

Abstract
The rapid increase in high-throughput single-nucleotide polymorphism data has led to a great interest in applying genome-wide evaluation methods to identify an individual's genetic merit. Genome-wide evaluation combines statistical methods with genomic data to predict genetic values for complex traits. Considerable uncertainty currently exists in determining which genome-wide evaluation method is the most appropriate. We hypothesize that genome-wide methods deal differently with the genetic architecture of quantitative traits and genomes. A genomic linear method (GBLUP), and a genomic nonlinear Bayesian variable selection method (BayesB) are compared using stochastic simulation across three effective population sizes and a wide range of numbers of quantitative trait loci (NQTL). GBLUP had a constant accuracy, for a given heritability and sample size, regardless of NQTL. BayesB had a higher accuracy than GBLUP when NQTL was low, but this advantage diminished as NQTL increased and when NQTL became large, GBLUP slightly outperformed BayesB. In addition, deterministic equations are extended to predict the accuracy of both methods and to estimate the number of independent chromosome segments (Me) and NQTL. The predictions of accuracy and estimates of Me and NQTL were generally in good agreement with results from simulated data. We conclude that the relative accuracy of GBLUP and BayesB for a given number of records and heritability are highly dependent on Me, which is a property of the target genome, as well as the architecture of the trait (NQTL).
THE rapid progress and reducing costs of genome sequencing and high-throughput DNA techniques have led to a great interest in applying genome-wide evaluation methods to identify individuals of high genetic merit. Genome-wide evaluation uses associations of a large number of SNP (single nucleotide polymorphism) markers across the whole genome with phenotypes to produce accurate estimates of breeding values (EBVs) for candidates to selection (Meuwissen et al. 2001). The accuracy of genome-wide selection (i.e., selection based on genomic EBVs) is expected to be substantially higher than that of traditional best linear unbiased prediction (BLUP) selection, which is based on pedigree and phenotypic data (Daetwyler et al. 2008; Goddard 2009; Hayes et al. 2009c). In addition, genome-wide selection has the potential to reduce inbreeding rates because of the increased emphasis on own rather than family information (Woolliams et al. 2002; Daetwyler et al. 2007; Dekkers 2007). Furthermore, the application of genome-wide evaluation approaches can significantly aid our understanding of quantitative trait genetic architecture.
The genome-wide evaluation methods suggested to date can be broadly categorized into groups according to whether there is an assortment of the SNP by magnitude of effect or contribution to the variance. One group treats SNP homogeneously and includes variants of genomic best linear unbiased prediction (GBLUP). This group includes a form of ridge regression (Meuwissen et al. 2001) and the use of a realized relationship matrix computed from the markers instead of the traditional pedigree matrix (NejatiJavaremi et al. 1997; Villanueva et al. 2005; Hayes et al. 2009c). Both approaches have been shown to be equivalent (Habier et al. 2007; Goddard 2009). A second group provides for heterogeneity among SNP contributions to the variance, with some contributions permitted to be large while the remainder are small, possibly zero. This assortment is helped by Bayesian approaches, which place priors on numbers of SNP with major contributions (e.g., BayesA and BayesB; see Meuwissen et al. 2001, 2009; Lee et al. 2008), or with some penalty based on functions of the magnitude of effect for each SNP (e.g., Lasso; see Tibshirani 1996; Yi and Xu 2008) or with other smoothing metrics (Long et al. 2007). A third group attempts to reduce dimensionality by using principal components or partial least squares (Raadsma et al. 2008; Solberg et al. 2009) to identify an informative subset of SNP genotypes. The main two methods currently used in real data sets are a linear prediction method, GBLUP, and variants of nonlinear Bayesian variable selection approaches such as BayesB.
In most simulated published data, the accuracy of BayesB outperformed that of GBLUP (e.g., Meuwissen et al. 2001; Habier et al. 2007; Lund et al. 2009). However, real data results have not consistently supported this conclusion. Two reviews of empirical results in dairy cattle to date have shown that GBLUP and BayesB result in very similar accuracies for most traits (Hayes et al. 2009a; Vanraden et al. 2009). One reason for the disagreement between simulated and real data results could be that the genetic architecture simulated is significantly different from what is found in real populations. Most studies published to date that compare methods using simulated architectures have considered only 50 or fewer QTL affecting the trait (e.g., Meuwissen et al. 2001; Habier et al. 2007; Lund et al. 2009). In this article we hypothesize that the relative utility of genome-wide evaluation methods depends significantly on both the genomic structure of the population and the genetic trait architecture.
The main objective of this study was to compare a linear method, GBLUP, and a nonlinear variable selection method, BayesB, using simulated data across a range of population and trait genetic architectures to further understand the mechanics of genome-wide evaluation methods. An important secondary objective was to extend deterministic prediction models to predict the accuracy of both methods. Theoretical models complement stochastic simulation by helping the understanding of the factors involved in genome-wide evaluation performance and, in return, stochastic simulation is used to confirm theoretical derivations.
METHODS
Theoretical development:
Daetwyler et al. (2008) derived equations for predicting the accuracy of a simple least-squares genome-wide evaluation approach for continuous and dichotomous traits. The original formula for genome-wide accuracy for a continuous trait is 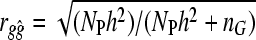 , where rgĝ is the correlation between true and estimated additive genetic values (i.e., accuracy), NP is the number of individuals in the training population, h2 is the heritability, and nG is the number of independent loci (Daetwyler et al. 2008). The accuracy was independent of how large the subset of loci was that make nonzero contributions. Thus, it did not matter whether there were many nonzero loci effects of small magnitude or only a few nonzero loci effects of large magnitude. In Daetwyler et al. (2008), the formulae were derived by considering the regression of phenotypes on one locus at a time as loci were assumed independent. Therefore the formula will work for small numbers of dispersed loci in a genome but the accuracy will tend to zero as nG becomes large; erroneously, because loci cannot be added independently in a finite genome due to linkage. Daetwyler et al. (2008) discussed that an empirical value for the number of independent chromosome segments (
, where rgĝ is the correlation between true and estimated additive genetic values (i.e., accuracy), NP is the number of individuals in the training population, h2 is the heritability, and nG is the number of independent loci (Daetwyler et al. 2008). The accuracy was independent of how large the subset of loci was that make nonzero contributions. Thus, it did not matter whether there were many nonzero loci effects of small magnitude or only a few nonzero loci effects of large magnitude. In Daetwyler et al. (2008), the formulae were derived by considering the regression of phenotypes on one locus at a time as loci were assumed independent. Therefore the formula will work for small numbers of dispersed loci in a genome but the accuracy will tend to zero as nG becomes large; erroneously, because loci cannot be added independently in a finite genome due to linkage. Daetwyler et al. (2008) discussed that an empirical value for the number of independent chromosome segments (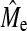 ) could be used in place of nG, because nG was assumed independent. Goddard (2009) also proposed accuracy predictions for GBLUP, which used the concept of predicted Me. His derivation builds on work by Visscher et al. (2006) in which the variance of identical-by-descent sharing for full sibs was developed and provides a prediction for Me, which is Me = 2NeL/log(4NeL), where L is the genome length in Morgans (Goddard 2009). Substituting Me in place of nG in the original formula of Daetwyler et al. (2008) results in
) could be used in place of nG, because nG was assumed independent. Goddard (2009) also proposed accuracy predictions for GBLUP, which used the concept of predicted Me. His derivation builds on work by Visscher et al. (2006) in which the variance of identical-by-descent sharing for full sibs was developed and provides a prediction for Me, which is Me = 2NeL/log(4NeL), where L is the genome length in Morgans (Goddard 2009). Substituting Me in place of nG in the original formula of Daetwyler et al. (2008) results in
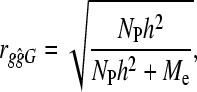 |
(1) |
which also predicts the accuracy of GBLUP. At no time does the argument moving from the original formula of Daetwyler et al. (2008) to Equation 1 depend on the distribution of the effects of loci, so we come to the first hypothesis in this study that states that GBLUP accuracy is independent of the number of quantitative trait loci (NQTL) associated with the phenotypic trait.
Our second hypothesis was that the accuracy of BayesB when NQTL is high would tend to that of GBLUP. If our first hypothesis is confirmed, then the dependence of GBLUP on Me is an advantage at high NQTL, even though NQTL may be higher than Me. Heuristically, if GBLUP delivers accuracy as if there are a Me number of QTL, the benefit from prior information that there are approximately Me (or more) QTL is unclear given Equation 1. On the other hand, it is a clear disadvantage if NQTL < Me because GBLUP cannot adapt the model to suit the data. In contrast, BayesB is a variable selection method that attempts to determine the “optimum dimensionality” given the data and prior information. When NQTL is high this optimum is likely to be Me in both methods. Hence, the accuracy of BayesB at high NQTL can be predicted in the same way as GBLUP, but if NQTL < Me, variable selection may deliver an advantage in accuracy because choosing a subset of variables will reduce the dimensionality of the model. Thus, substituting NQTL for Me is likely to better predict the accuracy of BayesB. This results in the following equation,
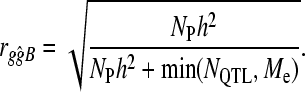 |
(2) |
Further rearrangement of Equations 1 and 2 allows for empirical estimates of Me (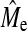 ) to be made in the following way,
) to be made in the following way,
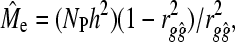 |
(3) |
where 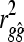 is the squared accuracy of estimates of genetic values using GBLUP or BayesB (when NQTL ≥ Me) for individuals without phenotypes. Predicting
is the squared accuracy of estimates of genetic values using GBLUP or BayesB (when NQTL ≥ Me) for individuals without phenotypes. Predicting 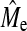 with GBLUP requires molecular relatedness to be known, whereas this is not required when using BayesB. This result gives a further subhypothesis that the empirical Me is predicted by the formula for independent chromosome segments given by Goddard (2009). Also, if NQTL < Me, additional information on NQTL can be gathered using BayesB accuracy because it can choose a subset of loci or variables, by applying the following formula:
with GBLUP requires molecular relatedness to be known, whereas this is not required when using BayesB. This result gives a further subhypothesis that the empirical Me is predicted by the formula for independent chromosome segments given by Goddard (2009). Also, if NQTL < Me, additional information on NQTL can be gathered using BayesB accuracy because it can choose a subset of loci or variables, by applying the following formula:
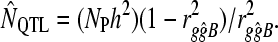 |
(4) |
Therefore, additional insight into quantitative traits can be gained by combining genome-wide evaluation and deterministic prediction. The accuracy of BayesB is of course influenced by the priors used in the analyses (especially priors on the proportion of loci with no effect); hence it is important to use appropriate priors to get accurate  .
.
Simulations:
Our study consisted of three main steps. First, populations of individuals were simulated to be in mutation drift equilibrium. Second, effects were assigned to a number of QTL that were randomly selected from the whole set of segregating loci, and true genetic values and phenotypes were generated for each individual. The third step consisted of the genetic evaluations of the individuals generated with both GBLUP and BayesB.
Populations and genome:
Populations in mutation drift equilibrium were simulated by random mating individuals for many generations with recombination and mutation. The number of male and female parents was 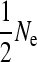 across generations. A total of 1000, 5000, and 10,000 generations were simulated until linkage disequilibrium and heterozygosity values were stable for Ne = 200, Ne = 1000, and Ne = 2000, respectively. In the final generation, a set of training individuals (of variable size) in which the loci effects were to be estimated was generated by random mating. Using the same parents, a set of validation individuals of size equal to the training set was produced whose genetic values were to be predicted. In scenarios where the size of the training sets (NP) was larger than Ne, population size was increased by increasing the number of offspring per mating in the final generation.
across generations. A total of 1000, 5000, and 10,000 generations were simulated until linkage disequilibrium and heterozygosity values were stable for Ne = 200, Ne = 1000, and Ne = 2000, respectively. In the final generation, a set of training individuals (of variable size) in which the loci effects were to be estimated was generated by random mating. Using the same parents, a set of validation individuals of size equal to the training set was produced whose genetic values were to be predicted. In scenarios where the size of the training sets (NP) was larger than Ne, population size was increased by increasing the number of offspring per mating in the final generation.
The total genome size was 10 M (10 chromosomes of 1 M each). In generation zero all individuals were completely homozygous for the same allele and mutations were applied at a rate of 2.5 × 10−5 per locus per meiosis in the following generations. Mutations switched allele one to two and vice versa. The number of randomly distributed mutations per chromosome was sampled from a Poisson distribution with mean corresponding to the product of the number of loci per chromosome and the mutation rate. Similarly, recombinations per chromosome were sampled from a Poisson distribution with a mean of one per morgan and were then randomly placed along the chromosome. Linkage disequilibrium (LD) statistics, i.e., R2 (Hill and Robertson 1968), between adjacent segregating loci were averaged among all pairs exceeding a minor allele frequency of 0.05 and matched expected R2 values (Sved 1971; Tenesa et al. 2007). Allele frequency distributions were found to follow a U-shaped distribution.
The number of loci at the start of the simulation (generation zero) required several considerations concerned with obtaining an appropriate number of segregating loci (Ne) and NQTL in the final generation. The realized relationship matrix used in GBLUP can be singular if NL is less than the number of individuals in the matrix (Vanraden 2008), preventing the inversion needed to compute solutions. Thus, NL at mutation drift equilibrium was made larger than the maximum sum of training and validation individuals to be used, and a similar NL was used across all scenarios. However, as Ne increased, the proportion of segregating loci in the last generation also increased and for Ne = 200, Ne = 1000, and Ne = 2000, approximately 0.04, 0.28, and 0.52 of initial loci were segregating at mutation drift equilibrium, respectively. This required the adjustment of the number of initial loci. Across all scenarios, NL varied between 4576 and 4721 loci (SE < 9.5 loci). To obtain NQTL, Me in a random mating population, as derived by Goddard (2009), was used as a guide to allow comparisons to be made across Ne. The following NQTL scenarios were simulated: 0.03, 0.05, 0.15, 0.30, 0.50, 0.75, and 1 Me. Table 1 outlines the corresponding NQTL for these proportions of Me for the three Ne, whereas Table 2 shows all scenarios that were carried out. Note that throughout this study our use of the terms “low” or “high” NQTL may refer to different actual NQTL across the three Ne because NQTL was scaled to be proportional to Me (Table 1).
TABLE 1.
Number of QTL simulated for each proportion of independent chromosome segments (Me) for three values of effective population size (Ne)
| Ne | 0.03 Me | 0.05 Me | 0.15 Me | 0.3 Me | 0.5 Me | 0.75 Me | 1 Me |
|---|---|---|---|---|---|---|---|
| 200 | 13 | 22 | 67 | 133 | 223 | 334 | 445 |
| 1000 | 57 | 94 | 283 | 566 | 944 | 1415 | 1887 |
| 2000 | 106 | 177 | 531 | 1063 | 1772 | 2657 | 3543 |
TABLE 2.
Parameter values for the simulated scenarios, where Ne is the effective population size; NP is the number of individuals in the training set; h2 is the heritability; Prior is the prior used for π in BayesB; and Me is the number of independent chromosome segments
| Scenario | Ne | NP | h2 | Prior | Me |
|---|---|---|---|---|---|
| 1 | 200 | 200 | 0.3 | Informed | 445 |
| 2 | 200 | 1000 | 0.3 | Informed | 445 |
| 3 | 1000 | 1000 | 0.5 | Informed | 1887 |
| 4 | 1000 | 1000 | 0.3 | Informed | 1887 |
| 5 | 1000 | 1000 | 0.1 | Informed | 1887 |
| 6 | 1000 | 500 | 0.3 | Informed | 1887 |
| 7 | 1000 | 2000 | 0.3 | Informed | 1887 |
| 8 | 1000 | 1000 | 0.3 | 57 QTL | 1887 |
| 9 | 2000 | 2000 | 0.3 | Informed | 3543 |
The desired NQTL were randomly chosen from NL. True allele substitution effects were sampled from N (0, 1). True breeding values for 2NP (i.e., training and validation set) individuals were calculated for each QTL as 2(1 − p j)βj (where p j is the major allele frequency at locus j), −2pjβj, and ((1 − pj) − pj)βj for the major and minor homozygote and heterozygote genotype, respectively (Falconer and Mackay 1996). Total breeding values were obtained by summing over NQTL. Breeding values were scaled to have the variance of h2 (i.e., phenotypic variance is 1). Phenotypic records were simulated for NP (training set) animals by adding independent environmental terms drawn from N(0, 1 − h2) to true breeding values. In addition, sampling βj from a Laplace (double exponential) distribution was investigated in scenario 4. The accuracy of both genome-wide evaluation methods was computed as the correlation between true and estimated breeding values.
GBLUP analysis:
The evaluation with GBLUP applied the following model, which was fit in ASReml (Gilmour et al. 1995): y = μ1 + Za + e, where y is the vector of phenotypic values, μ is the population mean, Z is an incidence matrix for random individual effects, a is a vector of random individual additive genetic values, and e is the residual. Random effects a and e were assumed normally distributed as 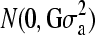 and
and 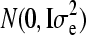 , respectively, where G was the realized relationship matrix computed using the NL loci. In G, the relationship between a pair of individuals was based on identical-by-state probabilities and included all training individuals with phenotypes and validation individuals without phenotypes. The total allelic relationship at a locus between a pair of individuals was calculated as
, respectively, where G was the realized relationship matrix computed using the NL loci. In G, the relationship between a pair of individuals was based on identical-by-state probabilities and included all training individuals with phenotypes and validation individuals without phenotypes. The total allelic relationship at a locus between a pair of individuals was calculated as 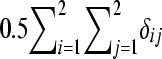 , where δij is 1 if allele i in the first individual is identical to allele j in the second individual and 0 otherwise. Averaging over loci as
, where δij is 1 if allele i in the first individual is identical to allele j in the second individual and 0 otherwise. Averaging over loci as 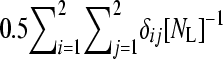 yields the numerator relationship between all individual pairs required for G (NejatiJavaremi et al. 1997). Breeding values were obtained by solving the mixed model equations (Henderson 1975).
yields the numerator relationship between all individual pairs required for G (NejatiJavaremi et al. 1997). Breeding values were obtained by solving the mixed model equations (Henderson 1975).
BayesB analysis:
We implemented a variant of the original BayesB (Meuwissen et al. 2001). The model applied was 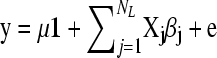 , where μ was the mean, X was an incidence equal to −1, 0, and 1 for 11, 12/21, and 22, respectively, β is the allele substitution effect for locus j, and e was the vector of residuals distributed as
, where μ was the mean, X was an incidence equal to −1, 0, and 1 for 11, 12/21, and 22, respectively, β is the allele substitution effect for locus j, and e was the vector of residuals distributed as 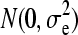 . Allele substitution effects were assumed to come for a mixture distribution where a proportion (π) had no effect and a proportion (1 − π) had effects distributed as
. Allele substitution effects were assumed to come for a mixture distribution where a proportion (π) had no effect and a proportion (1 − π) had effects distributed as 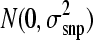 .
.
The prior used for μ was uniform, over a long range (i.e., 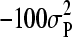 to
to 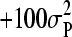 ), and for
), and for 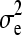 was uniform (i.e., 0 to
was uniform (i.e., 0 to 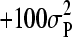 ), where
), where 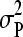 is the phenotypic variance of the data set analyzed. The prior for
is the phenotypic variance of the data set analyzed. The prior for 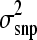 was taken from a scaled inverted chi-square distribution of the form
was taken from a scaled inverted chi-square distribution of the form 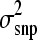 ∼
∼ 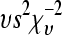 , where s2 represented the prior value for
, where s2 represented the prior value for 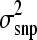 and υ was its degree of belief (Wang et al. 1994). A weak prior was chosen to be 1 for both υ and s2 across all scenarios. The influence of our s2 on
and υ was its degree of belief (Wang et al. 1994). A weak prior was chosen to be 1 for both υ and s2 across all scenarios. The influence of our s2 on 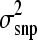 estimates from BayesB was investigated and was found to be small. A critical discussion of the impact of priors on BayesB can be found in Gianola et al. (2009).
estimates from BayesB was investigated and was found to be small. A critical discussion of the impact of priors on BayesB can be found in Gianola et al. (2009).
To test effect of π on breeding value accuracy, two types of scenarios were carried out. In the majority of scenarios, 1 − π was fixed to true values simulated for a particular scenario, 1 − π = NQTL[NL]−1, which we call the “informed prior.” However, in scenario 8, 1 − π was fixed to 57 QTL for all NQTL scenarios, 1 − π = 57 QTL[NL]−1, which we term the low prior. While not done in this study, one could estimate 1 − π instead of using a prior and this may be an advantage in real data with unknown NQTL (e.g., Pong-Wong and Hadjipavlou 2010).
Our implementation of sampling loci variances was slightly different from that of Meuwissen et al. (2001). They performed a haplotype analysis and therefore several haplotype effects would need estimating per locus and having more than one effect per locus required sampling of locus variances. In this study, biallelic loci were simulated and, because only one effect was estimated per locus, it was not necessary to sample a per-locus variance. This process of sampling from one variance is more similar to that of Xu (2003). However, his implementation assumed all loci had an effect, which is comparable to BayesA of Meuwissen et al. (2001). In contrast, our model assumed that some loci will not have an effect (π), which is more similar to BayesB. In addition, while Xu (2003) used a naïve prior for 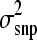 , we used an informative prior (i.e., υ and s2 = 1) as proposed by Ter Braak et al. (2005).
, we used an informative prior (i.e., υ and s2 = 1) as proposed by Ter Braak et al. (2005).
The length of the Gibbs chain was 105,000 iterations and the first 5000 iterations were discarded as burn-in. Estimates at every 20th iteration were stored as a sample resulting in a total of 5000 samples. Several analyses were carried out to test if the sampling protocol was appropriate. Autocorrelations of effects and estimated breeding values were found to be close to zero in stored samples, which showed that they were almost independent, so Monte Carlo error was < 0.0003 in all scenarios (Geyer and Thompson 1992). This allowed for shortening of the chain length to 45,000 iterations (2000 samples) for scenarios with NP = 2000 to reduce running time. Furthermore, convergence was investigated by using a variety of starting values for s2 in the BayesB. Using different starting values resulted in nearly identical estimates of 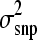 and breeding value accuracy suggesting that our Gibbs chains were converging. In addition, a long chain of 160,000 iterations was run and
and breeding value accuracy suggesting that our Gibbs chains were converging. In addition, a long chain of 160,000 iterations was run and 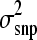 estimates and accuracy values were very similar to the shorter chain. While convergence cannot be guaranteed in any MCMC study, evidence suggests that our MCMC protocol converged.
estimates and accuracy values were very similar to the shorter chain. While convergence cannot be guaranteed in any MCMC study, evidence suggests that our MCMC protocol converged.
RESULTS
GBLUP accuracy:
The accuracy of GBLUP for a given set of values for NP and h2 stayed constant regardless of NQTL in all scenarios simulated (Figure 1 and Table 3). This confirmed our first hypothesis. The constant accuracy results from the unique Me of a random mating population, which, in turn, depends on Ne and L (Goddard 2009). The plateau of GBLUP accuracy increased when more phenotypic records were used in the estimation of breeding values and when h2 increased (Figure 1). Sampling βj from a Laplace (double exponential) distribution resulted in the same GBLUP accuracy as sampling from N(0, 1).
Figure 1.—

Accuracy of GBLUP and BayesB (informed priors for π) in validation individuals for different numbers of QTL and heritabilities (h2) when the effective population size is 1000 and the number of individuals in the training set is 1000. SE < 0.018 in all scenarios.
TABLE 3.
Accuracy of GBLUP and BayesB (informed priors for π) for different effective population sizes (Ne), numbers of QTL expressed as proportions of Me, and numbers of individuals in the training set (NP) when the heritability is 0.3. SE < 0.023 in all scenarios
| Method | Ne | NP | 0.03 Me | 0.05 Me | 0.15 Me | 0.3 Me | 0.50 Me | 0.75 Me | 1 Me |
|---|---|---|---|---|---|---|---|---|---|
| GBLUP | 200 | 200 | 0.405 | 0.450 | 0.429 | 0.414 | 0.444 | 0.416 | 0.398 |
| 1000 | 1000 | 0.505 | 0.501 | 0.508 | 0.502 | 0.507 | 0.501 | 0.511 | |
| 2000 | 2000 | 0.575 | 0.579 | 0.571 | 0.568 | 0.571 | 0.571 | 0.568 | |
| BayesB | 200 | 200 | 0.739 | 0.649 | 0.463 | 0.400 | 0.398 | 0.365 | 0.344 |
| 1000 | 1000 | 0.865 | 0.772 | 0.601 | 0.516 | 0.480 | 0.451 | 0.445 | |
| 2000 | 2000 | 0.886 | 0.812 | 0.646 | 0.573 | 0.544 | 0.522 | 0.506 |
BayesB accuracy:
In contrast to GBLUP, with BayesB the accuracy was highest at low NQTL and then decreased as NQTL increased (Figure 1 and Table 3). Once NQTL was high, BayesB reached a plateau where the accuracy does not decrease anymore despite increasing NQTL. This plateau was observed in all BayesB scenarios and the value of the accuracy at this plateau depended on Ne, h2, and NP (Tables 3 and 4). The plateau decreased when Ne increased. An increase in h2 and NP influenced the accuracy in two ways: first, it raised the overall accuracy in all NQTL scenarios, and second, it slightly shifted the onset of the accuracy plateau to higher NQTL. Sampling effects from a Laplace distribution instead of N(0, 1) raised BayesB accuracy slightly, but when NQTL was equal to 0.03 Me or NQTL > 0.5 Me no difference in accuracy was observed between sampling from both distributions.
TABLE 4.
Accuracy of GBLUP and BayesB (informed priors for π) in training (T) and validation (V) individuals for different effective population sizes (Ne), numbers of QTL (NQTL) expressed as proportions of Me, and numbers of individuals in the training set (NP) when the heritability is 0.3. SE < 0.023 in all scenarios
|
Ne = 200 |
Ne = 1000 |
||||||
|---|---|---|---|---|---|---|---|
| NQTL | Population | NP = 200 | NP =1000 | NP = 500 | NP =1000 | NP =2000 | |
| BayesB | 0.03 Me | T | 0.794 | 0.958 | 0.801 | 0.883 | 0.933 |
| V | 0.739 | 0.952 | 0.757 | 0.865 | 0.924 | ||
| 0.05 Me | V | 0.649 | 0.905 | 0.602 | 0.772 | 0.870 | |
| 0.15 Me | V | 0.463 | 0.803 | 0.421 | 0.601 | 0.744 | |
| 0.30 Me | V | 0.400 | 0.709 | 0.371 | 0.516 | 0.656 | |
| 0.50 Me | T | 0.599 | 0.778 | 0.583 | 0.657 | 0.741 | |
| V | 0.398 | 0.654 | 0.373 | 0.480 | 0.613 | ||
| 1 Me | V | 0.344 | 0.591 | 0.342 | 0.445 | 0.567 | |
| GBLUP | All | T | 0.656 | 0.771 | 0.625 | 0.682 | 0.747 |
| V | 0.444 | 0.622 | 0.407 | 0.507 | 0.612 | ||
The use of low NQTL priors for 1 − π yielded a lower accuracy than informed priors (Table 2, scenario 8, and Figure 2). The gap between the accuracy of informed and low priors increased as NQTL increased because the proportion of the genetic variance explained by the low NQTL prior became smaller.
Figure 2.—

Accuracy of BayesB in validation individuals with informed priors on π (BayesB Prior Informed) and a low prior of 57 QTL regardless of the actual number of QTL (BayesB Prior 57 QTL) when the effective population size is 1000, the number of individuals in the training set is 1000, and heritability is 0.3. Accuracy of GBLUP is included for reference. SE < 0.009 in all scenarios.
Comparison of GBLUP and BayesB:
The comparison of GBLUP and BayesB leads to several key observations. BayesB always performed better than GBLUP at low NQTL. However, as NQTL increased, the difference between the two methods became smaller and eventually both approaches achieved very similar accuracy. The NQTL at which this equivalence occurred was increased with increasing Ne, NP, and h2 (Figure 1 and Tables 3 and 4). Once NQTL increased past the equivalence point, BayesB had a slightly lower accuracy than GBLUP and settled at a constant accuracy (Table 3). The difference between GBLUP and BayesB at high NQTL decreased when NP was increased. Sampling effects from a Laplace instead of a Normal distribution did not affect these general trends.
In Figure 2, the maximum x-value of NQTL plotted is equal to the predicted Me from Goddard (2009) and one observes that BayesB accuracy approaches the plateau and becomes similar to GBLUP accuracy well below Me. A first inspection therefore suggests that the second hypothesis does not hold. However the argument for the second hypothesis is based upon the empirical 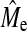 .
. 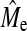 calculated with Equation 3 for h2 = 0.1, 0.3, and 0.5 by averaging over the values of NQTL gives values of 890, 900, and 700. In this context, hypothesis 2 is shown to be broadly valid, in that superiority of BayesB over GBLUP disappears when NQTL approaches
calculated with Equation 3 for h2 = 0.1, 0.3, and 0.5 by averaging over the values of NQTL gives values of 890, 900, and 700. In this context, hypothesis 2 is shown to be broadly valid, in that superiority of BayesB over GBLUP disappears when NQTL approaches 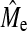 , although there is a trend for BayesB accuracy to become similar to GBLUP accuracy sooner than Me. These observations held for other scenarios. The comparison between Me and
, although there is a trend for BayesB accuracy to become similar to GBLUP accuracy sooner than Me. These observations held for other scenarios. The comparison between Me and 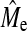 is addressed in more detail below.
is addressed in more detail below.
Decay in accuracy:
The decay in accuracy between training and validation individuals was also greater for GBLUP than that of BayesB at low NQTL (Table 4) as observed by Habier et al. (2007). However, this trend diminished as NQTL increased and the decay of accuracy reached similar levels in both methods at high NQTL.
Predictions of accuracy:
Figure 3 shows the accuracies of GBLUP and BayesB predicted with Equations 1 and 2, respectively, and the accuracies from simulations in the validation set. Predictions of GBLUP and BayesB (at high NQTL) accuracy were generally accurate. The accuracy of the predictions was highly dependent on Me. In BayesB, the drop in accuracy as NQTL increased was predicted well. Equation 2 tended to overpredict BayesB accuracy, particularly in scenarios where NQTL was a low proportion of Me, using Goddard (2009), and low h2 and NP.
Figure 3.—

Predicted (solid bars) and simulated (shaded bars) accuracy of GBLUP and BayesB for (A) a heritability (h2) of 0.3 and varying effective population size (Ne) and number of individuals in the training set (NP) and for (B) Ne = 1000 and varying h2 and NP. Different numbers of QTL expressed as proportions of Me were considered for BayesB.
Empirical 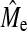 and
and  :
:
We estimated Me using the accuracy of GBLUP or BayesB (when NQTL = Me) (Equation 3). When GBLUP accuracy was used, we averaged the accuracy across all NQTL scenarios simulated for a given set of values for h2 and NP. This was done for each population replicate to obtain a standard error. It is a subhypothesis that Me as predicted by Goddard (2009) approximates 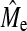 . Empirical estimates of Me using GBLUP were always lower than those using BayesB (Table 5) due to the higher GBLUP accuracy when NQTL is high. The estimates using BayesB accuracy were more variable than GBLUP as shown by the larger SE of BayesB. A general trend was apparent showing that
. Empirical estimates of Me using GBLUP were always lower than those using BayesB (Table 5) due to the higher GBLUP accuracy when NQTL is high. The estimates using BayesB accuracy were more variable than GBLUP as shown by the larger SE of BayesB. A general trend was apparent showing that 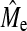 increased as NP increased, which suggests that
increased as NP increased, which suggests that 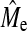 has not reached a bound; however, the change in
has not reached a bound; however, the change in 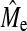 is small in relation to the difference from Me. Furthermore,
is small in relation to the difference from Me. Furthermore, 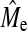 does not increase linearly with NP and this may indicate that it may be approaching asymptotic values.
does not increase linearly with NP and this may indicate that it may be approaching asymptotic values.
TABLE 5.
Estimated (M̂e) and predicted (Me and MeH) number of independent chromosome segments. Estimates were obtained from Equation 3 with mean squared accuracy of GBLUP or BayesB from 50 replicates of simulated data (±SE). The number of QTL is 1 Me and predictions are Me = 2NeL/log(4NeL) as in Goddard (2009) and MeH = 2NeL as in Hayes et al. (2009c)
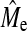 |
||||||
|---|---|---|---|---|---|---|
| Ne | NP | h2 | GBLUP | BayesB | Me | MeH |
| 200 | 200 | 0.3 | 294 ± 29 | 579 ± 73 | 445 | 4,000 |
| 200 | 1,000 | 0.3 | 487 ± 17 | 584 ± 25 | 445 | 4,000 |
| 1,000 | 1,000 | 0.5 | 883 ± 24 | 1,103 ± 27 | 1,887 | 20,000 |
| 1,000 | 1,000 | 0.3 | 890 ± 28 | 1,243 ± 33 | 1,887 | 20,000 |
| 1,000 | 1,000 | 0.1 | 822 ± 41 | 1,551 ± 111 | 1,887 | 20,000 |
| 1,000 | 500 | 0.3 | 803 ± 39 | 1,187 ± 48 | 1,887 | 20,000 |
| 1,000 | 2,000 | 0.3 | 1,014 ± 21 | 1,280 ± 26 | 1,887 | 20,000 |
| 2,000 | 2,000 | 0.3 | 1,253 ± 24 | 1,769 ± 42 | 3,543 | 40,000 |
The number of QTL controlling the trait (NQTL) was estimated using Equation 4 with reliability values from BayesB when NQTL < Me. As shown in Figure 4 for scenario 7, the estimated NQTL do follow the actual NQTL well and are predictive of the trend. Empirical  were better estimated with higher NP. Note that incorrect priors will reduce
were better estimated with higher NP. Note that incorrect priors will reduce  accuracy.
accuracy.
Figure 4.—

Actual number of QTL simulated and number of QTL predicted with Equation 4 using BayesB accuracy when the effective population size is 1000, the number of individuals in the training set is 2000, and the heritability is 0.3.
DISCUSSION
We have compared GBLUP and BayesB at various population and trait genetic architectures and at various NP. We demonstrated that GBLUP had a constant accuracy, for a given NP and h2, regardless of NQTL. The accuracy of BayesB was greatest at low NQTL, decreased with increasing NQTL, and eventually reached a lower accuracy plateau below which the accuracy did not fall even when NQTL was further increased. BayesB has an advantage over GBLUP at low NQTL, but this advantage decreased as NQTL increased and it finally diminished completely or, in some cases, the advantage switched to GBLUP depending on Ne and NP. The point at which GBLUP and BayesB accuracy became equal was related to the empirical number of independent segments estimated from the GBLUP accuracy, 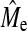 , which was less than the theoretical prediction of Me provided by Goddard (2009). It is clear from this study that quantifying the superiority of GBLUP over BayesB or vice versa depends upon three sets of attributes: the population genome structure (e.g., Ne), the trait genetic architecture (e.g., NQTL, h2), and the size of the training set. Superiority is, therefore, not a property of the method and general statements to that effect should be avoided. Furthermore, we have proposed and tested equations for the prediction of GBLUP and BayesB accuracy and the estimation of
, which was less than the theoretical prediction of Me provided by Goddard (2009). It is clear from this study that quantifying the superiority of GBLUP over BayesB or vice versa depends upon three sets of attributes: the population genome structure (e.g., Ne), the trait genetic architecture (e.g., NQTL, h2), and the size of the training set. Superiority is, therefore, not a property of the method and general statements to that effect should be avoided. Furthermore, we have proposed and tested equations for the prediction of GBLUP and BayesB accuracy and the estimation of 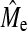 and
and  . Our predictions follow achieved GBLUP and BayesB accuracy well. Empirical
. Our predictions follow achieved GBLUP and BayesB accuracy well. Empirical 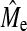 values seem to be approaching an asymptote with increasing NP and our estimates of
values seem to be approaching an asymptote with increasing NP and our estimates of  follow the trend of true NQTL.
follow the trend of true NQTL.
The constant accuracy of GBLUP, for a given h2 and NP, confirmed our first hypothesis and clearly shows that this accuracy depends crucially on genomic properties, and not on properties of the trait, and is summarized in the concept of Me. In turn, Me will depend on Ne and L, which can be viewed, in the short term, as constants in a random mating population (Goddard 2009). In a wider sense, Me and the more commonly known haplotype blocks are both measures resulting from population history and as such are related, although not interchangeable. Haplotype blocks are physical segments of the genome within which haplotype diversity is low, bounded by areas where evidence for historical recombination exists (e.g., Goldstein 2001; Gabriel et al. 2002; Frazer et al. 2007). While haplotype blocks are a physical measure, Me is a more statistical concept related to the behavior of the genome in genomic evaluations. It is theoretically derived from variation in relationship between relatives and from the continuum of variation in linkage disequilibrium across the genome (Goddard 2009). Both the number of haplotype blocks and Me increase with increasing Ne and in close relatives haplotype blocks will be long and Me will be small (Hayes et al. 2009c). It should be noted that the dependence of GBLUP on Me shown in this study does not support the conclusion that GBLUP assumes an infinitesimal model in which there are a very large number of genes each contributing a small portion to the genetic variance. In fact, GBLUP is indifferent to NQTL, unless NQTL is very small (i.e., one or < 10 QTL; unpublished results), as demonstrated in this study.
While it is clear that in GBLUP the accuracy depends on Me and not on NQTL, in BayesB the accuracy depends on the interplay of both features of genetic architecture (NQTL and Me). Our results follow our second hypothesis that the behavior of BayesB accuracy at high NQTL is similar to that of GBLUP. The accuracy of BayesB declines as NQTL increases and eventually becomes similar to GBLUP as NQTL increases. The point at which this occurs approaches Me with increasing NP. Therefore, we show that the accuracy of BayesB at high NQTL is also dependent on Me just like in GBLUP. This is also supported by the accuracy plateau being observed across similar proportions of Me and that near equivalence is approximated closely by NQTL = 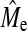 , where
, where 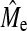 is the empirical estimate of Me obtained from GBLUP. Therefore, the plateau is not a function of actual NQTL but of Me. Another argument for
is the empirical estimate of Me obtained from GBLUP. Therefore, the plateau is not a function of actual NQTL but of Me. Another argument for 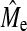 to be a major determinant in BayesB accuracy at high NQTL is that it can be accurately predicted with Equation 3. In addition, a recent study of Barley using real data has also shown that GBLUP accuracy exceeded that of BayesB when NQTL was large (Zhong et al. 2009).
to be a major determinant in BayesB accuracy at high NQTL is that it can be accurately predicted with Equation 3. In addition, a recent study of Barley using real data has also shown that GBLUP accuracy exceeded that of BayesB when NQTL was large (Zhong et al. 2009).
The difference in accuracy between GBLUP and BayesB may be explained by the way both methods process and model information. With GBLUP, each independent segment is estimated irrespective of whether it has an effect whereas with BayesB, an additional step is involved in which for each locus it is estimated if the locus has an effect or not. The fact of choosing a near-correct subset of loci with effect with BayesB increases accuracy but there is also an error associated with determining π, which depends on NL. When NQTL < Me the advantage of choosing a subset is clear but when NQTL ≥ Me this diminishes (heuristically, it is likely that each independent segment contains QTL). Thus, under the latter scenario, GBLUP performs slightly better than BayesB. This argument is further supported by the decreasing difference in accuracy between GBLUP and the BayesB accuracy plateau at high NQTL when NP is increased, because BayesB can identify the loci with effect better with more information.
The use of QTL effects sampled from a Laplace distribution instead of a Normal distribution resulted in no quantitative change to GBLUP accuracy and in only minor changes in accuracy to BayesB. Overall, the trends observed were fully consistent with the conclusions obtained with normally distributed effects, confirming other studies that have compared these effect distributions (Habier et al. 2007; Daetwyler et al. 2008; Meuwissen 2009).
It is challenging to compare statistical approaches that embody different statistical philosophies. In the genomic and animal breeding context, comparing approaches using point estimates of breeding values, such as their accuracy, is common. Breeding itself is an application of decision theory (e.g., Woolliams and Meuwissen 1993) and in model terms the value of a breeding decision may be measured as the expected rate of genetic gain (ΔG) arising from the decision. The focus on accuracy in this article arises from the “breeders equation” E[ΔG] = irσA/L, where i is standardized intensity, σA is the additive genetic standard deviation, L is generation interval, and r is the accuracy. The concept of accuracy is therefore central to the infrastructure of genetic evaluation.
This does not mean that accuracy as presented here will remain the key comparison in the future, and the richer information available from Bayesian methods may at some point overturn this paradigm. The expectation of the posterior distribution for an individual's breeding value is a natural estimate arising from a Bayesian analysis (e.g., Goddard 2009; Meuwissen et al. 2009) and also gives a natural comparison with the estimates from BLUP approaches. In addition, alternative measures of comparison to expected gain (in a Bayesian sense, minimizing the squared loss) have been considered, such as percentiles of the posterior, which will be influenced by (co)variances of candidates (e.g., Woolliams and Meuwissen 1993), or other approaches to selection such as minimax regret.
Genome-wide evaluation methods are popular because they seem to offer a solution to predicting a large numbers of parameters (i.e., loci effects) from a limited number of phenotypes, but a number of concerns have been expressed. For example, Bayesian methods without an influential prior may experience convergence problems. Investigation with noninformative priors for scale factors have resulted in BayesB accuracy similar to those using priors in this study (unpublished results). However, more investigation of convergence of genome-wide methods is needed, including minimum length of Gibbs chains, influence of priors on convergence, and optimal protocols for parallel computing. At the same time, there is concern that the priors on degree of belief and scale factors used in the BayesB (Meuwissen et al. 2001) exert too much influence, thereby preventing Bayesian learning (Gianola et al. 2009). Gianola et al. (2009) clearly show that priors influence estimates.
The findings that both GBLUP and BayesB depend significantly on Me are given more weight by the fact that the accuracy of both methods can be predicted with Equations 1 and 2, respectively. The predictions were generally accurate but limitations have also been highlighted, especially in predicting BayesB accuracy. Extensions to the formulae may be needed to predict BayesB more accurately at low NQTL relative to Me when h2 or NP are also low, and there is also a need to review whether Me as formulated by Goddard (2009) is a good predictor of 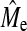 . However, being able to predict the trend in BayesB accuracy is a significant step forward (Figure 3). One of the assumptions in the original derivation (Daetwyler et al. 2008) was that all of the genetic variance was tagged by the loci used in the analysis. This represents a complication when applying our equations to predict the accuracy using a commercially available SNP chip, because the current chips are likely to miss a portion of the genetic variance. First, it is likely that the number of SNP on current chips is not high enough to tag all the genetic variance and variation not associated with SNP (e.g., copy number variation; Redon et al. 2006) will also be missed. Second, SNP with higher than average heterozygosity are selected for developing the chips and therefore loci with low minor allele frequency are proportionally underrepresented (i.e., ascertainment bias). The result of this missing genetic variance in the analysis of real populations is that our deterministic equations are likely to overpredict the accuracy in both methods.
. However, being able to predict the trend in BayesB accuracy is a significant step forward (Figure 3). One of the assumptions in the original derivation (Daetwyler et al. 2008) was that all of the genetic variance was tagged by the loci used in the analysis. This represents a complication when applying our equations to predict the accuracy using a commercially available SNP chip, because the current chips are likely to miss a portion of the genetic variance. First, it is likely that the number of SNP on current chips is not high enough to tag all the genetic variance and variation not associated with SNP (e.g., copy number variation; Redon et al. 2006) will also be missed. Second, SNP with higher than average heterozygosity are selected for developing the chips and therefore loci with low minor allele frequency are proportionally underrepresented (i.e., ascertainment bias). The result of this missing genetic variance in the analysis of real populations is that our deterministic equations are likely to overpredict the accuracy in both methods.
The fact that our equations account for the entire genetic variance will, however, be a clear advantage as the scientific community moves toward the analysis of sequence data for which our formulae are appropriate in their current form. In sequence data analysis, all basepairs are included and therefore no rare alleles would be missing. Thus, all the genetic variance is contained in the sequence and the prediction does not rely on capturing LD with the true mutation.
Additional insight into quantitative traits can be gained by combining genome-wide evaluation and deterministic prediction. We have shown that Me can be estimated with Equation 3 if the accuracy of GBLUP or BayesB is known. Two theoretical values for Me have been proposed to date, Me = 2NeL/log(4NeL) (Goddard 2009) and 2NeL (Hayes et al. 2009c). Our estimates of Me (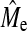 ), even though still increasing with increasing NP, remain lower than both theoretical values but were of the right order of magnitude when using Goddard's equation (Goddard 2009) rather than 2NeL (Table 5). In real data using 2NeL in Equation 1 appears to predict GBLUP accuracy well (Hayes et al. 2009b), but this may be due to the fact that SNP arrays miss a significant proportion of the genetic variance. Once more of the genetic variance is captured with new technology we would expect that estimates of
), even though still increasing with increasing NP, remain lower than both theoretical values but were of the right order of magnitude when using Goddard's equation (Goddard 2009) rather than 2NeL (Table 5). In real data using 2NeL in Equation 1 appears to predict GBLUP accuracy well (Hayes et al. 2009b), but this may be due to the fact that SNP arrays miss a significant proportion of the genetic variance. Once more of the genetic variance is captured with new technology we would expect that estimates of 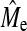 from real data would likely tend toward the derivation of Goddard (2009), or possibly lower. In addition to
from real data would likely tend toward the derivation of Goddard (2009), or possibly lower. In addition to 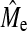 , NQTL can be estimated with BayesB accuracy if NQTL < Me. As Figure 4 shows, this can be a coarse measure of NQTL, because small changes in accuracy can cause relatively large fluctuations in
, NQTL can be estimated with BayesB accuracy if NQTL < Me. As Figure 4 shows, this can be a coarse measure of NQTL, because small changes in accuracy can cause relatively large fluctuations in  . A complication in estimating NQTL is that several SNP may be in partial LD with a particular QTL and this could lead to overestimates of NQTL. In addition, BayesB requires knowledge of the true NQTL in its prior. Nevertheless, estimates of NQTL could aid investigations into complex trait architectures, perhaps through examining the correspondence between the assumed prior on NQTL and the resulting estimate.
. A complication in estimating NQTL is that several SNP may be in partial LD with a particular QTL and this could lead to overestimates of NQTL. In addition, BayesB requires knowledge of the true NQTL in its prior. Nevertheless, estimates of NQTL could aid investigations into complex trait architectures, perhaps through examining the correspondence between the assumed prior on NQTL and the resulting estimate.
The trends observed in this study are supported by experiences in real data. Results in dairy cattle genotyped with a 50K SNP chip show that GBLUP and BayesB lead to very similar accuracies in most traits (Hayes et al. 2009a; Vanraden et al. 2009). Vanraden et al. (2009) report correlations between linear and nonlinear methods of >0.99 in a vast majority of traits. This suggests that in real animal populations quantitative traits are controlled by a large number of QTL and for most traits NQTL ≥ Me. There are of course exceptions to the rule and, for example, in dairy cattle BayesB performed better than GBLUP in milk fat content (Vanraden et al. 2009). This is likely due to a significant portion of the variation being explained by few genes of large effect, such as DGAT (Grisart et al. 2004). Hence, in this trait it is likely that NQTL < Me or that a relatively small number of QTL explain the majority of the genetic variance in the trait. We have investigated a scenario where one QTL explained 25% of the genetic variance and 1000 very small QTL explained the rest of the variance (results not shown) and the results confirmed our hypothesis.
The principles established in this study should be transferable to other populations as the trends have been confirmed across three different Ne. In our view, investigators need to gather evidence to answer two questions. First, what is the population's Me and, second, how many NQTL are likely contributing to the genetic variance in a particular trait? When NQTL ≥ Me GBLUP will result in higher accuracy than BayesB, but when NQTL < Me BayesB will outperform GBLUP.
Acknowledgments
We are grateful to Piter Bijma, Jesús Fernández, and two anonymous reviewers for their helpful and constructive comments. H.D.D. was supported by the SABRETRAIN Project, which is funded by the Marie Curie Host Fellowships for Early Stage Research Training, as part of the Sixth Framework Programme of the European Commission. B.V. received support from the Scottish Executive Environment and Rural Affairs Department (SEERAD) and Instituto Nacional de Investigación y Technologica Agraria y Alimentaria, and J.A.W. received funding from the Biotechnology and Biological Sciences Research Council (BBSRC). This work has made use of the resources provided by the Edinburgh Compute and Data Facility (ECDF, http://www.ecdf.ed.ac.uk/). The ECDF is partially supported by the eDIKT initiative (http://www.edikt.org.uk).
References
- Daetwyler, H. D., B. Villanueva, P. Bijma and J. A. Woolliams, 2007. Inbreeding in genome-wide selection. J. Anim. Breed. Genet. 124 369–376. [DOI] [PubMed] [Google Scholar]
- Daetwyler, H. D., B. Villanueva and J. A. Woolliams, 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS One 3 e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers, J. C. M., 2007. Prediction of response from marker-assisted and genomic selection using selection index theory. J. Anim. Breed. Genet. 124 331–341. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, Harlow, UK.
- Frazer, K. A., D. G. Ballinger, D. R. Cox, D. A. Hinds, L. L. Stuve et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449 851–8U3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy et al., 2002. The structure of haplotype blocks in the human genome. Science 296 2225–2229. [DOI] [PubMed] [Google Scholar]
- Geyer, C. J., and E. A. Thompson, 1992. Constrained Monte-Carlo maximum-likelihood for dependent data. J. R. Stat. Soc. Ser. B Methodological 54 657–699. [Google Scholar]
- Gianola, D., G. de los Campos, W. G. Hill, E. Manfredi and R. L. Fernando, 2009. Additive genetic variability and the Bayesian alphabet. Genetics 183 347–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, A. R., R. Thompson and B. R. Cullis, 1995. Average information REML: an efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51 1440–1450. [Google Scholar]
- Goddard, M. E., 2009. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136 245–252. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., 2001. Islands of linkage disequilibrium. Nat. Genet. 29 109–111. [DOI] [PubMed] [Google Scholar]
- Grisart, B., F. Farnir, L. Karim, N. Cambisano, J. J. Kim et al., 2004. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc. Natl. Acad. Sci. USA 101 2398–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. M. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, B. J., P. J. Bowman, A. J. Chamberlain and M. E. Goddard, 2009. a Invited review: Genomic selection in dairy cattle: progress and challenges. J. Dairy Sci. 92 433–443. [DOI] [PubMed] [Google Scholar]
- Hayes, B. J., H. D. Daetwyler, P. J. Bowman, G. Moser, B. Tier et al., 2009. b Accuracy of genomic selection: comparing theory and results. Proc. Assoc. Advmt. Anim. Breed. 17 352–355. [Google Scholar]
- Hayes, B. J., P. M. Visscher and M. E. Goddard, 2009. c Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 91 47–60. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1975. Best linear unbiased estimation and prediction under a selection model. Biometrics 31 423–447. [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38 226–231. [DOI] [PubMed] [Google Scholar]
- Lee, S. H., J. H. van der Werf, B. J. Hayes, M. E. Goddard and P. M. Visscher, 2008. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 4 e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, N., D. Gianola, G. J. Rosa, K. A. Weigel and S. Avendano, 2007. Machine learning classification procedure for selecting SNPs in genomic selection: application to early mortality in broilers. J. Anim. Breed. Genet. 124 377–389. [DOI] [PubMed] [Google Scholar]
- Lund, M. S., G. Sahana, D. J. de Koning, G. Su and O. Carlborg, 2009. Comparison of analyses of the QTLMAS XII common dataset. I. Genomic selection. BMC Proc. 3 (Suppl. 1): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., 2009. Accuracy of breeding values of ‘unrelated’ individuals predicted by dense SNP genotyping. Genet. Select. Evol. 41 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., T. R. Solberg, R. Shepherd and J. A. Woolliams, 2009. A fast algorithm for BayesB type of prediction of genome-wide estimates of genetic value. Genet. Select. Evol. 41 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NejatiJavaremi, A., C. Smith, and J. P. Gibson, 1997. Effect of total allelic relationship on accuracy of evaluation and response to selection. J. Anim. Sci. 75 1738–1745. [DOI] [PubMed] [Google Scholar]
- Pong-Wong, R., and G. Hadjipavlou, 2010. A two-step approach combining the Gompertz growth model with genomic selection for longitudinal data. BMC Proc. 4 (Suppl.): S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raadsma, H. W., G. Moser, R. E. Crump, M. S. Khatkar, K. R. Zenger et al., 2008. Predicting genetic merit for mastitis and fertility in dairy cattle using genome wide selection and high density SNP screens. Anim. Genomics Anim. Health 132 219–223. [DOI] [PubMed] [Google Scholar]
- Redon, R., S. Ishikawa, K. R. Fitch, L. Feuk, G. H. Perry et al., 2006. Global variation in copy number in the human genome. Nature 444 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg, T. R., A. K. Sonesson, J. A. Woolliams and T. H. E. Meuwissen, 2009. Reducing dimensionality for prediction of genome-wide breeding values. Genet. Select. Evol. 41 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved, J. A., 1971. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2 125–141. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Braak, C. J. F., M. P. Boer and M. C. A. M. Bink, 2005. Extending Xu's Bayesian model for estimating polygenic effects using markers of the entire genome. Genetics 170 1435–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B Methodological 58 267–288. [Google Scholar]
- VanRaden, P. M., 2008. Efficient methods to compute genomic predictions. J. Dairy Sci. 91 4414–4423. [DOI] [PubMed] [Google Scholar]
- VanRaden, P. M., C. P. Van Tassell, G. R. Wiggans, T. S. Sonstegard, R. D. Schnabel et al., 2009. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92 16–24. [DOI] [PubMed] [Google Scholar]
- Villanueva, B., R. Pong-Wong, J. Fernandez and M. A. Toro, 2005. Benefits from marker-assisted selection under an additive polygenic genetic model. J. Anim. Sci. 83 1747–1752. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., S. E. Medland, M. A. R. Ferreira, K. I. Morley, G. Zhu et al., 2006. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet.s 2 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. S., J. J. Rutledge and D. Gianola, 1994. Bayesian-analysis of mixed linear-models via Gibbs sampling with an application to litter size in Iberian pigs. Genet. Select. Evol. 26 91–115. [Google Scholar]
- Woolliams, J. A., and T. H. E. Meuwissen, 1993. Decision rules and variance response in breeding schemes. Anim. Prod. 56 179–186. [Google Scholar]
- Woolliams, J. A., R. Pong-Wong and B. Villanueva, 2002. Strategic optimisation of short- and long-term gain and inbreeding in MAS and non-MAS schemes, CD ROM communication no. 23–02 in Proceedings of the 7th World Congress of Genetics Applied to Livestock Production, Montpellier, France.
- Xu, S. Z., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N. J., and S. H. Xu, 2008. Bayesian LASSO for quantitative trait loci mapping. Genetics 179 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, S. Q., J. C. M. Dekkers, R. L. Fernando and J. L. Jannink, 2009. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: a barley case study. Genetics 182 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]