

Abstract
The rising prevalence of complex disease suggests that alterations to the human environment are increasing the proportion of individuals who exceed a threshold of liability. This might be due either to a global shift in the population mean of underlying contributing traits, or to increased variance of such underlying endophenotypes (such as body weight). To contrast these quantitative genetic mechanisms with respect to weight gain, we have quantified the effect of dietary perturbation on metabolic traits in 146 inbred lines of Drosophila melanogaster and show that genotype-by-diet interactions are pervasive. For several metabolic traits, genotype-by-diet interactions account for far more variance (between 12 and 17%) than diet alone (1–2%), and in some cases have as large an effect as genetics alone (11–23%). Substantial dew point effects were also observed. Larval foraging behavior was found to be a quantitative trait exhibiting significant genetic variation for path length (P = 0.0004). Metabolic and fitness traits exhibited a complex correlation structure, and there was evidence of selection minimizing weight under laboratory conditions. In addition, a high fat diet significantly increases population variance in metabolic phenotypes, suggesting decreased robustness in the face of dietary perturbation. Changes in metabolic trait mean and variance in response to diet indicates that shifts in both population mean and variance in underlying traits could contribute to increases in complex disease.
METABOLIC syndrome (MetS) is a complex disease that is promoted by interactions between genetic and environmental effects (O'Rahilly and Farooqi 2006), and seems to be increasing in prevalence in response to a transition from traditional toward Westernized lifestyles (Lee et al. 2004; Schulz et al. 2006). MetS is a constellation of metabolic symptoms including insulin resistance, abdominal obesity, and dyslipidemia, and is predictive of cardiovascular disease and type-2 diabetes (Alberti et al. 2006). The condition has reached epidemic proportions in many Westernized countries (Isomaa et al. 2001; Ford and Giles 2003; Lorenzo et al. 2003; Alberti et al. 2006). Not all individuals are susceptible to the deleterious effects of a Westernized lifestyle, but some individuals are very sensitive to the effects of their environment (Schulz et al. 2006).
We argued previously that environmental perturbation contributes to the recent increases in chronic disease in Westernized societies by exposing cryptic genetic variation, a phenomenon that may be particularly evident in metabolic syndrome (Gibson 2009). Increases in complex disease after an environmental shift can be caused by both a change in the population mean or increased variance in a predisposing underlying trait, or endophenotype, causing a larger portion of the population to exceed a disease threshold (Gibson and Reed 2008). Endophenotypes can be molecular, such as rate of uptake of glucose into cells, but also include visible disease covariates, such as body mass. The transition from traditional diets and lifestyles may have perturbed our metabolic homeostasis, thereby promoting increased susceptibility to, and in turn prevalence of, obesity, hyperlipidemia, diabetes, and cardiovascular illness.
The complexity of genetic and environmental interactions leads to major challenges in successful disease treatment and prevention strategies, in that it is very difficult to accurately model the relative contributions of nature and nurture to disease susceptibility in a human population. Dietary factors have been demonstrated to interact with specific genetic variants to increase the risk of metabolic disease in humans (Corella and Ordovas 2005; Ordovas 2006; Corella et al. 2009; Warodomwichit et al. 2009), but the relative contribution of overall genotype and environmental effects on human variation is difficult to determine. Modeling population level genotype-by-environment interactions using a model organism like Drosophila can compensate for the research challenges of parameter estimation in human populations.
Drosophila share great homology to humans in a number of systems including central metabolism, insulin-signaling pathways, and organs responsible for physiological homoeostasis (e.g., heart, liver, and kidney) (Rizki 1978; Bodmer 1995; Nation 2002; Rulifson et al. 2002; Denholm et al. 2003; Wessells et al. 2004). It has been shown that Drosophila with ablated insulin-producing neurons have elevated hemolymph trehalose levels, considered to parallel a diabetic phenotype (Rulifson et al. 2002). Loss of insulin signaling also restores normal rhythmicity of adult heart rate in old flies (Wessells et al. 2004), providing a link between the obesity and cardiac components of MetS. We have used 146 natural genetic isolates of Drosophila melanogaster to model the relative contributions of genetics, diet, and other environmental effects on the MetS-like phenotypes of larval weight gain, blood sugar concentration, lipid storage, and survival. Individuals from each of these genetic lines were raised on four different diets: their normal lab food, a calorie restricted (0.75% glucose) food, a high (4%) glucose food, and a high fat diet containing (3%) coconut oil.
Using this approach, we sought evidence pertaining to two major hypotheses. There are six general types of phenotypic reaction scenarios that a genetically variable population can exhibit in response to an environmental transition: (1) no phenotypic variation in response to genetic or environmental factors, (2) genetic variation in mean phenotype but no change across environments, (3) an additive change in phenotypic mean across genotypes between environments, (4) an interaction effect between genetic and environment leading to a crossing of line means, and (5) a decrease or (6) an increase in variance in the new environment (Gibson and Vanhelden 1997). First, we considered whether the predominant source of metabolic variation within a Drosophila population is genetic, environmental, or the interaction between genetic and environmental effects. Our null was that none of these factors significantly influence weight gain or other phenotypes (scenario 1 above), but it was expected that genetic variation would be prevalent. The more fundamental issue is which of two alternate hypotheses apply: that dietary effects are essentially additive across genotypes (3) or that they are largely genotype dependent (4), possibly also with contributions of behavior and the external environment. Second, we considered whether the transition from a standard laboratory diet to a perturbing diet would change the environmental and/or genetic variance observed in the population: a decrease (5) indicating physiological limitation to the variation or an increase (6) indicating decanalization of the metabolic phenotypes due to loss of physiological buffering.
MATERIALS AND METHODS
Experimental lines:
The 146 experimental lines used in this study were derived from inbred isofemale lines originally collected from two wild populations in the summer of 2004, one from West End, North Carolina, and the other collected by Martin Kreitman near Cherryfield, Maine. Lines were inbred by 15–20 generations of full-sib mating. The specific estimates of genetic effects in this model are not directly applicable to outbred populations because inbreeding has eliminated dominance effects. Nevertheless, the broad patterns of genetic variation observed in this model do estimate the additive and a subset of epistatic effects present in outbred populations.
Dietary treatments:
Larvae were raised from early first instars through late third instars (for lipid and hemolymph carbohydrate measurements) and mature pupae for weight and survival measurements at a density of 50 per vial on their respective treatment diet. Sets of five vials were started at the same time for each treatment (genetic line and diet) and replicate, from which three vials were used for the larval phenotypes and two for pupal phenotypes.
The maximum rate of development in lab-based strains is achieved with a sugar concentration by weight of 0.75% (Sang 1956). A diet of 4% sugar causes a decrease in development rate indicating decreased larval health (Sang 1956), and a diet of 3% fat causes a dramatic decrease in adult life span (Driver and Cosopodiotis 1979). The primary sugar in most laboratory Drosophila diets is sucrose (a disaccharide of fructose and glucose) in the form of molasses, but the sugars commonly available to flies in the wild in the fruits they eat are glucose and fructose. We chose glucose to modify sugar content to address the direct role of dietary glucose on the insulin-signaling pathway, as well as to reflect the more natural dietary condition. Coconut oil was used for the fat due to its high (90%) concentration of saturated fatty acids and because insects naturally ingest most of their lipids as triglycerides (Nation 2002), which is the form of the lipids in coconut oil. Based on these criteria, treatment diets were: Normal (standard cornmeal-molasses lab food, 4% sucrose), calorie restricted (cornmeal-based food with 0.75% glucose by weight), high glucose (cornmeal-based food with 4% glucose by weight), high fat (cornmeal-based food with 0.75% glucose by weight and 3% coconut oil by weight added) (supporting information, Table S1). The tests of varied sugar concentrations on weight gain, subsequently referred to as the sugar titration experiment, were conducted with simple sugar (glucose, fructose, or sucrose) concentrations of 0, 0.25, 0.75, 2, 4, 6, 8, 10, 12, 15, 17.5, and 20% by weight substituted for the molasses in the cornmeal-based diet. For the nutritional geometry experiment (Lee et al. 2008), diets of varied fat and simple sugar concentration consisted of all possible combinations of glucose or sucrose concentrations of 0, 0.25, 0.75, 2, 4, 6, and 12% by weight substituted for molasses in the cornmeal-based diet with the added coconut oil concentrations of 0, 1.5, and 3% by weight for a total of 42 different diets.
Phenotypic measurements:
For larval phenotypic measurements, third instar larvae pooled from three experimentally identical vials were fasted for 6 hr before being frozen for assay of triglyceride and hemolymph carbohydrates (in three independent replicates per treatment for each phenotype). Total triglyceride content was determined on homogenates of six randomly selected larvae using the Sigma Triglyceride Determination kit and a 96-well spectrophotometer (Clark and Keith 1988; De Luca et al. 2005). Insects use trehalose sugar, circulating in the hemolymph, as the primary molecule to store and deliver potential energy to tissues (Wyatt 1961). Trehalose is a disaccharide that is easily broken down into two glucoses by trehalase to make glucose available for glycolysis (Nation 2002). Hemolymph carbohydrate concentration was determined on pooled hemolymph from greater than 20 larvae per sample, collected by capillary tube, and then treated with trehalase, and the resulting glucose concentration was determined by Sigma Glucose Determination kit and a 96-well spectrophotometer (Rulifson et al. 2002).
Pupal wet weight is quite constant throughout the pupal stage (Church and Robertson 1966) and is the weight of the late third instar larva, which has purged its gut contents (Ashburner et al. 2005). It is a reliable indicator of body weight accumulated during larval development before the confounding factors of adult feeding and reproduction. Pupae wet weights were determined individually for up to 15 clean and mature (within 12 hr of eclosion when sex combs can be easily observed) male pupae per vial for each of the two vials per treatment and replicate, using a Mettler Toledo XS105 balance. Larval survival was calculated as the proportion of the original 50 first instar larvae per vial to reach pupation while pupal survival was the proportion of pupae to reach maturity in each vial. Survival to pupation has been shown to be a function of diet quality and is not strictly dependent on larval weight (de Moed et al. 1999). Development time was calculated as the time from first instar larvae to the first mature pupa and was only measured in the spring replicate. Larval foraging behavior, captured here as “path length,” was measured on a subset of 22 lines as the distance a mid-third instar larva traveled on a yeast paste in 5 min with 20 larvae per strain replicated over 5 consecutive days (Osborne et al. 1997).
All parental lines were raised on the normal diet. Parental lines and experimental vials were maintained on a 12 hr:12 hr light:dark cycle at 25°. Measurements were obtained for all 146 lines in a randomized block design, spread over a 6-month period in April through September 2007, in which each week we set up blocks of 7 to 12 lines that were tested on all four diets, with five replicate vials per treatment. Thirty lines across all four diets were replicated in the same manor (randomly blocked into weeks) the following spring 2008. Phenotypic variation did correlate with external humidity so we controlled for external environmental variation by including dew point as a covariate using the proxy measurements from local weather station data from the Raleigh-Durham International Airport (KRDU, 35–54N 78–46W 130M). All datasets are available in File S1.
Data analysis:
No phenotypic variance for the measured traits due to population was observed and so both populations (North Carolina and Maine) were pooled for all analyses. There were no significant main effects of season (summer vs. spring) between the two large replicate groups. Hemolymph carbohydrate concentration was log transformed for normality while other phenotypic measures were untransformed. Variance components were estimated using Proc Mixed in SAS/STAT software. The linear model for phenotypic variance partitioning was
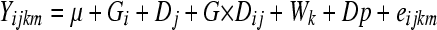 |
for measurements taken from the mth individual sample in the ith genetic line (G) raised on the jth diet (D), taken in the kth week (W), with seasonal effects such as dew point (Dp) included as a random covariate binned by similar measures to make a categorical variable. Exploratory models were also considered with Dp as a continuous variable without greatly impacting the estimates. Similarly, models with Vial as a random level of replication nested within time of sampling were constructed, and while vial effects were significant, they made only a minimal contribution to the variance and are omitted here for clarity. Variance due to larval foraging behavior was estimated using the model Yi = μ + Pli + ɛi, where Pl is the mean path length for ith genetic line and is a random effect. There was no apparent deviation from normality for the residuals of these models.
Heritability estimates were made for the normal diet only (since by definition heritability is the genetic contribution to total phenotypic variance in a single environment). Within-line (environmental) variance (VE) is the variance associate with the error term (ɛim) and between-line (genetic) variance (VG) is the variance associated with the genetic term (Gi), determined as the variance components from the simple model in Proc Varcomp in the SAS/STAT software. Total genetic variance (VG) in a fully inbred line model equals 2FVA, assuming F = 1 (inbreeding coefficient) and VA is the additive genetic variance (Falconer and Mackay 1996). Heritability (h2) was calculated as VA/VTotal where VTotal = VG + VE (Lynch and Walsh 1998), and in this experiment is largely narrow sense since there is little variation segregating in the highly inbred lines and no crosses were performed. The coefficient of genetic variation (CVA), an estimate of evolvability, is 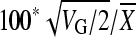 (Houle 1992; Lynch and Walsh 1998). The coefficient of residual variation (CVR) is
(Houle 1992; Lynch and Walsh 1998). The coefficient of residual variation (CVR) is 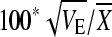 (Houle 1992). Note that the within-line variance will be an underestimate of individual variation for all of the traits except weight (where individuals were weighed) since measurements were, by necessity, based on pools of individuals. Parallel plots of genotype-by-diet interactions and contour plots of nutritional geometry were drawn using JMP, version 7 (SAS Institute).
(Houle 1992). Note that the within-line variance will be an underestimate of individual variation for all of the traits except weight (where individuals were weighed) since measurements were, by necessity, based on pools of individuals. Parallel plots of genotype-by-diet interactions and contour plots of nutritional geometry were drawn using JMP, version 7 (SAS Institute).
Significance in sugar titration and nutritional geometry experiments was determined using the F-test for fixed effects in Proc Mixed in SAS/STAT software. For the sugar titration experiment the model was
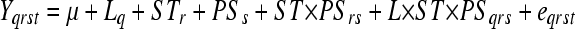 |
for data obtained from the tth pupae of the qth line (L), rth sugar-type (ST), and sth percent sugar (PS), in this case modeling all factors as fixed effects since they were deliberately chosen to represent specific lines or treatments. For the nutritional geometry analysis, an additional term representing the uth level of percent fat (PF) was added.
Pair wise phenotypic trait correlations were calculated in JMP, version 7 based on trait means for each line on each diet grouped by replicate week. Increased variance due to diet was tested using JMP, version 7 and the Levene test for unequal variance, applied across diets for between-line variance (genetic variance), while the within-line environmental variance was calculated as the within-line variance of residuals after accounting for the main effect of line (R. Wolfinger, personal communication). Genetic correlation across pairs of diets, which is the contribution to total phenotypic correlation across diets that is attributable to genetics alone, was calculated using SAS Proc Mixed in SAS/STAT software by specifying genetic line as the subject to estimate the genetic variance and covariance for each pair of diets for each trait, while diet was modeled as a random effect (Holland 2006). Genetic correlation was calculated as 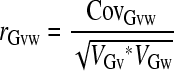 , where CovGvw is the genetic covariance between diets v and w, and VGv and VGw are the genetic variances on each diet, respectively.
, where CovGvw is the genetic covariance between diets v and w, and VGv and VGw are the genetic variances on each diet, respectively.
RESULTS
Sources of phenotypic variation:
We found significant genetic variation for seven measured phenotypes (Table 1, Figure 1); pupal wet weight (P < 0.0001), triglyceride content (P < 0.0001), hemolymph carbohydrate content (P < 0.0001), larval survival (P < 0.0001), pupal survival (P = 0.026), development time (P = 0.003), and foraging behavior (P = 0.0004). Genetic variation accounted for a large portion of the total phenotypic variation, between 11 and 40% for all of the traits except pupal survival, where genetics accounted for only 3% of the variation (Figure 1, Table 1).
TABLE 1.
Proportion of variance explained across traits for individual sample measures
| Trait | Genetic | Diet | Genetic × diet | Dew point | Week | Residual | |
|---|---|---|---|---|---|---|---|
| Weighta | Variance estimate | 0.0083**** | 0.0008 | 0.0053**** | 0.0001 | 0.0069*** | 0.0224**** |
| Proportion of total variance | 0.189 | 0.019 | 0.120 | 0.003 | 0.157 | 0.511 | |
| Triglycerideb | Variance estimate | 0.0018**** | 0.0002 | 0.0019**** | 0.0008 | 0.0080*** | 0.0039**** |
| Proportion of total variance | 0.109 | 0.013 | 0.115 | 0.048 | 0.481 | 0.234 | |
| Hemolymphb | Variance estimate | 0.3603**** | 0.0152 | 0.2697**** | 0.1005 | 0.2663* | 0.5449**** |
| Proportion of total variance | 0.231 | 0.010 | 0.173 | 0.065 | 0.171 | 0.350 | |
| Larval survival | Variance estimate | 0.0101**** | 0.0046 | 0.0027*** | 0.0007 | 0.0207*** | 0.0143**** |
| Proportion of total variance | 0.190 | 0.086 | 0.051 | 0.014 | 0.390 | 0.269 | |
| Pupal survival | Variance estimate | 0.0012* | 0.0186 | 0.0080**** | 0.0003 | 0.0027** | 0.0089**** |
| Proportion of total variance | 0.031 | 0.467 | 0.202 | 0.008 | 0.069 | 0.224 | |
| Development timec | Variance estimate | 1.54E9** | 1.94E8 | NA | NA | NA | 2.11E9**** |
| Proportion of total variance | 0.401 | 0.051 | NA | NA | NA | 0.548 |
*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, respectively.
Fifteen pupae per vial for weight.
Three replicates per vial for lipid and hemolymph.
There was insufficient power to estimate G × D interaction and other effects for development time.
Figure 1.—

Genetic variation. X-axis indicates individual inbred isofemale lines ordered by increasing trait mean. Error bars indicate one standard error. In A–E, * indicates the Oregon R laboratory strain, while in A, D, and E, # indicates Canton S laboratory strain. (A) Significant genetic variation for weight. (B) Significant genetic variation for lipid content. (C) Significant genetic variation for hemolymph carbohydrate concentration on log transformed data. (D) Significant genetic variation for larval survival. (E) Significant genetic variation for pupal survival. (F) Significant genetic variation for foraging behavior. R indicates the control “rover” line and S indicates the control “sitter” line.
Diet effects made a less substantial contribution to the variance, explaining only between 1 and 8% of the total phenotypic variance, with the exception of pupal survival, where diet explained 47% of the total variation (Table 1, Figure S1). Despite the low variance due to diet, there were some counterintuitive patterns, most notably elevated pupal weight on the calorie restricted diet relative to the normal diet (Figure S1). Such a pattern could imply that the larvae are modulating their food intake to compensate for restricted calories or that there is a fundamental difference in physiological response to the type of sugar in the normal (sucrose) vs. the calorie restricted diet (glucose).
The most interesting variance finding was that the interaction effect of genotype-by-diet was much greater than the effect of diet alone for weight, triglyceride storage, and hemolymph carbohydrate concentration, explaining between 12 and 17% of the phenotypic variance, as demonstrated by extensive crossing of line means (Figure 2, Table 1). Pupal survival also showed a high proportion of genotype-by-diet variance (20%), though it was not greater than the variance due to diet (47%). The presence of significant interaction terms compromises the interpretation of the main effects alone. In these cases, the effect of genotype can be different for each level of diet. These results indicate that diet on its own is only a minor overall contributor to most metabolic phenotypes, but that most of the variation in metabolic phenotypes is driven by genetics and the genetic interaction with diet (Figure 3).
Figure 2.—

Genetic-by-diet interaction. X-axis indicates each experimental diet. A black line and * indicate Oregon R in all panels, while black line and # indicates Canton S laboratory strain in A, D, and E. All other lines are colored by K-means clustering of reaction norms for the weight phenotype. Note that there is little correlation between reaction norms across different metabolic phenotypes as indicated by the scrambling of colors in B–E relative to A. All genetic-by-diet interaction terms are significant at P < 0.0001. (A) Significant genetic-by-diet interaction variation for weight. (B) Significant genetic-by-diet interaction variation for lipid content. (C) Significant genetic-by-diet interaction variation for hemolymph carbohydrate concentration. (D) Significant genetic-by-diet interaction variation for larval survival. (E) Significant genetic-by-diet interaction variation for pupal survival. Note the dramatic increase in genetic variation on a high fat diet for pupal survival.
Figure 3.—

Variance partition. “Week” indicates blocked week effect. Note that for many traits, the genetic and genetic-by-diet interaction effects explain far more of the variance than diet alone. (A) Variance partition for weight. (B) Variance partition for lipid content. (C) Variance partition for hemolymph carbohydrate concentration. (D) Variance partition for larval survival. (E) Variance partition for pupal survival.
We also measured the effects of larval foraging behavior (Osborne et al. 1997) on these metabolic phenotypes and found that 8.5% of the phenotypic variation in weight and triglyceride content on a normal diet is associated with innate larval behavior. Despite all lines having a “sitter-like” phenotype, larval foraging behavior in these lines exhibits significant genetic variation, 10.7% of the total phenotypic variation (P = 0.0004), showing that foraging behavior is a quantitative polygenic trait in addition to having a strong Mendelian factor in natural populations (Debelle and Sokolowski 1987). Dew point effects were also substantial, explaining up to 6% of the phenotypic variance of each trait (Table 1).
Heritabilities and correlations:
Estimated heritabilities were moderate for all traits and ranged from 0.053 to 0.261 (Table 2), which is consistent with most estimates of morphometric, behavioral, and life history traits in other natural populations (Mousseau and Roff 1987; Lynch and Walsh 1998; Kruuk et al. 2000). The coefficients of genetic variation (CVA), however, argued to be a better predictor of the potential of a trait to evolve (Houle 1992), are more intriguing. For most of the traits the values are in the range of 3.8–25.7%, consistent with CVA typically observed in other Drosophila studies (Houle 1992), but for the trait of hemolymph carbohydrate concentration the value is unusually large at 101.8% (Table 2). A high coefficient of genetic variation implies that the trait is not experiencing efficient selection to reduce genetic variation in the wild. Possible reasons of inefficient selection are that selection on the trait is relatively weak and/or the environmental variance in the trait is relatively large. In the case of hemolymph concentration, the coefficient of residual variation (CVR) was also large (Table 2), thus we speculate that hemolymph carbohydrate has intrinsically high environmental variance and that genetic regulation of it evolves under stabilizing selection toward a heritability that is on a par with the other metabolic traits.
TABLE 2.
Within (environmental) and between-line (genetic) variation, heritability, and coefficient of genetic variation for phenotypic traits on a normal diet based on an inbred line model
| Trait | Within-line variance (Ve) | Between-line variance (VG) | Heritability (h2) | Coefficient of genetic variation (CVA) | Coefficient of residual variation (CVR) |
|---|---|---|---|---|---|
| Weight | 0.025 | 0.012 | 0.164 | 7.90 | 16.02 |
| Triglycedrides | 0.006 | 0.006 | 0.261 | 25.71 | 34.81 |
| Hemolymph carbohydrate | 0.622 | 0.510 | 0.225 | 101.75 | 158.79 |
| Larval survival | 0.022 | 0.008 | 0.127 | 7.93 | 19.18 |
| Pupal survival | 0.003 | 0.003 | 0.228 | 3.82 | 5.91 |
| Path length | 2.661 | 0.319 | 0.053 | 6.33 | 25.84 |
Across all of the diets and genotypes, weight was negatively correlated with hemolymph carbohydrate concentration and larval survival, but weight was positively correlated with pupal survival and foraging path length (Table 3, trait correlations by diet are available in Table S2). Total triglyceride content was negatively correlated with both survival measures, indicating that the environmental or genetic characteristics that increase fat storage will have a detrimental effect on juvenile and adult fitness. Larval and pupal survival were positively correlated with each other. The positive correlation between weight and pupal survival is consistent with the well-documented positive correlation between weight and fitness seen in other insect studies (Honek 1993). A lack of correlation between weight and triglyceride content is consistent with previous findings (Wang et al. 2005).
TABLE 3.
Pairwise correlations between phenotypic traits across all diets
| Weight | Triglycerides | Hemolymph carbohydrate | Larval survival | Pupal survival | Path length | |
|---|---|---|---|---|---|---|
| Weight | — | 0.091 | −0.218 | −0.134 | 0.225 | 0.324 |
| Triglycerides | NS | — | −0.127 | −0.134 | −0.188 | 0.165 |
| Hemolymph carbohydrate | 3.5 | NS | — | −0.156 | −0.102 | −0.112 |
| Larval survival | 3.4 | 2.5 | NS | — | 0.165 | −0.113 |
| Pupal survival | 8.7 | 4.5 | NS | 4.9 | — | −0.094 |
| Path length | 6.4 | NS | NS | NS | NS | — |
Negative Log10 P below the diagonal, and NS, not significant at a Bonferonni corrected significance level of 2.48; correlation coefficient above.
The positive correlation between weight and larval foraging path length is perhaps counterintuitive, but may indicate an overall physiological health allowing for greater activity levels and more efficient resource acquisition as larvae, allowing for healthy weight gain. Relative to sitters, it has been shown rovers eat less but absorb calories more efficiently and also tend to store more of their calorie intake as lipid than carbohydrate under plentiful food conditions (Kaun et al. 2007, 2008). Our finding of a nonsignificant positive correlation between path length and lipid content are consistent with patterns previously observed contrasting rovers and sitters. These results, taken together, suggest that there is not a simple relationship between foraging behavior, body weight, and fitness.
Sugar titration and nutritional geometry:
Nutritional geometry is the empirical mapping of an organism's physiological response onto two or more dimensions of nutritional variation (Lee et al. 2008). These authors found that lifespan and fecundity in a single genotype of flies are optimized at different relative concentration of protein and carbohydrate. Given the large genotype-by-diet effects we observed for weight across our 146 lines on our four experimental diets, we were interested in whether those effects could be mapped to specific sugar types or sugar and fat concentrations.
We observed that genotype-by-diet interactions are sensitive to the type of sugar in the food. In the sugar titration experiment, there was a significant interaction (P < 0.0001) between genotype, sugar type (sucrose, glucose, and fructose), and sugar concentration, measured across six different genotypes, and 12 simple sugar concentrations between 0 and 20% (Figure 4). Maximal weight was achieved on relatively low sugar concentrations of 0.75% for all three sugars and there was a dramatic decease in weight on higher sugar concentrations, especially for glucose. Wang and Clark (1995) observed a similar pattern of reduced weight on a high (10%) sucrose food and attributed such loss in weight to stress-induced reduction in metabolic enzyme activity (Wang and Clark 1995).
Figure 4.—

Sugar titration. Measured across six different genotypes and 12 simple sugar concentrations between 0 and 20%, there was a significant interaction effect between genotype, sugar type (sucrose, glucose, and fructose), and sugar concentration (P < 0.0001), as well as significant main effects of % sugar (P < 0.0001), and genetic line (P < 0.0001).
In the nutritional geometry experiment, two genotypes were further evaluated across seven sucrose and glucose concentrations between 0 and 12% and also varying fat concentrations of 0, 1.5, and 3%. The greatest weight gain occurred at sugar concentrations greater or less than the concentration found in the normal lab food (4%), suggesting that even recently domesticated lines are adapted to reduce their weight under laboratory conditions (Figure 5). There was also generally weight loss with increased fat content in the diet regardless of the sugar type or sugar concentration (Figure 5), indicating that the weight loss observed on the high fat food in Figure 1A is probably also due primarily to increased fat rather than to a change in sugar content. The negative correlation between weight and larval survival reported above could be the mechanism of selection for lower weight on a laboratory diet. This observation is consistent with observed rapid adaptation to laboratory diets seen in other systems (Warbrick-Smith et al. 2006). However, also note that the canonical lab strains (Oregon R and Canton S, Figures 1 and 2) tended to have relatively high weights and pupal survival and low triglyceride content, hemolymph sugar concentration, and larval survival, which cautions against the assumption that metabolic regulation in standard lab strains is fully representative of the normal range of physiologies found in nature.
Figure 5.—

Nutritional geometry. Isoclines indicating average weight (mg) of flies raised on varying fat and sugar concentrations for two inbred lines. Low weight gain is generally exhibited on 4% sugar concentration indicating a possible adaptation of these lines to the 4% sugar lab food. There is significant variation in weight due to genetic line (P = 0.0164), % sugar (P < 0.0001), and % fat (P < 0.0001).
Decanalization:
Canalization refers to an evolved decreased variance of traits under normal circumstances; if a mutation or change in environment results in an increase in trait variance, it is decanalizing. We detected decanalization of metabolic phenotypes in flies grown on high fat food. Increased within-line variation for pupal weight, lipid content, and larval and pupal survival on a high fat diet (Figure 6, Table S3) indicates environmental decanalization. This means that flies grown on a high fat diet have metabolic phenotypes that are more difficult to predict from their genotype than they are on other diets. Larval and pupal survival also shows increased genetic (among line) variance on high fat food (Figure 6, Table S4), and hence genetic decanalization as well. This indicates that the high fat environment exposes cryptic genetic variation for these traits (Gibson and Dworkin 2004; Gibson 2009). Interestingly, relative to the normal diet, two of these traits, weight and total triglycerides (Figure S1), showed no net increase in mean phenotype but did show a change in the trait variance on a high fat diet, while the survival traits showed both a decreased mean and increased variance on the high fat diet.
Figure 6.—

Genetic and environmental decanalization of various metabolic phenotypes on a high fat diet. (A) Genetic decanalization of larval survival depicted as increased between-line standard deviation on a high fat diet. (B) Genetic decanalization of pupal survival depicted as increased between-line standard deviation on a high fat diet. (C) Environmental decanalization of weight depicted as increased within-line standard deviation of residuals of line effects on a high fat diet. (D) Environmental decanalization of pupal survival depicted as increased within-line standard deviation of residuals of line effects on a high fat diet.
We also examined the genetic correlations for each trait across the four diets (Table 4). These range from a low of 0.097 for larval survival on the normal and high fat diets to a high of 0.97 for the same trait between normal and high glucose diets. In general, the genetic correlations between diets involving the high fat diet are lower than among the other three diets (P < 0.0004, by two-tailed t-test), consistent with the observation that the metabolic traits are most genetically variable on high fat. This is particularly apparent for larval survival, and to a lesser extent pupal weight. Triglyceride and hemolymph sugar content tend to show lower genetic correlations overall.
TABLE 4.
Genetic correlation within traits across diets
| Diet 2 |
|||||
|---|---|---|---|---|---|
| Trait | Diet 1 | Normal | Calorie restricted | High glucose | High fat |
| Weight | Normal | — | 0.668 | 0.672 | 0.541 |
| Calorie restricted | 0.668 | — | 0.643 | 0.389 | |
| High glucose | 0.672 | 0.643 | — | 0.406 | |
| High fat | 0.541 | 0.389 | 0.406 | — | |
| Triglyceride | Normal | — | 0.561 | 0.213 | 0.395 |
| Calorie restricted | 0.561 | — | 0.634 | 0.539 | |
| High glucose | 0.213 | 0.634 | — | 0.233 | |
| High fat | 0.395 | 0.539 | 0.233 | — | |
| Hemolymph | Normal | — | 0.929 | 0.490 | 0.542 |
| Calorie restricted | 0.929 | — | 0.504 | 0.579 | |
| High glucose | 0.490 | 0.504 | — | 0.318 | |
| High fat | 0.542 | 0.579 | 0.318 | — | |
| Larval survival | Normal | — | 0.938 | 0.970 | 0.097 |
| Calorie restricted | 0.938 | — | 0.963 | 0.186 | |
| High glucose | 0.970 | 0.963 | — | 0.232 | |
| High fat | 0.097 | 0.186 | 0.232 | — | |
| Pupal survival | Normal | — | 0.504 | 0.508 | Undefined |
| Calorie restricted | 0.504 | — | 0.577 | 0.504 | |
| High glucose | 0.508 | 0.577 | — | Undefined | |
| High fat | Undefined | 0.504 | Undefined | — | |
DISCUSSION
Diet clearly contributes to metabolic phenotypes in D. melanogaster, but it does so in a manner that is highly genotype dependent. Most phenotypic variation observed is due primarily to genetic (scenario 2 above) and genotype-by-diet effects (scenario 4) rather than due to diet alone (scenario 3), and the average effect of each diet across all genotypes, though significant, is a small proportion of the total variance. These findings at a population-wide level are consistent with past findings of substantial genotype-by-diet interaction effects on physiological traits seen in a few distinct genotypes (Falconer 1960; Clark and Fucito 1998; Vieira et al. 2000). The high heritabilities and coefficients of genetic variation observed, especially for hemolymph carbohydrate concentration and total triglycerides, suggest that these traits not only show large genetic contribution to their variation but also that they should be able to evolve in response to the selective forces of dietary or other environmental perturbation. Such adaptation in lipid storage has been seen in Lepidoptera (Warbrick-Smith et al. 2006).
Regarding our second question as to whether a perturbing diet changes the phenotypic variance in metabolic traits, we found that both genetic and phenotypic variance increased on a high fat diet for most of the trait measures (scenario 6 from above), thus they were decanalized. The increase in trait variance in these cases is consistent with the notion that the prevalence of chronic disease can increase even in the absence of an overall mean phenotypic deviation, due to a larger portion of the population exceeding a disease threshold due principally to an increase in phenotypic variance (Gibson and Reed 2008; Gibson 2009).
Decanalization, or the loss of physiological homeostasis in the face of an external stimulus, suggests lack of adaptation to the perturbing stimulus due to insufficient evolutionary exposure to the perturbation or to evolutionary genetic constraint. The large amount of observed genetic variance as well as the evidence of genetic decanalization for the survival traits, hint that the cause of the loss of homeostasis is more likely due to a lack of evolutionary experience with a high fat diet, since there is sufficient genetic variance available for selection to act on if it had been given the opportunity. Looking for recanalization of the metabolic phenotypes in response to extended exposure to the high fat diet over many generations could formally test this hypothesis.
These findings show both why identifying the specific mechanisms of MetS disease in humans will be difficult and how the use of model organisms can inform the population level models of complex human disease. The experimental ease offered by a population-based metabolic model, such as we have begun to develop here in Drosophila, gives the opportunity to explore the underlying molecular and evolutionary mechanisms contributing to the genotype-by-environment effects on population variation in MetS-like symptoms. Lessons from such models could inform productive avenues of research into these phenomena in human populations.
Acknowledgments
We thank R. Wolfinger, P. Visscher, E. Stone, J. Osborn, and T. Mackay for statistical advice. This research was supported by National Institutes of Health (NIH)-National Research Service Award fellowship F32 GM082021-03 to L.K.R., NIH 2-R01-GM61600 and Australian Research Council DP0880204 to G.G., and National Institute of Diabetes and Digestive and Kidney Diseases DK70141-2 to M.B.S.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.113571/DC1.
References
- Alberti, K. G. M. M., P. Zimmet and J. Shaw, 2006. Metabolic syndrome: a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet. Med. 23 469–480. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., K. G. Golic and R. S. Hawley, 2005. Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Bodmer, R., 1995. Heart Development in Drosophila and Its relationship to vertebrates. Trends Cardiovasc. Med. 5 21–28. [DOI] [PubMed] [Google Scholar]
- Church, R. B., and F. W. Robertson, 1966. A biochemical study of growth of Drosophila melanogaster. J. Exp. Zool. 162 337. [Google Scholar]
- Clark, A. G., and C. D. Fucito, 1998. Stress tolerance and metabolic response to stress in Drosophila melanogaster. Heredity 81 514–527. [DOI] [PubMed] [Google Scholar]
- Clark, A. G., and L. E. Keith, 1988. Variation among extracted lines of Drosophila melanogaster in triacylglycerol and carbohydrate storage. Genetics 119 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corella, D., and J. M. Ordovas, 2005. Single nucleotide polymorphisms that influence lipid metabolism: interaction with dietary factors. Annu. Rev. Nutr. 25 341–390. [DOI] [PubMed] [Google Scholar]
- Corella, D., G. Peloso, D. K. Arnett, S. Demissie, L. A. Cupples et al., 2009. APOA2, dietary fat, and body mass index: replication of a gene-diet interaction in 3 independent populations. Arch. Intern. Med. 169 1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelle, J. S., and M. B. Sokolowski, 1987. Heredity of Rover Sitter: alternative foraging strategies of Drosophila melanogaster larvae. Heredity 59 73–83. [Google Scholar]
- De Luca, M., N. Yi, D. B. Allison, J. Leips and D. M. Ruden, 2005. Mapping quantitative trait loci affecting variation in Drosophila triacylglycerol storage. Obes. Res. 13 1596–1605. [DOI] [PubMed] [Google Scholar]
- de Moed, G. H., C. L. J. J. Kruitwagen, G. de Jong and W. Scharloo, 1999. Critical weight for the induction of pupariation in Drosophila melanogaster: genetic and environmental variation. J. Evol. Biol. 12 852–858. [Google Scholar]
- Denholm, B., V. Sudarsan, S. Pasalodos-Sanchez, R. Artero, P. Lawrence et al., 2003. Dual origin of the renal tubules in Drosophila: mesodermal cells integrate and polarize to establish secretory function. Curr. Biol. 13 1052–1057. [DOI] [PubMed] [Google Scholar]
- Driver, C. J. I., and G. Cosopodiotis, 1979. Effect of dietary fat on longevity of Drosophila melanogaster. Exp. Gerontol. 14 95–100. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., 1960. Selection of mice for growth on high and low planes of nutrition. Genet. Res. 1 91–113. [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Pearson Education, Harlow, UK.
- Ford, E. S., and W. H. Giles, 2003. A comparison of the prevalence of the metabolic syndrome using two proposed definitions. Diabetes Care 26 575–581. [DOI] [PubMed] [Google Scholar]
- Gibson, G., 2009. Decanalization and the origin of complex disease. Nat. Rev. Genet. 10 134–140. [DOI] [PubMed] [Google Scholar]
- Gibson, G., and I. Dworkin, 2004. Uncovering cryptic genetic variation. Nat. Rev. Genet. 5 681–690. [DOI] [PubMed] [Google Scholar]
- Gibson, G., and L. K. Reed, 2008. Cryptic genetic variation. Curr. Biol. 18 R989–R990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, G., and S. vanHelden, 1997. Is function of the Drosophila homeotic gene Ultrabithorax canalized? Genetics 147 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, J. B., 2006. Estimating genotypic correlations and their standard errors using multivariate restricted maximum likelihood estimation with SAS Proc MIXED. Crop Sci. 46 642–654. [Google Scholar]
- Honek, A., 1993. Intraspecific variation in body size and fecundity in insects: a general relationship. Oikos 66 483–492. [Google Scholar]
- Houle, D., 1992. Comparing evolvability and variability of quantitative traits. Genetics 130 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomaa, B., P. Almgren, T. Tuomi, B. Forsen, K. Lahti et al., 2001. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care 24 683–689. [DOI] [PubMed] [Google Scholar]
- Kaun, K. R., C. A. L. Riedl, M. Chakaborty-Chatterjee, A. T. Belay, S. J. Douglas et al., 2007. Natural variation in food acquisition mediated via a Drosophila cGMP-dependent protein kinase. J. Exp. Biol. 210 3547–3558. [DOI] [PubMed] [Google Scholar]
- Kaun, K. R., M. Chakaborty-Chatterjee and M. B. Sokolowski, 2008. Natural variation in plasticity of glucose homeostasis and food intake. J. Exp. Biol. 211 3160–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruuk, L. E. B., T. H. Clutton-Brock, J. Slate, J. M. Pemberton, S. Brotherstone et al., 2000. Heritability of fitness in a wild mammal population. Proc. Natl. Acad. Sci. USA 97 698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. P., S. J. Simpson, F. J. Clissold, R. Brooks, J. W. O. Ballard et al., 2008. Lifespan and reproduction in Drosophila: new insights from nutritional geometry. Proc. Natl. Acad. Sci. USA 105 2498–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W., J. Park, S. Noh, E. Rhee, S. K Im et al., 2004. Prevalence of the metabolic syndrome among 40,698 Korean metropolitan subjects. Diabetes Res. Clin. Pract. 65 143–149. [DOI] [PubMed] [Google Scholar]
- Lorenzo, C., M. Okoloise, K. Williams, M. P. Stern and S. M. Haffner, 2003. The metabolic syndrome as predictor of type 2 diabetes: the San Antonio Heart Study. Diabetes Care 26 3153–3159. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetic and the Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Mousseau, T. A., and D. A. Roff, 1987. Natural-selection and the heritability of fitness components. Heredity 59 181–197. [DOI] [PubMed] [Google Scholar]
- Nation, J. L., 2002. Insect Physiology and Biochemistry. CRC Press, Boca Raton, FL.
- O'Rahilly, S., and I. S. Farooqi, 2006. Genetics of obesity. Philos. Trans. R Soc. B Biol. Sci. 361 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas, J. M., 2006. Genetic interactions with diet influence the risk of cardiovascular disease. Am. J. Clin. Nutr. 83 443S–446S. [DOI] [PubMed] [Google Scholar]
- Osborne, K. A., A. Robichon, E. Burgess, S. Butland, R. A. Shaw et al., 1997. Natural behavior polymorphism due to a cGMP-dependent protein kinase of Drosophila. Science 277 834–836. [DOI] [PubMed] [Google Scholar]
- Rizki, T. M., 1978. Fat body, pp. 561–601 in The Genetics and Biology of Drosophila, edited by M. Ashburner and T. R. F. Wright. Academic Press, London.
- Rulifson, E. J., S. K. Kim and R. Nusse, 2002. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296 1118–1120. [DOI] [PubMed] [Google Scholar]
- Sang, J. H., 1956. The quantitative nutritional requirements of Drosophila melanogaster. J. Exp. Biol. 33 45–72. [Google Scholar]
- Schulz, L. O., P. H. Bennett, E. Ravussin, J. R. Kidd, K. K. Kidd et al., 2006. Effects of traditional and Western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care 29 1866–1871. [DOI] [PubMed] [Google Scholar]
- Vieira, C., E. G. Pasyukova, Z. B. Zeng, J. B. Hackett, R. F. Lyman et al., 2000. Genotype–environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., and A. G. Clark, 1995. Physiological genetics of the response to a high-sucrose diet by Drosophila melanogaster. Biochem. Genet. 33 149–165. [DOI] [PubMed] [Google Scholar]
- Wang, M. H., L. G. Harshman and S. V. Nuzhdin, 2005. Quantitative trait loci for lipid content in Drosophila melanogaster. Obes. Res. 13 1891–1897. [DOI] [PubMed] [Google Scholar]
- Warbrick-Smith, J., S. T. Behmer, K. P. Lee, D. Raubenheimer and S. J. Simpson, 2006. Evolving resistance to obesity in an insect. Proc. Natl. Acad. Sci. USA 103 14045–14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warodomwichit, D., J. Shen, D. K. Arnett, M. Y. Tsai, E. K. Kabagambe et al., 2009. ADIPOQ polymorphisms, monounsaturated fatty acids, and obesity risk: the GOLDN study. Obesity 17 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessells, R. J., E. Fitzgerald, J. R. Cypser, M. Tatar and R. Bodmer, 2004. Insulin regulation of heart function in aging fruit flies. Nat. Genet. 36 1275–1281. [DOI] [PubMed] [Google Scholar]
- Wyatt, G. R., 1961. Biochemistry of insect hemolymph. Annu. Rev. Entomol. 6 75. [Google Scholar]