Scheme 2.

Reactions of thymine 7-hydroxylase
Efforts to characterize T7H have focused on the enzymes from fungi such as Neurospora crassa, R. glutinis, and Aspergillus nidulans that can grow on thymine as their sole nitrogen source. These microorganisms possess a salvage pathway enabling the host to utilize thymine as a pyrimidine source in the absence of the common “de novo” pathway of uracil biosynthesis (Fig. 2). The gene encoding T7H was recently identified,18 and sequence comparisons reveal many homologues outside of the fungal kingdom including bacteria (e.g., Pseudomonas species and Xanthomonas campestris) and protists (e.g., Trypanosoma brucei). It is not known whether these gene products possess T7H activity.
Fig. 2.

Pyrimidine salvage pathway. Selected fungi are capable of salvaging thymine from dTMP by a series of reactions catalyzed by (1) pyrimidine deoxynucleoside-2′-hydroxylase, (2) nucleoside hydrolase or phosphorylase, (3) T7H, (4) uracil-5-carboxylate decarboxylase (also named iso-orotate decarboxylase), and (5) uracil phosphoribosyltransferase. The resulting UMP is available for conversion to other nucleotides by standard reactions.
The kinetic properties of T7H were extensively studied,19–21 and thymine was shown to be greatly preferred as a primary substrate over HMU and FU. For example, T7H purified from R. glutinis exhibits a Km of about 60 μM for thymine, twice that for HMU, and over 60-fold greater for FU.21 Furthermore, the kcat of the reaction for thymine, estimated to be in the range of 1000 min−1 for the R. glutinis enzyme,21 is greater than that for HMU or FU. Similarly, the ratios of reaction rates of the three substrates with the N. crassa enzyme are estimated to be 4.6:2.3:1.16 Thymine and HMU are bound at the same active site as shown by studies demonstrating that HMU competitively inhibits thymine hydroxylation with an observed Ki that matches the Km for HMU utilization.19,21 On the basis of the high Km and low kcat for FU, hydroxylation of this substrate by T7H is suggested to be physiologically irrelevant; the presence of an NAD-dependent FU dehydrogenase in N. crassa is consistent with this proposal.21,22 No evidence exists for processivity of the enzyme; rather, it has been argued that each oxidation requires product release in order to free the T7H active site for another turnover.21
The reactivity of T7H with various thymine analogues nicely illustrates the remarkable chemical versatility associated with the FeII/αKG family of enzymes. In addition to the three reactions shown above in Scheme 2, T7H oxidizes 5-vinyluracil, 5-methylthiouracil, 5,6-dihydrothymine, 1-methylthymine, 1-methyluracil, 1-ethyluracil, 6-azathymine, and others some of which are shown in Scheme 3.20,23 In addition, ethynyluracil (EU) is metabolized by the enzyme to form 5-carboxymethyluracil and uracil-5-acetylglycine along with a reactive intermediate that acts as a mechanism-based inactivator which covalently modifies a phenylalanine of the protein.24 Overall, the reactivity of T7H is reminiscent of that observed with cytochrome P450s and includes epoxidations, sulfur oxidations, hydroxylation of non-activated carbon-hydrogen bonds, and N-demethylations.21
Scheme 3.

Diversity of alternative reactions catalyzed by thymine 7-hydroxylase
In contrast to the relaxed specificity for its primary substrate, T7H is very specific for its co-substrate αKG and no reaction occurs without O2. The Km values of these substrates (24 and 36 μM, respectively) for the R. glutinis enzyme are lower than that of thymine. In the presence of ascorbate plus 5-fluorouracil or uracil (substrate analogues that are incapable of hydroxylation), T7H catalyzes an uncoupled reaction in which O2 is reduced at about 1–6% of the thymine-dependent catalytic rate.21,25,26 This uncoupling reaction does not involve oxidative decomposition of αKG and thus differs from that exemplified by prolyl 4-hydroxylase.27–29
Kinetic isotope effect studies of T7H have provided useful insights into the general reaction mechanism of αKG dependent dioxygenases.21,25 Experiments using (trideuteriomethyl)thymine as substrate revealed a smaller catalytic rate constant than measured with the standard substrate, consistent with cleavage of the carbon-hydrogen bond being at least partially rate limiting. Because Vmax/Km did not exhibit an isotope effect for this enzyme, even when measured under reduced O2 concentration, an irreversible step prior to oxygen binding was postulated to occur. These results agree with the classic prolyl 4-hydroxylase mechanism proposed by Hanauske-Abel & Gunzler,5 and recent studies with other family members establish that αKG decarboxylation and oxygen-oxygen bond cleavage precede the cleavage of the carbon-hydrogen bond.
2.2 Xanthine hydroxylase
Xanthine dehydrogenase and xanthine oxidase are well studied and highly conserved molybdopterin cofactor (Moco)-containing enzymes that transform xanthine into uric acid.30 These reactions enable many microorganisms to utilize the purine bases xanthine and hypoxanthine as N-sources. Mutants of A. nidulans defective in xanthine dehydrogenase activity were shown to retain the ability to grow on xanthine as sole nitrogen source, suggesting the presence of a second mechanism for xanthine metabolism.31 An FeII/αKG-dependent xanthine hydroxylase (Scheme 4), encoded by xanA, was shown to be responsible for this novel activity.32
Scheme 4.

Reaction of FeII/αKG-dependent xanthine hydroxylase
The enzymatic properties of XanA were investigated as purified from A. nidulans and as heterologously expressed as a His6-tagged fusion protein in E. coli.33 The two forms differ in their quaternary structure (dodecamer versus monomer, respectively), posttranslational modifications (e.g., the fungal form is highly glycosylated), and length (the fungal protein is significantly truncated at the N-terminus); however, they are very similar in their enzymatic properties (kcat of 30–70 s−1 and Km of 45 μM for xanthine). XanA is highly specific for its primary substrate with only trace activity observed for 9-methylxanthine, while 6,8-dihydroxypurine was found to be a slow-binding competitive inhibitor.33,34 FeII cannot be replaced by any other divalent metal, and α-ketoadipic acid is the only alternate co-substrate that confers activity.33 Kinetic isotope effect studies using xanthine deuterated at C8 demonstrate that carbon-hydrogen bond cleavage is not rate limiting in the overall reaction. In contrast, Vmax is significantly decreased in 2H2O indicating that a chemical group possessing an exchangeable proton is important in the rate-limiting step.33
No crystal structure of XanA is available; however, a homology model was constructed (Fig. 3) by using TauD as a template and several features of this model were confirmed by mutational studies.33,34 Drastic activity losses were noted upon mutation of the putative FeII-binding ligands (His 149, Asp 151 and His 340). Lys 122 was shown to be important in stabilizing αKG as demonstrated by an increase of the Kd for αKG according to fluorescence quenching studies, and Arg 352 is predicted to form a salt bridge with the C5 carboxylate of the α-keto acid. The kcat/Km value of the N358A variant was reduced 23-fold compared to the wild-type enzyme, suggesting an important role for Asn 358 in catalysis. Cys 357, Glu 137, and Asp 138 were predicted to lie close to the N9 position of xanthine on the basis of activity measurements of the Ala mutants when comparing xanthine and 9-methylxanthine as substrates. Gln 101 and Gln 356 were tentatively identified as additional residues that line the active site and likely to be important for catalysis. The model provides a reasonable explanation of the narrow substrate specificity of XanA in terms of steric clashes, incorrect tautomeric preferences, or disruption of critical interactions for other substrate-like compounds.
Fig. 3.
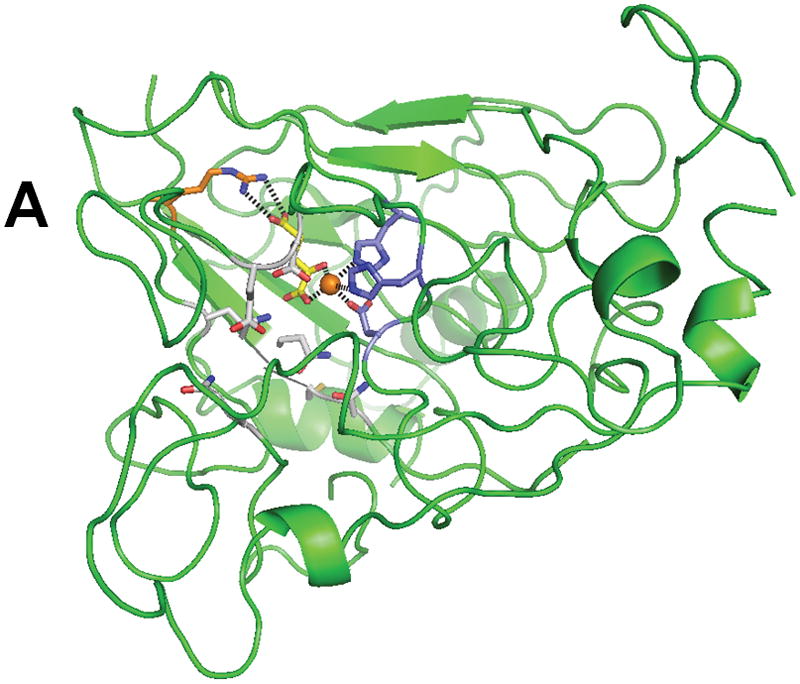
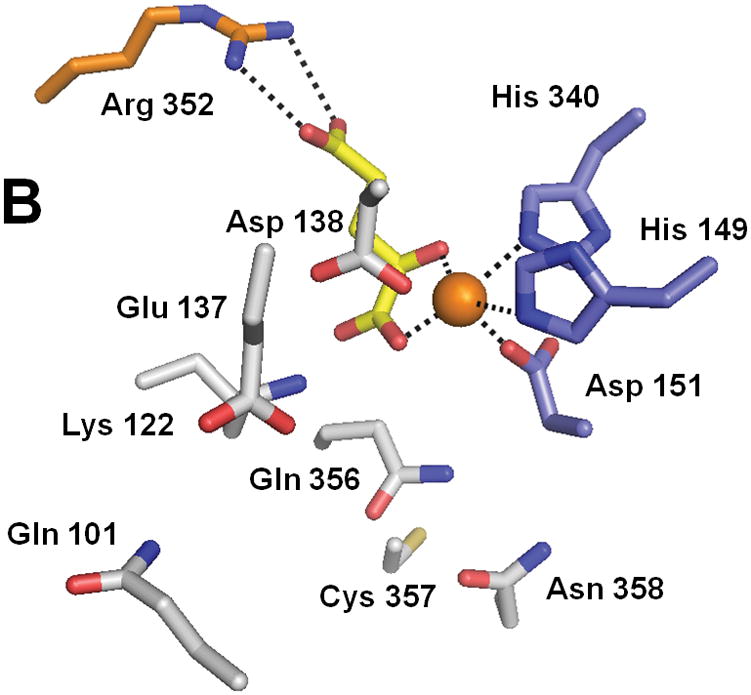
Structure of XanA according to a homology model. (A) The predicted overall fold of the XanA protein is depicted as a green ribbon. (B) A blow-up of the proposed active site. In both panels, FeII is an orange sphere, αKG has yellow carbon atoms, metal ligands have blue carbon atoms, a cofactor-stabilizing residue (Arg 352) is shown with orange carbon atoms, and other postulated active site residues are indicated with white carbon atoms.
3.0 Nucleoside hydroxylases
Two types of Fe(II)/αKG-dependent hydroxylases have been shown to utilize nucleosides as their substrates. First, a pyrimidine deoxyribonucleoside 2′-hydroxylase activity (reaction 1 in Figure 2) was noted in N. crassa, A. nidulans, and R. glutinis and shown to convert deoxythymidine or deoxyuridine to their respective ribosides (Scheme 5).35–38 Secondly, R. glutinis was shown to contain an enzyme that hydroxylates the 1′ position of uridine or deoxyuridine to release uridine as illustrated in Scheme 6.39 The genes encoding these enzymes have not been identified and the proteins have not been further characterized.
Scheme 5.

Reaction of pyrimidine deoxyribonucleoside 2′-hydroxylase with thymidine (X = CH3) or deoxyuridine (X = H)
Scheme 6.

Reaction of deoxyuridine (uridine) 1′-hydroxylase
4.0 Polynucleotide hydroxylases
4.1 Thymidine hydroxylase of protists
Another case of thymine modification is found in kinetoplastids. These protists exhibit two distinct life stages; i.e., a procyclic form within insect vectors and the bloodstream form responsible for several diseases, the most well known being African Sleeping Sickness. This disease results from the mammalian host being infected by Trypanosoma brucei which persists due to the parasite’s ability to switch its expressed surface coat glycoprotein, a process termed antigenic variation.40 T. brucei contains many variable surface glycoprotein genes that are organized into about 20 subtelomeric expression sites. Up to one percent of the thymine in the telomeric and subtelomeric repeats of nuclear DNA in the bloodstream form of T. brucei is replaced with β-D-glucosyl-hydroxymethyluracil (base J) but no modification is found in the insect form of the parasite where antigenic variation is absent.41,42 The modified base is thought to induce chromatin compaction and thereby stabilize the chromosome from rearrangements within these repetitive sequences.43,44 Moreover, base J is suggested to play a role in gene silencing related to the process of antigenic variation.45
Indirect evidence suggests that base J is synthesized in two steps (Scheme 7).46,47 First, the methyl group of thymidine must be hydroxylated to create hydroxymethyluridine (HOMedU), as evidenced by the presence of low levels of HOMedU in DNA of bloodstream form trypanosomes.48 Next, the hydroxymethyl moiety must be glucosylated. This reaction is not specific to the sequence or life cycle-stage as demonstrated by the random placement of base J in the bloodstream and procyclic forms of cells grown with HOMedU included in the medium.43 A J binding protein (JBP1) was identified in T. brucei and shown to specifically bind to base J in DNA.49 Subsequently, another trypanosomal protein was discovered to share some N-terminal sequence identity with JBP1 (along with exhibiting homology to the SWI2/SNF2 chromatin remodeling protein family) and named JBP2 even though it does not bind to J-containing DNA.50 A portion of the JBP1 and JBP2 sequences exhibit weak similarity to proteins in the FeII/αKG dioxygenase family, leading to the hypothesis that these proteins may function as thymidine hydroxylases in trypanosomes.51
Scheme 7.

Biosynthesis of base J in kinetoplastids
Recent evidence supports the proposal that JBP1 and JBP2 function as FeII/αKG-dependent hydroxylases during J biosynthesis. The developmentally regulated JBP1 is produced only in the bloodstream form of trypanosomes, whereas the insect form is devoid of this protein or base J.52 A double knockout of the gene encoding JBP1 in T. brucei, while causing no phenotype, reduces base J levels to 5% of the level found in the wild-type microorganism.52 A JBP2 knockout reduces base J levels to 20% and abolishes the trypanosome’s ability to synthesize base J in new telomeric DNA.53 Cells grown with HOMedU lose base J more slowly in the presence of JBP1 than do cells not containing this protein, consistent with a role in maintaining J.52 Secondary structure predictions suggest an all-β core sharing features with several members of the FeII/αKG dioxygenase family according to fold-recognition searches.51 Mutation of putative active-site residues in JBP1 yielded protein that was unable to complement a JBP1 null mutant to restore base J levels.51 Finally, anaerobic incubation of purified JBP1 with FeII and αKG yielded a weak absorbance feature at ~500 nm (unpublished observations) that was consistent with the metal-to-ligand charge-transfer transition observed in several other members of this enzyme family.54–56
4.2 Escherichia coli AlkB and repair of alkylated DNA/RNA
E. coli alkB has been studied for its role in the adaptive response to alkylation damage since the 1980’s.57,58 This gene was long known to confer resistance to certain methylating agents, and in 2002 the encoded protein was discovered to be a member of the FeII/αKG dioxygenases.59,60 The enzyme catalyzes N-dealkylation of 1-methyladenine (1meA) and 3-methylcytosine (3meC) in DNA by the mechanism shown in Scheme 8, creating an unstable intermediate that spontaneously releases formaldehyde to regenerate the native base. AlkB repairs the analogous lesions in RNA,61 including mRNA and tRNA.62 Furthermore, the protein dealkylates 1-methylguanine (1meG), 3-methylthymidine (3meT) and several etheno adducts of DNA.63–66
Scheme 8.

Reactions of AlkB with 1meA and 3meC in DNA
Crystal structures have been reported for E. coli AlkB in complex with FeII, αKG, and the tri-deoxynucleotide substrate T-1meA-T (Fig. 4) and for protein with bound MnII, αKG, and covalently-tethered ds-DNA (not shown).67,68 These structures reveal details of the metallocenter active site and help to clarify how the enzyme can accommodate its various substrates (ss-DNA, ds-DNA, and various forms of RNA). Residues His 131, Asp 133, and His 187 bind the metal, αKG coordinates FeII in a bidentate manner (via the C1 carboxylate and C2 keto groups), and a salt bridge from Arg 204 stabilizes the αKG C5 carboxylate. The open coordination site for oxygen binding does not point toward the substrate (a situation termed “offline” geometry);1 thus requiring a shift in position of the C1 carboxylate prior to oxygen binding or a “ferryl flip” to occur after generating the FeIV-oxo intermediate.4 The trimer substrate is bent so as to extend the damaged base deep into the active-site pocket, making ten identified hydrogen bonds and many more van der Waals contacts with the protein. The N-terminus is responsible for about half of the identified contacts with the substrate via a highly flexible nucleotide recognition domain or lid made up of 3 β-strands. This protein region is flexible based upon the superposed structures of the bound and nucleotide-free states, thus allowing movement and reorientation without losing critical contacts. Substrate specificity at the base level is provided deep in the pocket; a hydrogen bond between Arg 210 and the adenine moiety could also be made with cytosine, but not with the other two bases, explaining why AlkB prefers 1meA and 3meC as substrates. Finally, AlkB’s modest preference for ss-DNA/RNA substrates61,69 is explained by the nucleotide polymer being oriented to extend into a hydrophobic pocket with stabilization provided by base stacking with nearby aromatic residues. Binding of ds-DNA involves an inversion of the sugar ring preceding the modified nucleotide, distortion of the complementary strand, and a flipping out of the lesion to extend the methylated base into the active site.68
Fig. 4.
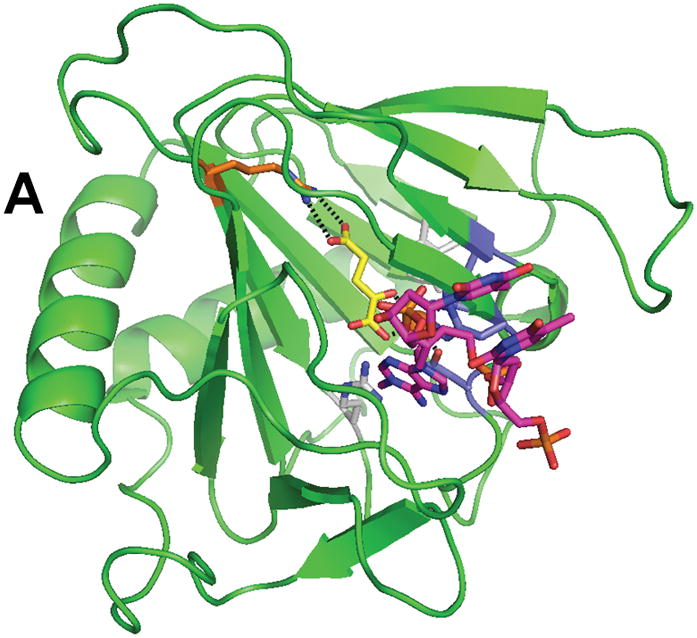
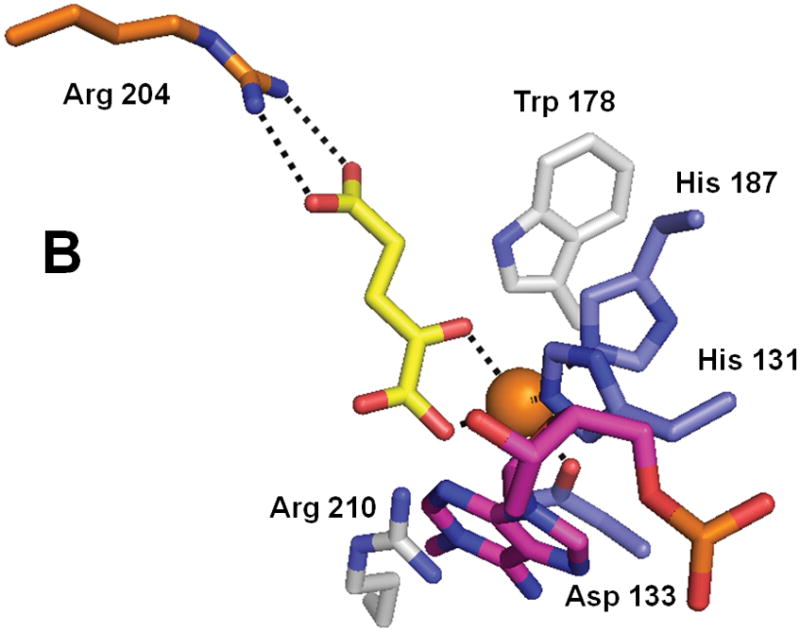
Structure of AlkB (PDB 2fd8). (A) The overall fold of AlkB. (B) The active site residues of AlkB. The coloring scheme is the same as for Figure 3, and the bound T-1meA-T substrate is shown with pink carbon atoms (the substrate is abbreviated to just 1meA in B).
Kinetic studies have examined AlkB activity using a wide range of substrates. A rate of 12 min−1 was reported for randomly methylated poly(dA),70 or about 4 min−1 for two specifically mono-methylated oligonucleotides.71 Catalytic rates with the tri-deoxynucleotide T-1meA-T used in the crystal structure studies were determined separately by two groups and found to be 7.4 and 4.5 min−1, respectively.67,70 A phosphate 5′ of the methylated base is critical for proper recognition based upon a 6-fold increase in the apparent Km when it is missing,70 and this result is compatible with the crystal structure which shows both a hydrogen bond and a salt bridge involving this 5′ phosphate.67
In addition to the dealkylation reactions observed with the above substrates, AlkB is known to exhibit additional types of reactivity. The minimal substrate that could be demethylated was identified as 1-me-dAMP,70 whereas the nucleosides 1-meA, 1-me-2′-dA, 3-meC, and 3-me-2′-dC were shown to stimulate αKG decomposition without undergoing demethylation.72 In the absence of nucleotide, but the presence of αKG and O2, AlkB hydroxylates its own Trp 178 (located near the active site, see Fig. 4) to generate a hydroxy-tryptophan side chain. A ligand-to-metal charge-transfer transition between this modified residue and the oxidized metal site results in a blue protein that absorbs maximally at 595 nm.73
4.3 Mammalian AlkB homologues
Eight human homologues of AlkB, named ABH1 through ABH8, have been shown to exist.74 The best studied of these, ABH2 and ABH3, catalyze the same reaction as AlkB (Scheme 8), i.e., the oxidative repair of methylated polynucleotides with concomitant decarboxylation of αKG producing the native base, formaldehyde, succinate and CO2.61,62,69,75,76 Genes encoding these enzymes can complement an alkB defective E. coli strain to restore resistance to the methylating agent methylmethanesulfonate. In vitro assays with the purified dioxygenases show that ABH2 prefers ds-DNA, whereas ABH3 exhibits greater activity on RNA and ss-DNA. Kinetic measurements demonstrated that the enzymes are well adapted, with activity toward their substrates approaching the diffusion limit.76 The two enzymes differ slightly in their tissue distribution, but both human homologues are highly expressed in testis, whereas the corresponding mouse homologues are largely expressed in the heart.75,76 Of interest, their subcellular localization is different; ABH2 is exclusively nuclear while ABH3 was found both in the nucleus and in the cytosol.61 ABH2 and ABH3 knock-out mice were created, shown to be viable, and used to demonstrate that repair of double-stranded DNA is mostly dependent on ABH3 whereas ABH2 might have a role in mRNA repair.77 ABH2 also exists in a form lacking exon 2, resulting in a frame-shift deletion of the C-terminal FeII/αKG-binding domain; the function of the truncated ABH2 is unknown.78
ABH2 was crystallized with MnII, αKG, and covalently-tethered DNA,68 providing a 2.5 A resolution structure (Fig. 5). The enzyme retains the overall fold of AlkB, coordinates the metal ion using His 171, Asp 173, and His 236, binds the C5 carboxylate of αKG via Arg 248, and possesses extra loops that bind the complementary strand of ds-DNA. Of particular interest is Phe 102 which intercalates into the DNA to replace both the methylated base that is flipped out from the duplex DNA plus its complementary base that is disordered.
Fig. 5.


Structure of ABH2 (PDB 3buc). (A) The overall fold of ABH2 with tethered DNA containing 1meA. (B) The active site residues of ABH2 with 1meA. To reduce oxygen reactivity, the active site FeII was replaced by MnII (depicted as a gray sphere). The rest of the coloring scheme is the same as above.
The crystal structure of ABH3 was solved in complex with iron and αKG at 1.5 A resolution (Fig. 6).79 The structure contains the typical core of FeII/αKG-dependent dioxygenases, and includes the iron-binding ligands His 191, Asp 193 and His 257 as well as Arg 269 which forms a salt bridge with αKG. A hairpin formed by β4 and β5 creates a lid over the active site and a positively charged groove constitutes the DNA and RNA binding cleft. Superpositioning of the ABH3 structure with ABH2 or AlkB reveals significant structural differences for substrate binding.68,79 In particular, ABH3 lacks the extra DNA binding loops of ABH2 that are used to recognize the complementary strand of ds-DNA. Furthermore, ABH3 has a more polar substrate binding pocket than AlkB which could partly explain the more restricted substrate specificity of this protein. In addition, very few of the residues making contact with the sugar-phosphate backbone are conserved between the latter two enzymes. Whereas the tri-deoxynucleotide T-1meA-T adopts a rather unusual conformation at the active site of AlkB, this conformation is precluded by steric hindrances in ABH3. The authors suggest that ABH3 binds its DNA substrate in a direction opposite to that in AlkB.79 In contrast to the primary substrate, αKG is bound similarly in these dioxygenases. The crystal structure also reveals several new key residues important for ABH3 activity. One feature consists of three negatively charged residues, Glu 123, Asp 189 and Asp 194, located at the entrance to the DNA/RNA binding groove. Experiments with the E123A mutant showed 5-fold increased activity with ds-DNA but only 1.5-fold with ss-DNA suggesting that this residue discriminates against ds-DNA substrates. Another important residue is Arg 122 whose side-chain is ideally positioned to intercalate into the DNA and thereby take part in nucleotide flipping. Leu 177 occupies a central position in the active site and was proposed to act as a ‘buffer stop’ to prevent penetration of alkylated purines too deeply into the catalytic pocket. Of additional interest and perhaps parallel to the self-hydroxylation of Trp 187 in AlkB, Leu 177 of ABH3 was found to be partially oxidized at C4 (to a mixture of the hydroxyl and carbonyl species), even when purified anaerobically.
Fig. 6.
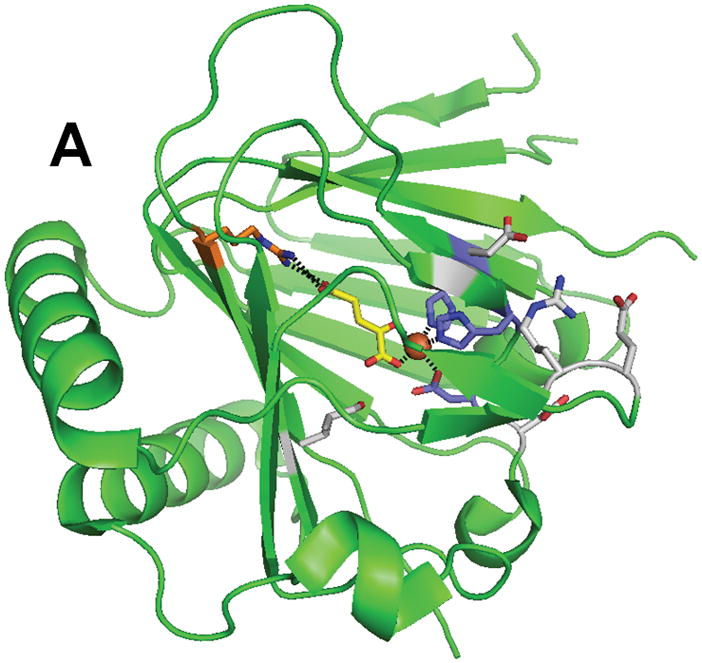
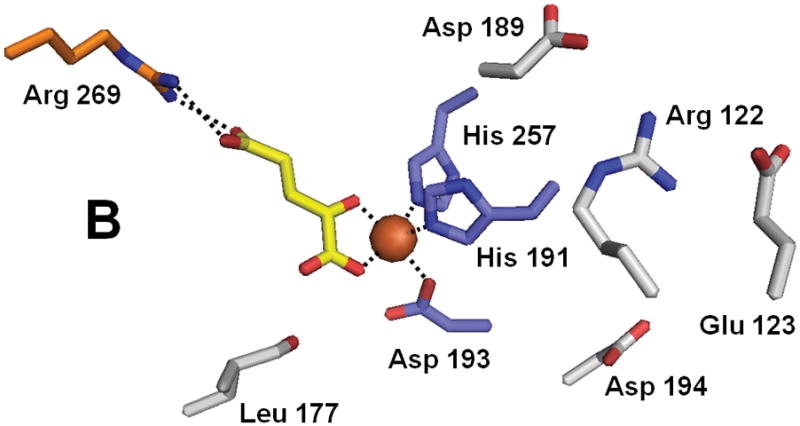
Structure of ABH3 (PDB 2iuw). (A) The overall fold of ABH3. (B) A blow-up of the ABH3 active site with coloring as indicated above. Note that Leu177 an oxidized species.
The functions of ABH1 and ABH4-ABH8 have not been elucidated. Of all the mammalian homologues ABH1 is closest in sequence to AlkB; however, demethylation activity was not detected.75–77 Similarly ABH4, ABH6, and ABH7 do not exhibit demethylation activity, while ABH5 and ABH8 were found to be hard to purify after being overproduced in E. coli, and, to our knowledge, no activity measurements have been reported.76 Just recently, a new, longer form of ABH8 was identified and shown to include an RNA recognition motif and a methyltransferase domain.78 In the same study, the cellular expression of all human AlkB homologues was studied with GFP-fusion proteins; ABH1 and ABH4-ABH7 were observed throughout the cell, whereas ABH8 was detected only in the cytoplasm. Real-time PCR experiments showed that all human AlkB homologues are expressed ubiquitously in the tissues examined. ABH1 is most highly expressed in spleen and all homologues showed elevated expression in pancreas and testis. In another study, an ABH1−/− knockout mouse was created.80 These knockout mice are viable and fertile, but show intra-uterine growth retardation and placental defects. It was further shown with yeast two hybrid assays that ABH1 interacts with Mrj, a mouse homolog of the E. coli DNA co-chaperone DnaJ. These data suggest that ABH1 might play a role in regulation of gene expression.
5.0 Chromatin hydroxylases
Several FeII/αKG-dependent enzymes have been shown to exploit oxidative chemistry to remove methylation tags on chromatin-associated proteins. The first published example of this activity involved JMDM1 (JmjC domain-containing histone demethylase 1) that demethylates dimethyl- and monomethyl-Lys at position 36 of histone 3.81 This work was quickly followed by a plethora of reports (not listed) describing related methyl-Lys demethylases that act at varied positions of selected histone tails, many capable of demethylating trimethyl-Lys. In addition, JMJD6 was shown to function as a methyl-Arg demethylase.82 In all cases, the enzymes are suggested to utilize a subset of the chemistries shown in Schemes 9 and 10. Reversal of monomethyl-Lys in histones also occurs by the flavoprotein amine oxidase LSD1.83
Scheme 9.

Reactions of methyl-Lys demethylases involved in removal of methyl groups from specific Lys residues of certain histones
Scheme 10.

Reactions of methyl-Arg demethylases involved in removal of methyl groups from specific Arg residues in selected histones
Structural studies have provided key insights into the mechanism of catalysis and specificity of the JMJD2A histone demethylase that recognizes dimethyl- and trimethyl-Lys at positions 9 or 36 of histone 3 (Fig. 7). The catalytic core of this protein folds into the jellyroll-like structure typical of this enzyme family, with additional domains including a zinc-finger motif.84 His 188, Glu 190, and His 276 serve as metal ligands, and αKG (or in the case shown, the αKG homologue N-oxalylglycine), stabilized at C5 by interactions with Tyr 132 and Lys 206 rather than by a salt bridge to an Arg, exhibits the expected bidentate coordination. Complexes of the catalytic domain with various peptides containing methylated Lys residues highlight the mechanism of substrate specificity, including the preference for a multiply-methylated peptide.85–87 The open coordination site for oxygen binding is directed towards the methyl group to be hydroxylated; i.e., this structure depicts an “on-line geometry” for hydroxylation.
Fig. 7.
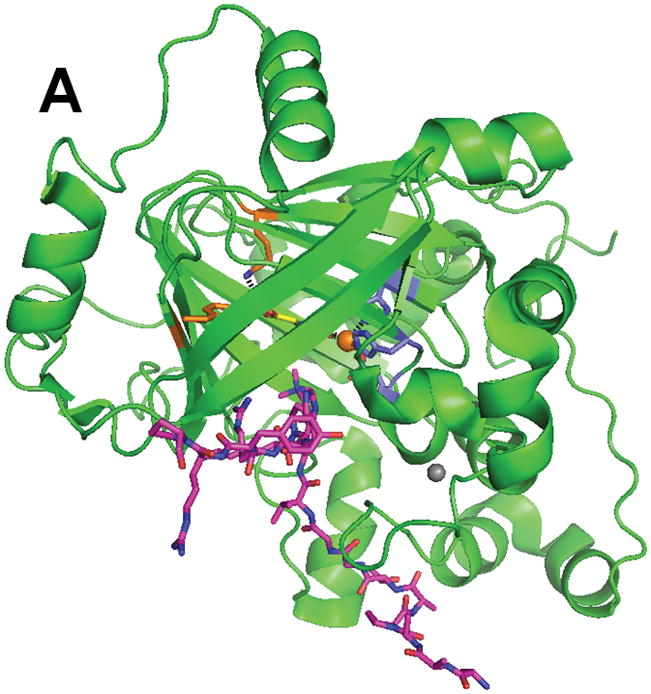
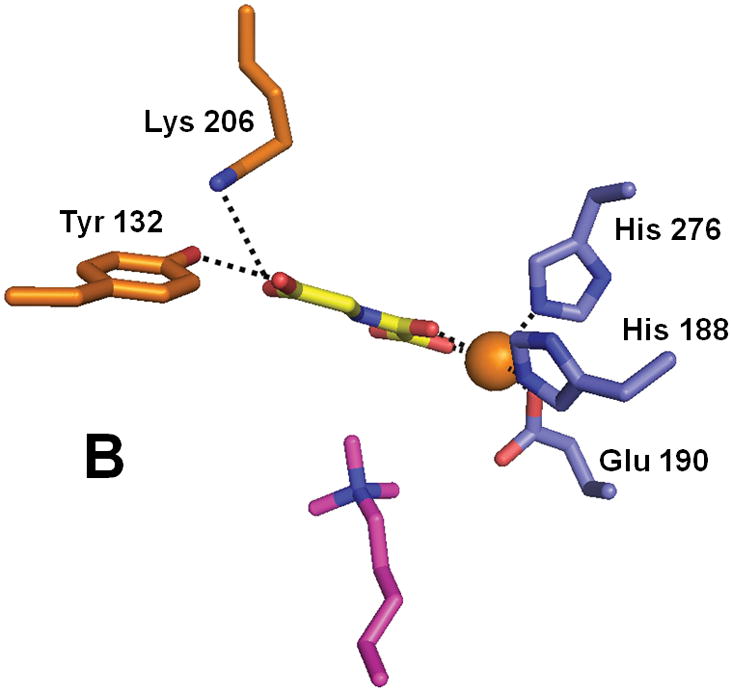
Structure of the histone demethylase JMJD2A (PDB 2p5b). (A) The overall view of the histone demethylase, with bound zinc shown as a grey sphere, the peptide substrate shown with pink carbons, and the αKG homologue N-oxalylglycine replacing the cosubstrate. (B) The active site of JMJD2A with just the trimethyl-Lys portion of the substrate shown. The coloring scheme is the same above.
6.0 Conclusions and outlook
The FeII/αKG-dependent dioxygenases represent a diverse family of enzymes that catalyze a wide range of oxidizing reactions,1,2 only a subset of which act on nucleobases, nucleosides, nucleotides, and chromatin. These proteins all contain a similar core fold and use analogous chemistry to hydroxylate their substrates. Additional representatives of this family are likely to be uncovered. For example, the FTO gene product, implicated in the onset of obesity in humans, was predicted by sequence analysis to be a member of this enzyme superfamily.88 Indeed, the murine FTO protein (found to be a nuclear protein with the highest expression in the hypothalamus, a part of the brain that regulates energy homeostasis) was shown to exhibit FeII, αKG, and oxygen-dependent demethylation activity with 1meT.89 The relevance of this dealkylation activity and the physiological function of this protein remain unknown. Other components of the chromatin, such as various regulatory proteins, are potential candidates for future inclusion in this protein family. In this regard, the hypoxia-inducible factor (HIF), a transcription factor involved in the hypoxic response, is regulated by several FeII/αKG-dependent hydroxylases that target specific prolyl and asparaginyl side chains in the proteins.90,91 The structures of a HIF-specific prolyl hydroxylase92 and a HIF-specific asparaginyl hydroxylase (also called factor-inhibiting HIF, FIH)93–95 have been elucidated.
Despite the prevalence of FeII/αKG-dependent hydroxylases in oxidation reactions of components of chromatin, nucleotides, nucleosides, and nucleobases, it is important to keep in mind that other classes of enzymes catalyze similar chemistry. One example already noted involves xanthine metabolism by the Moco-containing enzyme xanthine oxidoreductase; related Moco-hydroxylases potentially could be uncovered with specificity toward other related substrates.30 In the same manner, the oxygenase chemistry described here could alternatively be achieved by use of cytochrome P450s,96 di-iron hydroxylases,97 Rieske-type iron-containing oxygenases,98 pterin-dependent hydroxylases,99 flavoprotein monooxygenases,100 and other catalysts.101 Thus, biochemical and genetic studies will continue to be important to identify the hydroxylases responsible for such transformations in other organisms.
Acknowledgments
Studies related to this topic in the Hausinger laboratory are supported by NIH grant GM063584.
Biographies
Jana M. Simmons is a graduate student who received her B.S. in Biochemistry from Alma College in 2004. She examines the roles of putative thymine and thymidine hydroxylases from Trypanosoma brucei to identify whether these proteins participate in formation of the novel J base found in these protozoa.

Tina A. Müller received an M.S. in Environmental Sciences (1999) and a Ph.D. in Microbiology (2004) at the Swiss Federal Institute of Technology studying the degradation of phenoxypropionate herbicides. She has characterized several FeII/α-ketoglutarate hydroxylases since joining the Hausinger lab as a post-doctoral scientist in 2004 and is currently a Research Assistant Professor studying the function of the human AlkB homologue 1.

Robert P. Hausinger received his Ph.D. with Jim Howard at the University of Minnesota in 1982. After post-doctoral training with Chris Walsh at M.I.T. (1982–1984) he joined the faculty at Michigan State University where he is Professor of Biochemistry and Microbiology, and Director of the Quantitative Biology Program. His research focuses on the mechanisms of metalloenzyme biosynthesis and the catalytic mechanisms and versatility of FeII/α-ketoglutarate hydroxylases.

References
- 1.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Purpero V, Moran GR. J Biol Inorg Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- 3.Loenarz C, Schofield CJ. Nature Chemical Biology. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 4.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. J Inorg Biochem. 2006;100:644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 5.Hanauske-Abel HM, Günzler V. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 6.Hegg EL, Que L., Jr E J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 7.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 8.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 9.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 10.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 11.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Proc Natl Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. J Am Chem Soc. 2007;129:13408–13409. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 13.Abbott MT, Schandl EK, Lee RF, Parker TS, Midgett RJ. Biochim Biophys Acta. 1967;132:525–528. doi: 10.1016/0005-2744(67)90177-5. [DOI] [PubMed] [Google Scholar]
- 14.Holme E, Lindstedt G, Lindstedt S, Tofft M. Biochim Biophys Acta. 1970:212. doi: 10.1016/0005-2744(70)90177-4. [DOI] [PubMed] [Google Scholar]
- 15.Holme E, Lindstedt G, Lindstedt S, Tofft M. J Biol Chem. 1971;246:3314–3319. [PubMed] [Google Scholar]
- 16.Liu CK, Hsu CA, Abbott MT. Arch Biochem Biophysics. 1973;159:180–187. doi: 10.1016/0003-9861(73)90443-8. [DOI] [PubMed] [Google Scholar]
- 17.Wondrack LM, Hsu CA, Abbott MT. J Biol Chem. 1978;253:6511–6515. [PubMed] [Google Scholar]
- 18.Smiley JA, Kundracik M, Landfried DA, Barnes VR, Sr, Axhemi AA. Biochim Biophys Acta. 2005;1723:256–264. doi: 10.1016/j.bbagen.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Holme E. Biochemistry. 1975;14:4999–5003. doi: 10.1021/bi00693a033. [DOI] [PubMed] [Google Scholar]
- 20.Bankel L, Lindstedt G, Lindstedt S. Biochim Biophys Acta. 1977;481:431–437. doi: 10.1016/0005-2744(77)90276-5. [DOI] [PubMed] [Google Scholar]
- 21.Thornburg LD, Lai MT, Wishnok JS, Stubbe J. Biochemistry. 1993;32:14023–14033. doi: 10.1021/bi00213a036. [DOI] [PubMed] [Google Scholar]
- 22.Shaffer PM, Hsu CA, Abbott MT. J Bacteriol. 1975;121:648–655. doi: 10.1128/jb.121.2.648-655.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thornburg LD, Stubbe J. Biochemistry. 1993;32:14034–14042. doi: 10.1021/bi00213a037. [DOI] [PubMed] [Google Scholar]
- 24.Lai MT, Wu W, Stubbe J. J Am Chem Soc. 1995;117:5023–5030. [Google Scholar]
- 25.Holme E. Biochim Biophys Acta. 1982;707:259–266. doi: 10.1016/0167-4838(82)90359-4. [DOI] [PubMed] [Google Scholar]
- 26.Hsu CA, Saewert MD, Polsinelli LF, Jr, Abbott MT. J Biol Chem. 1981;256:6098–6101. [PubMed] [Google Scholar]
- 27.Myllylä R, Majamaa K, Günzler V, Hanauske-Abel HN, Kivirikko KI. J Biol Chem. 1984;259:5403–5405. [PubMed] [Google Scholar]
- 28.Puistola U, Turpeenniemi-Hujanen TM, Myllylä R, Kivirikko K. Biochim Biophys Acta. 1980;611:40–50. doi: 10.1016/0005-2744(80)90040-6. [DOI] [PubMed] [Google Scholar]
- 29.Tuderman L, Myllylä R, Kivirikko KI. Eur J Biochem. 1977;80:314–348. doi: 10.1111/j.1432-1033.1977.tb11889.x. [DOI] [PubMed] [Google Scholar]
- 30.Hille R. Arch Biochem Biophys. 2005;433:107–116. doi: 10.1016/j.abb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Darlington AJ, Scazzocchio C. Biochim Biophys Acta. 1968;166:569–571. doi: 10.1016/0005-2787(68)90244-x. [DOI] [PubMed] [Google Scholar]
- 32.Cultrone A, Scazzocchio C, Rochet M, Montero-Morán G, Drevet C, Fernàndez-Martìn R. Molec Microbiol. 2005;57:276–290. doi: 10.1111/j.1365-2958.2005.04686.x. [DOI] [PubMed] [Google Scholar]
- 33.Montero-Morán GM, Li M, Rendon-Huerta E, Jourdan F, Lowe DJ, Stumpff-Kane AW, Feig M, Scazzocchio C, Hausinger RP. Biochemistry. 2007;46:5293–5304. doi: 10.1021/bi700065h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Müller TA, Fraser BA, Hausinger RP. Arch Biochem Biophysics. 2007;470:44–53. doi: 10.1016/j.abb.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaffer PM, McCroskey RP, Palmatier RD, Midgett RJ, Abbott MT. Biochem Biophys Res Commun. 1968;33:806–811. doi: 10.1016/0006-291x(68)90232-5. [DOI] [PubMed] [Google Scholar]
- 36.Bankel L, Holme E, Lindstedt G, Lindstedt S. FEBS Lett. 1972;21:135–138. doi: 10.1016/0014-5793(72)80121-2. [DOI] [PubMed] [Google Scholar]
- 37.Shaffer PM, Cairns JR, Dorman DC, Lott AM. Int J Biochem. 1984;16:429–434. doi: 10.1016/0020-711x(84)90143-5. [DOI] [PubMed] [Google Scholar]
- 38.Warn-Cramer BJ, Macrander LA, Abbott MT. J Biol Chem. 1983;258:10551–10557. [PubMed] [Google Scholar]
- 39.Stubbe J. J Biol Chem. 1985;260:9972–9975. [PubMed] [Google Scholar]
- 40.Horn D, Barry JD. Chromosome Res. 2005;13:525–533. doi: 10.1007/s10577-005-0991-8. [DOI] [PubMed] [Google Scholar]
- 41.Gommers-Ampt JH, Van Leeuwen F, de Beer AL, Vliegenthart JF, Dizdaroglu M, Kowalak JA, Crain PF, Borst P. Cell. 1993;75:1129–1136. doi: 10.1016/0092-8674(93)90322-h. [DOI] [PubMed] [Google Scholar]
- 42.van Leeuwen F, Taylor MC, Mondragon A, Moreau H, Gibson W, Kieft R, Borst P. Proc Natl Acad Sci USA. 1998;95:2366–2371. doi: 10.1073/pnas.95.5.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Leeuwen F, Kieft R, Cross M, Borst P. Mol Cell Biol. 1998;18:5643–5651. doi: 10.1128/mcb.18.10.5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Leeuwen F, Kieft R, Cross M, Borst P. Mol Biochem Parasitol. 2000;109:133–145. doi: 10.1016/s0166-6851(00)00247-4. [DOI] [PubMed] [Google Scholar]
- 45.van Leeuwen F, Wijsman ER, Kieft R, van der Marel GA, van Boom JH, Borst P. Genes Dev. 1997;11:3232–3241. doi: 10.1101/gad.11.23.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulbert S, Cross M, Boorstein RJ, Teebor GW, Borst P. Nucleic Acids Res. 2002;30:3919–3926. doi: 10.1093/nar/gkf533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulbert S, Eide L, Seeberg E, Borst P. DNA Repair (Amst) 2004;3:145–154. doi: 10.1016/j.dnarep.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Gommers-Ampt JH, Teixeira AJ, van de Werken G, van Dijk WJ, Borst P. Nucleic Acids Res. 1993;21:2039–2043. doi: 10.1093/nar/21.9.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cross M, Kieft R, Sabatini R, Wilm M, de Kort M, van der Marel GA, van Boom JH, van Leeuwen F, Borst P. EMBO J. 1999;18:6573–6581. doi: 10.1093/emboj/18.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiPaolo C, Kieft R, Cross M, Sabatini R. Molec Cell. 2005;17:441–451. doi: 10.1016/j.molcel.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 51.Yu Z, Genest PA, ter Riet B, Sweeney K, DiPaolo C, Kieft R, Christodoulou E, Perrakis A, Simmons JM, Hausinger RP, van Luenen HG, Rigden DJ, Sabatini R, Borst P. Nucleic Acids Res. 2007;35:2107–2115. doi: 10.1093/nar/gkm049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cross M, Kieft R, Sabatini R, Dirks-Mulder A, Chaves I, Borst P. Molec Microbiol. 2002;46:37–47. doi: 10.1046/j.1365-2958.2002.03144.x. [DOI] [PubMed] [Google Scholar]
- 53.Kieft R, Brand V, Ekanayake DK, Sweeney K, DiPaolo C, Reznikoff WS, Sabatini R. Mol Biochem Parasitol. 2007;156:24–31. doi: 10.1016/j.molbiopara.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 1998;120:743–753. [Google Scholar]
- 55.Ryle MJ, Padmakumar R, Hausinger RP. Biochemistry. 1999;38:15278–15286. doi: 10.1021/bi9912746. [DOI] [PubMed] [Google Scholar]
- 56.Hegg EL, Whiting AK, Saari RE, McCracken J, Hausinger RP, Que L., Jr Biochemistry. 1999;38:16714–16726. doi: 10.1021/bi991796l. [DOI] [PubMed] [Google Scholar]
- 57.Kataoka H, Yamamoto Y, Sekiguchi M. J Bacteriol. 1983;153:1301–1307. doi: 10.1128/jb.153.3.1301-1307.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kondo H, Nakabeppu Y, Kataoka H, Kuhara S, Kawabata S, Sekiguchi M. J Biol Chem. 1986;261:15772–15777. [PubMed] [Google Scholar]
- 59.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 60.Falnes PO, Johansen RF, Seeberg E. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 61.Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, Sundheim O, Bjoras M, Slupphaug G, Seeberg E, Krokan HE. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 62.Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PO. Molec Cell. 2004;16:107–116. doi: 10.1016/j.molcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 63.Koivisto P, Robins P, Lindahl T, Sedgwick B. J Biol Chem. 2004;279:40470–40474. doi: 10.1074/jbc.M407960200. [DOI] [PubMed] [Google Scholar]
- 64.Delaney JC, Essigmann JM. Proc Natl Acad Sci USA. 2004:101. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mishina Y, Yang CG, He C. J Am Chem Soc. 2005;127:14594–14595. doi: 10.1021/ja055957m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaney JC, Smeester L, Wong C, Frick LE, Taghizdeh K, Wishnok JS, Drennan CL, Samson LD, Essigmann JM. Nature Struct Molec Biol. 2005;12:855–860. doi: 10.1038/nsmb996. [DOI] [PubMed] [Google Scholar]
- 67.Yu B, Edstrom WC, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 68.Yang CG, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Falnes PO, Bjoras M, Aas PA, Sundheim O, Seeberg E. Nucleic Acids Res. 2004;32:3456–3461. doi: 10.1093/nar/gkh655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koivisto P, Duncan T, Lindahl T, Sedgwick B. J Biol Chem. 2003;278:44348–44354. doi: 10.1074/jbc.M307361200. [DOI] [PubMed] [Google Scholar]
- 71.Roy TW, Bhagwat AS. Nucleic Acids Res. 2007;35:e147. doi: 10.1093/nar/gkm1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Welford RWD, Schlemminger I, McNeill LA, Hewitson KS, Schofield CJ. J Biol Chem. 2003;278:10157–10161. doi: 10.1074/jbc.M211058200. [DOI] [PubMed] [Google Scholar]
- 73.Henshaw TF, Feig M, Hausinger RP. J Inorg Biochem. 2004;98:856–861. doi: 10.1016/j.jinorgbio.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 74.Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. BMC Genomics. 2003;4:48. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Proc Natl Acad Sci USA. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O’Connor TR. J Biol Chem. 2005;280:39448–39459. doi: 10.1074/jbc.M509881200. [DOI] [PubMed] [Google Scholar]
- 77.Ringvoll J, Nordstrand LM, Vagbo CB, Talstad V, Reite K, Aas PA, Lauritzen KH, Liabakk NB, Bjork A, Doughty RW, Falnes PO, Krokan HE, Klungland A. EMBO J. 2006;25:2189–2198. doi: 10.1038/sj.emboj.7601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, Tsuchiya M, Makino Y, Furukawa T, Konishi N, Yamamoto H. J Cell Mol Med. 2007;11:1105–1116. doi: 10.1111/j.1582-4934.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sundheim O, Vagbo CB, Bjoras M, Sousa MM, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA, Slupphaug G. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pan Z, Sikandar S, Witherspoon M, Dizon D, Nguyen T, Benirschke K, Wiley C, Vrana P, Lipkin SM. Dev Dyn. 2007 doi: 10.1002/dvdy.21418. [DOI] [PubMed] [Google Scholar]
- 81.Tsukada Y-I, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 82.Chang B, Chen Y, Zhao Y, Bruick RK. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 83.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 84.Chen Z, Zang J, Whetstine JR, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 85.Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine JR, Dai S, Hansen K, Shi Y, Zhang G. Proc Natl Acad Sci USA. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 87.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Nature Struct Molec Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 88.Sanchez-Pulido L, Andrade-Navarro MA. BMC Biochem. 2007;8:23. doi: 10.1186/1471-2091-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dann CE, III, Bruick RK. Biochemical and Biophysical Research Communications. 2005;338:639–647. doi: 10.1016/j.bbrc.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 91.Schofield CJ, Ratcliffe PJ. Biochemical and Biophysical Research Communications. 2005;338:617–626. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 92.McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Liénard BMR, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, McNeill LA, Kurzeja RJM, Hewitson KS, Yang E, Jordan S, Syed RS, Schofield CJ. Proceedings of the National Academy of Sciences. 2006;103:9814–9819. doi: 10.1073/pnas.0601283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dann CE, III, Bruick RK, Deisenhofer J. Proceedings of the National Academy of Sciences. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Journal of Biological Chemistry. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 95.Lee C, Kim SJ, Jeong DG, Lee SM, Ryu SE. Journal of Biological Chemistry. 2003;278:7558–7563. doi: 10.1074/jbc.M210385200. [DOI] [PubMed] [Google Scholar]
- 96.Sono M, Roach MP, Coulter ED, Dawson JH. Chemical Reviews. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 97.Murray LJ, Lippard SJ. Accounts of Chemical Research. 2007;40:466–474. doi: 10.1021/ar600040e. [DOI] [PubMed] [Google Scholar]
- 98.Schneider D, Schmidt CL. Biochimica et Biophysica Acta. 2005;1710:1–12. doi: 10.1016/j.bbabio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 99.Fitzpatrick PF. Annual Review of Biochemistry. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 100.van Berkel WJ, Kamerbeek NM, Fraaije MW. Journal of Biotechnology. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 101.Ullrich R, Hofrichter M. Cell Mol Life Sci. 2007;64:271–293. doi: 10.1007/s00018-007-6362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]