

Abstract
Genetic linkage and association studies are empowered by proper modeling of relatedness among individuals. Such relatedness can be inferred from marker and/or pedigree information. In this study, the genetic relatedness among n inbred individuals at a particular locus is expressed as an n × n square matrix Q. The elements of Q are identity-by-descent probabilities, that is, probabilities that two individuals share an allele descended from a common ancestor. In this representation the definition of the ancestral alleles and their number remains implicit. For human inspection and further analysis, an explicit representation in terms of the ancestral allele origin and the number of alleles is desirable. To this purpose, we decompose the matrix Q by a latent class model with K classes (latent ancestral alleles). Let P be an n × K matrix with assignment probabilities of n individuals to K classes constrained such that every element is nonnegative and each row sums to 1. The problem then amounts to approximating Q by PPT, while disregarding the diagonal elements. This is not an eigenvalue problem because of the constraints on P. An efficient algorithm for calculating P is provided. We indicate the potential utility of the latent ancestral allele model. For representative locus-specific Q matrices constructed for a set of maize inbreds, the proposed model recovered the known ancestry.
HIGH-THROUGHPUT techniques allow extensive genotyping of individuals for thousands of SNP markers (Gibbs et al. 2003) and thereby provide accurate information about the genetic diversity within a population at many chromosomal loci. If two individuals within this population carry the same DNA sequence at a locus, and this sequence can be traced to the same common ancestor, the individuals are said to be identical by descent (IBD) for this segment (Chapman and Thompson 2003). Quite often, however, the ancestral source of a chromosomal segment is ambiguous and thus IBD relationships between haplotypes are given as probabilities. Various methods have been described to estimate the IBD probability of pairs of chromosomal segments (Meuwissen and Goddard 2001; Leutenegger et al. 2003). When pedigree relationships are known, these can be included to estimate IBD probabilities (Wang et al. 1995; Heath 1997; George et al. 2000; Meuwissen and Goddard 2000; Besnier and Carlborg 2007).
In quantitative genetic analysis we seek to find and characterize associations between the large number of SNPs that are now available for many organisms and phenotypic variation for traits of interest (e.g., grain yield and time to flowering). Many current methods developed for this purpose make use of IBD information. For example, a locus-specific matrix of IBD probabilities can be incorporated into restricted maximum-likelihood (REML) procedures for fine mapping quantitative trait loci (Bink and Meuwissen 2004) as well as for marker-based genetic evaluation (Fernando and Grossman 1989) using mixed models. The IBD matrix takes the role of a covariance matrix in the REML procedure.
Other approaches, however, require that chromosome segments (also referred to here as haplotypes or alleles) are assigned to independent ancestors. These approaches include regression approaches with genetic predictors (Malosetti et al. 2006) and Bayesian oligo-allelic approaches that sample the ancestral origin of each chromosomal segment (Heath 1997; Uimari and Sillanpaa 2001; Bink et al. 2008a). In the IBD matrix representation the ancestral alleles and their number remain implicit. For these approaches, the locus-specific matrix of IBD probabilities must therefore be decomposed into a matrix that links the chromosomal segments to independent ancestral alleles. This decomposition is addressed in this article.
The individuals that we consider in this article are inbred. For n inbred individuals the IBD matrix at a given chromosomal position is thus n × n, because there is no need to distinguish between identical chromosomes. In diploid, outbred populations, each individual would be represented by two haplotypes (alleles) and the matrix would be 2n × 2n (Fernando and Grossman 1989). This is feasible if any phase ambiguity can be resolved. From now on, the term “individual” thus means chromosomal segment or haplotype. Analogously, ancestor will be shorthand for ancestral allele (ancestral haplotype).
We propose two models of IBD matrix decomposition, a simple threshold model (TIBD) and a more sophisticated latent ancestral allele model (LAAM), that provide (1) an estimate of the number of independent ancestral alleles, (2) a concise, easy-to-interpret, summary of the relatedness, (3) an explicit (probabilistic) representation of the descent of alleles, and (4) the ability to sample alleles for each individual from a set of ancestral alleles in such a way that the probability that a pair of individuals shares the same allele corresponds to their IBD probability.
The last two features of the model are essential for its use in Bayesian oligo-allelic approaches to quantitative trait locus (QTL) analysis (Uimari and Sillanpaa 2001; Bink et al. 2008a).
STATISTICAL METHODS
Data and motivation:
For a set of n inbred individuals, let Q be an n × n square matrix with elements qij denoting the probability that individuals i and j are IBD. In our genetic context, the elements of Q could be the IBD probability for a specific gene, marker, chromosomal segment, or haplotype. Equivalently, the Q matrix could have values that are a weighted measure across specific genomic segments or the whole genome. For the scope of this article Q is taken to be measured at a specific chromosomal locus.
An example of a Q matrix constructed for six individuals is shown in Table 1A. Individual I1 has a unique allele. The alleles of individuals I2–I4 descend from a common ancestor. The individuals I5 and I6 are IBD with probability 0.7. The IBD relationships displayed in this matrix can arise if the individuals inherit their alleles from four common ancestral alleles, labeled A1–A4 in Table 1B. Individual I1 inherits from the unique ancestral allele A1 and individuals I2–I4 all inherit from the ancestral allele A2 in Table 1. The IBD probability of 0.7 between individuals I5 and I6 may arise if I5 always has a copy of the ancestral allele A3 and I6 has a copy of A3 with probability 0.7 and a copy of another ancestral allele (named A4 in Table 1) with probability 0.3. We note that the solution is not unique. For instance, an IBD probability of 0.7 also arises with I5 receiving a copy from A3 and A4 with probabilities 0.25 and 0.75 and I6 receiving a copy from A3 and A4 with probabilities 0.1 and 0.9, respectively, since 0.25 × 0.1 + 0.75 × 0.9 = 0.7. Furthermore, solutions with more than four ancestral alleles would also give a perfect fit.
TABLE 1.
Artificial 6 × 6 Q matrix for six individuals labeled I1–I6 (A) and a 6 × 4 matrix P with ancestor classes labeled A1–A4 (B), giving a perfect fit to the off-diagonal elements of Q by the formula PPT
| A. Q | B. P | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I1 | I2 | I3 | I4 | I5 | I6 | A1 | A2 | A3 | A4 | ||
| I1 | 1 | 0 | 0 | 0 | 0 | 0 | I1 | 1 | 0 | 0 | 0 |
| I2 | 0 | 1 | 1 | 1 | 0 | 0 | I2 | 0 | 1 | 0 | 0 |
| I3 | 0 | 1 | 1 | 1 | 0 | 0 | I3 | 0 | 1 | 0 | 0 |
| I4 | 0 | 1 | 1 | 1 | 0 | 0 | I4 | 0 | 1 | 0 | 0 |
| I5 | 0 | 0 | 0 | 0 | 1 | 0.7 | I5 | 0 | 0 | 1 | 0 |
| I6 | 0 | 0 | 0 | 0 | 0.7 | 1 | I6 | 0 | 0 | 0.7 | 0.3 |
The goal of this article is to develop a model that has an explicit, preferably probabilistic, representation for the descent of the allele of each individual from a common set of ancestral founders, but without further usage of the pedigree and/or marker data. Because there is no pedigree information beyond the information contained within the matrix Q, the ancestral founders of the intended model are unknown and therefore “latent” as they can only be hypothesized. The number of ancestral founders is also unknown, but we hypothesize K ancestors from now on for some value of K. The choice of the value of K is discussed later on.
We begin with a basic model of inheritance in which the allele of each individual descends from one out of K latent ancestral alleles. In this model the individuals can be partitioned into K classes (ancestral alleles) and the transitivity property applies: if the alleles for individuals I1 and I2 are inherited from the same ancestor, and the alleles for individuals I1 and I3 are inherited from the same ancestor, then the alleles for individuals I2 and I3 must be inherited from the same ancestor.
TIBD model:
The threshold model transforms the Q matrix into a discrete St matrix by applying the following rule
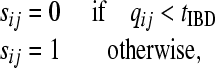 |
where tIBD is the threshold, sij is the IBD status for individuals i and j that can only take values 0 or 1, and as defined above qij is the probability that individuals i and j are IBD. By sliding tIBD between 0 and 1 we obtain different St, some of which define a partition of individuals with each class containing IBD individuals. The partition with the least-squares fit to Q is taken as the final model.
LAAM:
In the LAAM we extend the basic inheritance model with probabilities. Let P be an n × K matrix with K the number of latent ancestors (classes) and elements pik being the probability that the allele of individual i descends from ancestor k. Note that
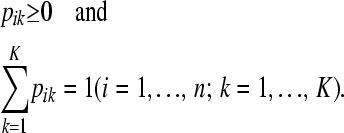 |
(1) |
In this model we do not know whether the allele of individual i is inherited from ancestor k, but only the probability of this inheritance. On assuming independence of inheritance for each pair of individuals, the probability that individuals i and j inherited from the same ancestor is, according to the model,
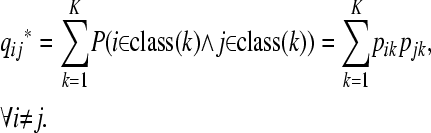 |
(2) |
Mathematically, the {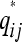 } are coincidence probabilities induced by a latent class model with membership probabilities P. Our aim is to find a matrix P such that
} are coincidence probabilities induced by a latent class model with membership probabilities P. Our aim is to find a matrix P such that 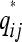 is as close as possible to qij for all i ≠ j in some well-defined sense. To do so we minimize the loss
is as close as possible to qij for all i ≠ j in some well-defined sense. To do so we minimize the loss
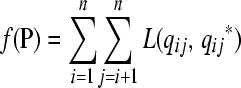 |
(3) |
with L(a, b) a nonnegative loss function, such as least-squares loss, L(a, b) = (a − b)2, and 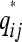 a function of P as defined in Equation 2. The best P, the one that minimizes f(P), is the latent ancestor approximation of the IBD matrix Q. Note that the columns of P can be reordered arbitrarily without changing the approximation to Q.
a function of P as defined in Equation 2. The best P, the one that minimizes f(P), is the latent ancestor approximation of the IBD matrix Q. Note that the columns of P can be reordered arbitrarily without changing the approximation to Q.
If the loss is small, we have thus obtained an explicit inheritance model for the alleles of the individuals that accurately approximate the IBD probabilities in Q, which was calculated from pedigree and/or marker information. The descent probabilities of alleles of individuals from latent ancestors are given in the matrix P and the key identity to arrive at IBD probabilities is Equation 2, which can also be written in matrix notation as Q* = PPT, while disregarding the diagonal elements of Q*. Here Q* is the approximation of Q. In shorthand, the latent ancestor model thus reads Q ≈ PPT. The decomposition cannot be obtained from an eigen analysis (Gourlay and Watson 1973; Press et al. 2002) because of the constraints on P.
A special case of Equation 2 is that the elements of P are 0 or 1, resulting in elements of Q* being 0 and 1. Then P represents a division of the individuals into disjoint groups and the elements are indicators of group membership. Such groups are easy to identify from Q directly as all its elements are then (up to approximation error) 0 or 1 and transitivity holds. By consequence, there is no need to apply more advanced methods such as eigen analysis of Q (Noy-Meir 1973) or of the Laplacian of Q (Newman 2006).
For overlapping groups, eigen analysis yields eigenvectors that cannot easily be transformed to probabilities. For overlapping groups some elements of P are between 0 and 1 (fuzzy or graded), and any form of fuzzy clustering could be applied. Many such methods, however, have no explicit underlying model. We interpret the graded elements as probabilities, explicitly use model (2), and develop methods to obtain the best P to approximate the IBD matrix Q. Additive fuzzy clustering (Sato and Sato 1994) has an explicit underlying model and can be interpreted as a latent class model by viewing the graded elements as probabilities (Ter Braak et al. 2009). LAAM is the genetic version of this model in which Q contains IBD probabilities and P contains descent probabilities from latent ancestors (classes).
Algorithm for the LAAM:
We use least squares for solving the latent ancestral allele model. The problem then is to minimize the loss function
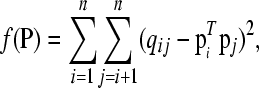 |
(4) |
where 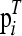 denotes the ith row of P, subject to the nK nonnegativity and n equality constraints in Equation 1. The loss can be reported in terms of the root mean squared error (RMSE), defined as
denotes the ith row of P, subject to the nK nonnegativity and n equality constraints in Equation 1. The loss can be reported in terms of the root mean squared error (RMSE), defined as 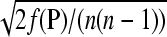 .
.
The loss function set forth in Equation 4 is not convex, which raises the possibility of multiple local minima, even beyond local minima generated by rearrangement of the columns of P. ter Braak et al. (2009) presented two algorithms to solve Equation 4. Both were able to find the best solution for n up to 100 and K up to 50. The first used a global optimization method known as differential evolution whereas the second, which was ∼O(n2) more efficient, used iterative row-wise quadratic programming (IRW), as follows.
IRW algorithm:
Step 1. Initialize P; for example, simply fill each row with random uniform numbers between 0 and 1, which are then divided by their sum, to satisfy the constraints of Equation 1.
Step 2. While f(P) decreases do the following: For i = 1, …, n minimize f(P) over the ith row pi, while keeping the other rows of P fixed.
The IRW algorithm is efficient because updating the ith row of P while keeping the other rows of P fixed leads to a quadratic program (Ter Braak et al. 2009). In appendix a we provide an algorithm for step 2 that is up to a factor 2 faster than that presented in ter Braak et al. (2009). It is based on an adaptation of the famous lasso path algorithm (Efron et al. 2004; Rosset and Zhu 2007) that we call the nonnegative least-squares (NNLS)-path algorithm (appendix b). It is a direct method for least-squares estimation of the coefficients of a linear regression model subject to both positivity and sum constraint on the coefficients. The algorithm (Bink et al. 2010) was implemented in Matlab and is freely available upon request for noncommercial purposes.
Methods to choose K:
The choice of K in the LAAM can be made in a variety of ways. ter Braak et al. (2009) minimize the Akaike information criterion (AIC), which for unknown variance is defined as AIC = N log(f (P)) + 2p* with N = n(n − 1)/2, the number of observations, and p* = n(K − 1), the number of parameters. An alternative approach, which we apply in this article, is to set the number of ancestral classes equal to its maximum (the number of individuals), estimate the best fitting matrix P, and then determine how many columns of the matrix P contain nonzero elements.
Summary statistics on P:
The number of columns (K) of P with positive column sum is the actual number of latent ancestors. Some column sums may be very small compared to others so that the effective number of the latent ancestors is lower than K. This is because the sum of the kth column of P, denoted by p+k, is the expected number of individuals that inherit from the kth latent ancestor. A measure for effective number of latent ancestors is
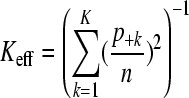 |
(Hill 1973), which gives values between 1 and K. If there is (almost) no genetic diversity among the individuals (all IBD probabilities close to 1), Keff is (close to) 1 and (almost) all individuals inherit from the same latent ancestor. In such a case, association to phenotypes cannot be detected. The other extreme is that all ancestors have the same number of descendants (p+k = n/K), yielding Keff = K. Note that 1/Keff is the Simpson index (Simpson 1949), which can be interpreted as the probability that two randomly chosen individuals inherit from the same ancestor.
The number of latent ancestors and the effective number of latent ancestors can also be usefully defined for the ith individual by the number of nonzero elements in the vector pi and by 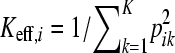 , respectively. The certainty about the inheritance of a particular individual in the set of n individuals under consideration is expressed on a 0-to-1 scale by 1/Keff,i.
, respectively. The certainty about the inheritance of a particular individual in the set of n individuals under consideration is expressed on a 0-to-1 scale by 1/Keff,i.
EXAMPLES
Two artificial examples:
We now discuss the decomposition of the two artificial examples in Tables 1 and 2.
TABLE 2.
Artificial 6 × 6 Q matrix for six individuals labeled I1–I6 (A) and threshold transformed matrices St for t = 0.6 (B) and 0.8 (C)
| A. Q | B. S0.6 | C. S0.8 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I1 | I2 | I3 | I4 | I5 | I6 | I1 | I2 | I3 | I4 | I5 | I6 | I1 | I2 | I3 | I4 | I5 | I6 | |||
| I1 | 1 | 0.9 | 0.2 | 0 | 0.1 | 0 | I1 | 1 | 1 | 0 | 0 | 0 | 0 | I1 | 1 | 1 | 0 | 0 | 0 | 0 |
| I2 | 0.9 | 1 | 0.1 | 0 | 0 | 0 | I2 | 1 | 1 | 0 | 0 | 0 | 0 | I2 | 1 | 1 | 0 | 0 | 0 | 0 |
| I3 | 0.2 | 0.1 | 1 | 0 | 0 | 0 | I3 | 0 | 0 | 1 | 0 | 0 | 0 | I3 | 0 | 0 | 1 | 0 | 0 | 0 |
| I4 | 0 | 0 | 0 | 1 | 0.8 | 0.7 | I4 | 0 | 0 | 0 | 1 | 1 | 1 | I4 | 0 | 0 | 0 | 1 | 1 | 0 |
| I5 | 0.1 | 0 | 0 | 0.8 | 1 | 0.9 | I5 | 0 | 0 | 0 | 1 | 1 | 1 | I5 | 0 | 0 | 0 | 1 | 1 | 1 |
| I6 | 0 | 0 | 0 | 0.7 | 0.9 | 1 | I6 | 0 | 0 | 0 | 1 | 1 | 1 | I6 | 0 | 0 | 0 | 0 | 1 | 1 |
For the example of Table 1, TIBD with tIBD = 0.6 results in an S matrix that is identical to Q except that the IBD probability between individuals I5 and I6 is 1. This yields a three-class solution with minimum RMSE (0.077). Each class is by definition a latent ancestor. With tIBD = 0.8 we obtain a four-class solution with I5 and I6 forming singleton classes, but this solution has higher RMSE (0.181). LAAM using the IRW algorithm was able to find a perfect fitting P (RMSE = 0) with four classes (Table 1B). IRW required between 5 and 10 iterations depending on the initial configuration.
Table 2 shows another 6 × 6 example of Q. TIBD with tIBD = 0.6 yields the minimum RMSE (0.118) and three groups of individuals, namely I1 + I2, I3, and I4 + I5 + I6, respectively (Table 2B). Note that TIBD does not yield a partition for some values of the threshold. For example, with tIBD = 0.8 we obtain an inconsistent S matrix (Table 2C); pair (I4, I5) and pair (I5, I6) are IBD while pair (I4, I6) is not (Table 2C). This transitivity problem may be solved by adaptation of the threshold. Increasing the threshold to 0.85 yields a four-class solution with RMSE = 0.284 whereas decreasing the threshold to 0.6 yields the best solution shown in Table 2B.
The minimum RMSE values that we found with LAAM were 0.254, 0.046, 0.022, 0.021, and 0.021 for two to six classes, respectively. IRW was thus not able to find a perfect fitting P, not even with six classes, which happens when Q is measured with error. Table 3 shows the solution with four classes. The classes A1 and A3 express the coancestry between individuals I1 and I2 and between I4, I5, and I6. Class A2 expresses the uniqueness of individual I3 and class A4 is needed to fit Q in more detail. The solution for K = 5 essentially splits class A4 in two, yielding a slightly better fit. Table 3 also illustrates the indexes derived from P. From the column sums of P (last row) the classes A1 and A3 show many more offspring than the other two classes. Because of this unevenness, the overall effective number of ancestors is not 4 but 2.9. The effective number of ancestors for individuals (Keff,i) varies between 1 and 1.5. The certainty of descent (last column of Table 3) is largest (1) for individual I2 that inherits from ancestor A1 only and smallest (0.67) for individual I4 that inherits from either A3 or A4. Individuals I1 and I6 may inherit from three different ancestors, but have a higher certainty than individual I4 because of their very uneven descent pattern.
TABLE 3.
Best-fitting 6 × 4 matrix P (A) with ancestors labeled A1-A4 for the Q matrix of Table 2, together with derived indexes (B)
| A. P |
B. No. ancestorsa |
||||||
|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | K0 | Keff | Cert | |
| I1 | 0.88 | 0.09 | 0.03 | 0 | 3 | 1.3 | 0.79 |
| I2 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| I3 | 0.12 | 0.88 | 0 | 0 | 2 | 1.3 | 0.80 |
| I4 | 0 | 0 | 0.79 | 0.21 | 3 | 1.5 | 0.67 |
| I5 | 0.02 | 0 | 0.98 | 0 | 2 | 1 | 0.97 |
| I6 | 0 | 0.01 | 0.90 | 0.10 | 3 | 1.2 | 0.81 |
| 2.02b | 0.98b | 2.7b | 0.31b | 4 | 2.9 | 0.34 | |
K0 (Keff), (effective) number of ancestors; Cert, certainty of descent.
Column sum equals the expected number of offspring from the latent ancestor.
Case study at 12 representative loci:
We also applied TIBD and the LAAM to 12 matrices expressing the IBD probabilities (Q) between 16 highly related elite inbred maize genotypes at 12 independent loci. Each Q matrix was calculated using a proprietary estimation method on the basis of the available pedigree and marker information. The pedigree that gave rise to the 16 inbreds totaled 142 inbred individuals and contained multiple complex loops. The longest lineage for any of the 16 individuals used in our study to its ancestral founders was nine generations. The markers that were used to calculate the IBD probabilities were selected from highly dense sets of markers of a variety of types, such as SSR and SNP. The markers spanned the entire genome and were positioned on proprietary genetic maps. Within ∼1 cM of the 12 loci we had on average 4.3 markers; the low value was 1 marker and the high value was 10 markers. Markers farther away also contributed in the calculation of Q. We used a proprietary estimation method to calculate Q, but numerous methods exist for creating such matrices from marker and/or pedigree data (see discussion).
This case study provides a unique opportunity to investigate whether the LAAM is able to reconstruct the allele flow in the pedigree from Q alone. For this purpose we compared the LAAM solution (P) consisting of descent probabilities of the genotypes from latent ancestors, with a matrix F consisting of descent probabilities of the genotypes from the known founders of the pedigree. Each F was calculated using the same methods and information as Q. In fact, F and Q are disjoint parts of the full IBD matrix for both founders and inbreds. At the 12 loci, there were between three and seven founders that contributed to the genotypes of the 16 inbreds.
Figure 1 shows the TIBD fit St, the LAAM fit 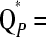 PP′, and the founder-based fit
PP′, and the founder-based fit 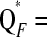 FF′. Clearly, TIBD fits the data much worse than the LAAM and is therefore disregarded in the next comparisons.
FF′. Clearly, TIBD fits the data much worse than the LAAM and is therefore disregarded in the next comparisons.
Figure 1.—

Error (RMSE) at 12 loci in the fit of Q by TIBD, the LAAM, and the descent probabilities to known founders in the pedigree.
The LAAM provided a perfect fit (RMSE < 0.001) in 9 of the 12 IBD matrices between the 16 maize genotypes (Figure 1). In 7 of these, the LAAM solution P is essentially equal to the founder-based one (F). One example of this is given in Figure 2; interchanging the first and the fourth column of F yields the LAAM solution matrix P. Note that the individuals were rearranged solely to improve readability of the figures. The matrix Q shows three major blocks of high IBD linked by individuals (numbered 5 and 11) that may be IBD with two of them (Figure 2). The LAAM solution matrix P represents the three blocks as latent ancestors 1, 2, and 4. Individual 5 inherits with probabilities 0.73 and 0.27 from the first and second, respectively, and individual 11 inherits with probabilities 0.15, 0.60, and 0.20 from latent ancestors 2, 3, and 4. Individual 11 thus introduces an extra latent ancestor (A3) as does individual 12 (last row), giving in total five ancestors, which can thus be perfectly matched with the known founders (Figure 2). The F matrix of Figure 2 shows that for many individuals the origin of the allele can be followed through the pedigree without much ambiguity (descent probability >0.9) whereas the allele origin for individuals 5, 11, and 12 remains uncertain so that some of their descent probabilities are intermediate between 0 and 1.
Figure 2.—

IBD matrix Q and associated descent probability matrices P and F at locus 1. Note that interchanging columns 1 and 4 in F gives matrix P.
We now consider a case (locus 4 in Figure 1) where the LAAM did not provide a perfect fit, but, judged on RMSE, fitted better than the founder-based model (Figures 3 and 4). The overall better fit is due to the central block consisting of nine genotypes that have unit IBD probabilities among one another. These individuals inherit from a single latent ancestor in the LAAM, whereas they inherit with probabilities 0.17 and 0.83 from founders 1 and 3, respectively. By consequence the fitted IBD probability is correct (1.0) in the LAAM and incorrect (0.72) in the founder-based model (Figure 4). The reason for the difference is that these individuals have a more recent common ancestor in the pedigree (Figure 5). The difference is maximum (0.5) with the descent probabilities given in Figure 5: the founder-based model gives an IBD probability of 2(0.5)2 = 0.5 whereas the true IBD is 1.0.
Figure 3.—

IBD matrix Q and associated descent probability matrices P and F at locus 4. Note that P and F are essentially different. Ancestors or founders with a column sum <0.05 are not shown.
Figure 4.—

Fitted IBD matrices at locus 4 corresponding to P (left) and F (right).
Figure 5.—

A more recent common ancestor (M) explains the mismatch between IBD between individuals I1 and I2 derived from the founders F1 and F2 in the pedigree via 2 × 2 matrix F with all entries equal to 0.5 and that derived from the latent ancestor A1 via 2 × 1 matrix P = (1, 1)′.
However, some IBD probabilities are fitted much better in the founder-based model, in particular those between individuals 16, 9, and 1 (top left block in Figures 3 and 4). We obtained a much better fit by representing the group of individuals that are IBD with probability 1 by a single individual. The LAAM applied to the reduced Q matrix gives a near perfect fit and yields latent ancestors that correspond well with the known founders (Figures 6 and 7). Interestingly, the central block in Figure 3, represented in Figures 6 and 7 by G2, may inherit from two latent ancestors. Such a solution was effectively ruled out as the LAAM solution of the full Q matrix since it would induce too low intragroup IBD probabilities.
Figure 6.—

Reduced IBD matrix Q and associated descent probability matrices P and F at locus 4, with G2 representing the central block of individuals in Figures 3 and 4. Note that P and F are similar, except for the descent of individuals 5 and 14. Ancestors or founders with a column sum <0.05 are not shown.
Figure 7.—

Fitted reduced IBD matrices at locus 4 corresponding to P (left) and F (right), giving RMSE 0.024 and 0.035, respectively.
We therefore also applied the LAAM to reduced Q matrices in which any group of IBD individuals is replaced by a single individual. Then, the LAAM gave a near perfect fit at all 12 loci. The latent ancestors found by the LAAM corresponded very well with known founders with P ≈ F, except at loci 10 and 12 where the LAAM identified a more recent common ancestor and so yielded fewer ancestors than founders (Figure 5).
DISCUSSION
This article proposes two models for approximating an IBD matrix for a population of n inbred individuals. The first model, the TIBD model, is straightforward to implement and simple to interpret but shows limitations in its ability to accurately approximate IBD matrices. The second model, the LAAM, corrects the deficiencies of the TIBD approach while still being computationally tractable and easy to interpret. Moreover, the LAAM was able to recover the known ancestors from real Q matrices with negligible error.
In this article we applied the LAAM to small examples that allowed us to verify the genetic validity of the decomposition. ter Braak et al. (2009) successfully applied the LAAM for n = 100 and K = 50 in simulations for both highly structured and ill-structured Q matrices. The estimated K differed by at most 3 from the true K. Our new algorithm achieved the same in less time. van Eeuwijk et al. (2010) analyzed 117 maize inbreds along a 1-cM grid throughout the genome using the LAAM and found good agreement with the known ancestry. The CPU time was ∼4 min per locus. The largest example so far had n = 600 and K = 27.
A typical data analyst will presumably start from marker data and possibly also from a genetic map and a pedigree. The first step is then to choose an appropriate method to estimate the relatedness among the individuals in terms of IBD probabilities, either genome-wide or locus specific, and the second step is to apply the method of this article, resulting in descent probabilities of latent ancestors. The first step is far from trivial although a number of methods exist for creating a similarity matrix between individuals, as well as genome-wide (Van De Casteele et al. 2001; Bink and Meuwissen 2004) and locus specific (Heath 1997; George et al. 2000; Meuwissen and Goddard 2001; Pong-Wong et al. 2001; Leutenegger et al. 2003; Besnier and Carlborg 2007). An advantage of our two-step approach is that the analyst is free to choose his own preferred method in the first step.
In association mapping numerous methods have been proposed to detect population structure, of which STRUCTURE (Pritchard and Rosenberg 1999; Pritchard et al. 2000), EIGENSTRAT (Patterson et al. 2006; Price et al. 2006), and multidimensional scaling (Zhu and Yu 2009) are important examples. What is the relationship with the LAAM and is there a role for the LAAM in association mapping? Let us first limit the discussion to STRUCTURE and the LAAM. STRUCTURE works directly from the marker data and, possibly, a genetic map (Pritchard et al. 2000), but not a pedigree, and produces latent ancestral populations, with linkage equilibrium and Hardy–Weinberg equilibrium within populations. The difference with the latent ancestral alleles of the LAAM is that populations have internal genetic variation whereas alleles have not. We note that the output of STRUCTURE looks very similar to our matrix P, but has a different meaning. In STRUCTURE it contains, for each individual, the proportions of its genome deriving from each of these populations, whereas in the LAAM it contains each individual's descent probabilities from the latent ancestral alleles. If STRUCTURE were applied on the chromosomal segment scale of our examples, it would produce close-to-crisp output as recombination is low on such a scale. The LAAM thus seems better suited than STRUCTURE for the chromosomal segment scale. STRUCTURE is thus primarily intended for the genome scale with latent classes representing admixture or genetic background, whereas the LAAM is designed for the chromosomal segment scale with latent classes representing different allele origins that potentially have different effects on the phenotype. The genome-wide kinship matrix can be used to adjust these effects for genetic background, even without decomposition (Kang et al. 2008; Van Eeuwijk et al. 2010).
In comparison with EIGENSTRAT, the LAAM allows the relationship matrix to be chosen, whereas it is predetermined in EIGENSTRAT (Zhu and Yu 2009). A comparison with (nonmetric) multidimensional scaling is more difficult. In general, the LAAM is called for if the output of the decomposition needs to be probabilities.
On the potential role for the LAAM in association mapping, we distinguish between the genome level (genetic background) and the chromosomal segment level (possible QTL effects). On the genome level, if we could directly estimate the probability that any two individuals are from the same population and collect the estimates in Q, then the LAAM would be the method of choice for finding the latent populations. However, in practice Q is a genome-wide relatedness matrix such as an identity-by-state allele-sharing kinship matrix (Bink et al. 2008b; Kang et al. 2008). Then LAAM could be useful for small K, but our method to choose K would not, as it would produce far too many clusters. The reason is that latent ancestors are assumed to be unique genotypes without internal variability. In this context K could be decided upon by another method, such as from a plot of RMSE against the number of classes, with K being the value where the decrease in RMSE tapers off. On the chromosomal segment level, the estimated Q is locus specific and integrates the information of a series of markers close to the locus. LAAM classes then replace the marker information in association mapping. The potential of this two-step approach over marker-based approaches such as fastPHASE (Scheet and Stephens 2006) will likely depend on the availability of pedigree information.
The key identity in the LAAM is Equation 2, which gives the IBD probability of two individuals, i.e., the probability that they inherit the allele from the same ancestor, as a function of descent probabilities from latent ancestors. The function is derived by assuming independence among the ancestors and among individuals given their ancestors. This assumption makes the model interpretable, but also constrains what can be fitted. This is the reason that a perfect fit is not always possible. In our application to maize genotypes we obtained a suboptimal fit when the data contained groups of IBD individuals. The group of closely related individuals forced the LAAM to consider them as a latent ancestor with unit descent probabilities for these individuals (Figure 3). A near-perfect fit was obtained when such groups were replaced by a single representative. After reduction the group can have nonzero descent probability for more than a single latent ancestor (Figure 6). We advise that this reduction should always be performed prior to analysis as it improves the fit and does not make sampling from the model more difficult. In the example of Figure 6 it just means that the draw of an ancestor for G2 applies to all the individuals of that group, so that they are always IBD. In practice, one may wish to merge close-to-IBD individuals, because of error in the IBD probability estimates.
In our current implementation of the LAAM, the reduction step is therefore slightly generalized as follows. We use UPGMA agglomerative clustering (Sneath and Sokal 1973) to merge individuals until the average between-cluster IBD is smaller than a predetermined threshold and then use the LAAM algorithm on the reduced Q. The generalization may be viewed as an integration of TIBD and the LAAM, with TIBD taking care of high IBD probabilities and the LAAM taking care of the intermediate ones. We also stress that the LAAM solution does not need to be perfectly fitting to be useful.
We believe that the utility of the LAAM is manifold. We name a few such utilities:
The matrix P is much smaller in size than the matrix Q if K ≪ n, which makes it easier to deal with both for human inspection and for computer representation.
The matrix P gives an explicit probabilistic representation of descent of alleles of individuals from a set of latent ancestral alleles. The elements of P have a clear meaning; they are the descent probabilities of the n individuals at a specified locus with the K latent ancestral alleles.
Each row of P is associated with a specified individual and indicates the number of ancestors that effectively contributed to the genotype of that individual at a specified locus.
The value of K (Keff) that gives a good approximation to Q indicates the (effective) number of ancestors that actually contribute to the genotype of the individuals at a specified locus.
In many cases in which a genotyped pedigree is available the latent ancestors can be identified as being the most recent common ancestors in the pedigree.
The matrix P makes it possible to sample or draw ancestors for each of the n individuals in such a way that the probability that individual i and j have a common ancestor is their identity-by-descent probability for all i ≠ j (i = 1, … , n; j = 1, … , n). Each such sample is an explicit possible way of descent of the individuals from the set of latent ancestors.
Utilities 2 and 6 are of foremost importance in regression approaches with genetic predictors (Malosetti et al. 2006) and in oligo-allelic Bayesian methods (Bink et al. 2008a; Van Eeuwijk et al. 2010) for quantitative trait locus identification that cannot work with the matrix Q directly.
APPENDIX A: ALGORITHM FOR SOLVING THE LATENT ANCESTRAL ALLELE MODEL
This appendix describes step 2 in the IRW algorithm in the main text for solving the latent ancestral allele model (Bink et al. 2010). We are given an n × n IBD matrix Q and wish to find an n × K matrix P such that Q ≈ PPT. The problem thus is to minimize the loss function
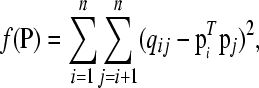 |
(A1) |
where 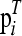 denotes the ith row of P, subject to the nK nonnegativity and n equality constraints
denotes the ith row of P, subject to the nK nonnegativity and n equality constraints
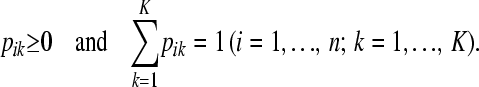 |
(A2) |
In fitting the ith row we minimize f(P) over pi, while keeping the other rows of P fixed. Let qi denote the ith column of Q without qii and P−i denote matrix P after deleting row i. The fitting of pi amounts to
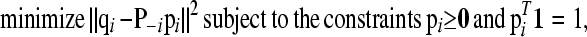 |
(A3) |
where 0 and 1 denote vectors of appropriate lengths with all zero and unit elements, respectively. This is a quadratic program but with the difficulty that P–i is singular, because each row of P–i sums to unity. Without the constraints the least-squares solution would not be unique. However, with the equality constraint, the number of independent parameters is reduced from K to K − 1. The difficulty can therefore be solved easily as follows.
As each row of P sums to unity, a column of P can be deleted as we show now. We delete the last column, i.e., column K. With the (K − 1) vector 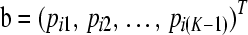 , we can write
, we can write
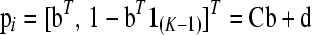 |
(A4) |
with K-vector 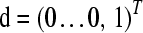 , with the “1” in position K, and K × (K − 1) matrix
, with the “1” in position K, and K × (K − 1) matrix
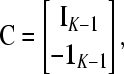 |
where IK−1 is a (K − 1) × (K − 1) identity matrix and 1K−1 is a (K − 1) vector of ones. Then by inserting (A4) into (A3) for both pi and each row of P–i and by defining the (N − 1) × (K − 1) matrix X with elements 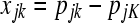 and the N − 1 vector y with elements
and the N − 1 vector y with elements 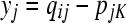 for j = 1, … , i − 1, i + 1, … , N and k = 1, … , (K − 1), we arrive at the following equivalent problem: find b to
for j = 1, … , i − 1, i + 1, … , N and k = 1, … , (K − 1), we arrive at the following equivalent problem: find b to
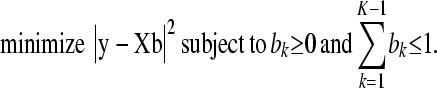 |
(A5) |
After having found the solution to problem (A5), we obtain the solution to problem (A3) by back transformation of (A4), namely 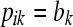 for k = 1, … , K − 1 and
for k = 1, … , K − 1 and 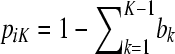 .
.
There are several ways to solve problem (A5) because it is a standard quadratic program (Gill et al. 1981). We mention in particular the Least Squares with Inequality constraints (LSI) algorithm by Lawson and Hanson (1974), which uses two other of their algorithms; LSI calls the Least Distance Programming (LDP) program that in its turn calls the NNLS program. This sequence of call appears rather inefficient as (A5) is almost a NNLS problem in itself. The only difference with an NNLS is the sum constraint (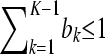 ). In appendix b we propose a new, direct algorithm for the NNLS problem with sum constraint. The algorithm (NNLS-path) is an adaptation of the lasso-path algorithm invented by Efron et al. (2004) and further improved and generalized by Rosset and Zhu (2007).
). In appendix b we propose a new, direct algorithm for the NNLS problem with sum constraint. The algorithm (NNLS-path) is an adaptation of the lasso-path algorithm invented by Efron et al. (2004) and further improved and generalized by Rosset and Zhu (2007).
The NNLS-path algorithm starts with b = 0, and thus with piK = 1, and step by step increases the sum over the b coefficients until the sum is equal to 1 or, if the unconstrained NNLS solution has sum t* < 1, to t*. By consequence, piK decreases to 0 or a positive value. The number of steps can be decreased by rearranging the P matrix such that piK is the maximum of all pik for a given i. This is done before each particular row is fitted. This completes the description of step 2 of the IRW algorithm.
APPENDIX B: NNLS-PATH ALGORITHM
This appendix describes a lasso-path approach to nonnegative least squares with sum constraint (Bink et al. 2010).
Some algorithms for finding lasso solutions (Tibshirani 1996) are based on nonnegative least squares with a sum constraint. This problem was originally solved using standard quadratic programming techniques (Tibshirani 1996). Efron et al. (2004) developed a very efficient new algorithm for finding lasso solutions, which was further improved and generalized by Rosset and Zhu (2007). This algorithm is known as the lasso-path algorithm. In this appendix we turn things around and use the lasso-path algorithm for obtaining an efficient algorithm for nonnegative least squares with a sum constraint. We take Rosset and Zhu (2007) as our starting point and use their notation:
The data are the n × p design matrix
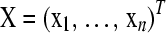 and response vector
and response vector 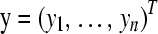 .
.The unknown regression coefficient vector is
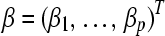 , which is required to be nonnegative; that is,
, which is required to be nonnegative; that is, 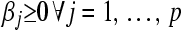 .
.L(., .) is a convex nonnegative loss functional.
J(.) is a convex nonnegative penalty functional with J(0) = 0. In this appendix we use
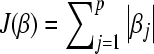 . Because βj ≥ 0, this is equivalent with
. Because βj ≥ 0, this is equivalent with 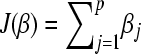 .
.
The problem we consider is to find
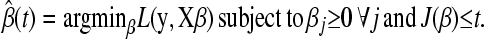 |
(B1) |
In the latent ancestral allele model t = 1. For the least-squares loss functional problem
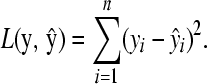 |
(B1) is the NNLS problem with a sum constraint.
We also need 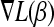 , the derivative of L with respect to β with
, the derivative of L with respect to β with 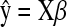 . In the least-squares case,
. In the least-squares case,
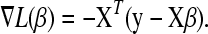 |
The proof of Theorem 2 of Rosset and Zhu (2007) shows the relation of the lasso solution with the NNLS problem with a sum constraint and can trivially be simplified to it by deleting (or zeroing) all 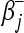 terms (which indicate negative regression coefficients). We modified their Algorithm 1 accordingly, using the notation that A is the set of active variables, AC is its complement, and
terms (which indicate negative regression coefficients). We modified their Algorithm 1 accordingly, using the notation that A is the set of active variables, AC is its complement, and 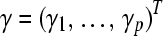 is a p-vector, with
is a p-vector, with 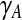 the elements of γ belonging to set A. As all active variables will have an equal gradient, we use for this common value also the shorthand
the elements of γ belonging to set A. As all active variables will have an equal gradient, we use for this common value also the shorthand 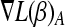 . Steps involving “d3” in Rosset and Zhu (2007) are removed as they deal with the cases beyond least squares.
. Steps involving “d3” in Rosset and Zhu (2007) are removed as they deal with the cases beyond least squares.
The algorithm for the nonnegative least-squares problem with a sum constraint (NNLSpath) is as follows:
- Initialize:
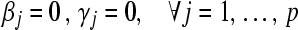 , so that
, so that 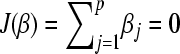 .
.- Calculate
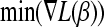 , the minimum of the gradient vector
, the minimum of the gradient vector 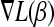 and the variable jmin for which the minimum is attained.
and the variable jmin for which the minimum is attained. - If
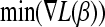 < 0, set λ = −
< 0, set λ = −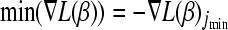 and
and 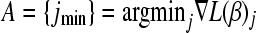 ; else set λ = 0.
; else set λ = 0.
- While (λ > 0 and
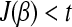 ):
):- Calculate a (new) direction
where XA is the matrix containing the columns of X corresponding to the variables in A, and 1A is a ones vector of the size of set A, and the elements of γ not belonging to set A are set to 0.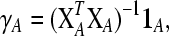
- Calculate the step length d to be taken in this direction:
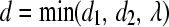 , where
, where 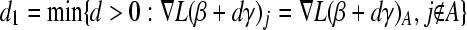 (equal gradient values attained); if no such variable is found
(equal gradient values attained); if no such variable is found 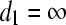 .
. 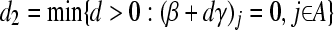 (hit 0); if no such variable is found
(hit 0); if no such variable is found 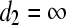 .
. - Take step
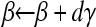 .
. - If
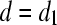 , then add to set A the variable attaining equality at d. If
, then add to set A the variable attaining equality at d. If 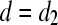 , then remove from set A the variable attaining 0 at d. If
, then remove from set A the variable attaining 0 at d. If 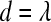 , then do nothing.
, then do nothing. - Modify λ:
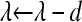 .
.
After step 2: if
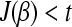 , exit; otherwise set
, exit; otherwise set 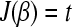 by changing β by
by changing β by
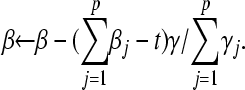 |
This is the end of the algorithm.
After each run we check numerically whether the algorithm yielded the global minimum by verifying the Karush–Kuhn–Tucker (KKT) conditions. These conditions are as follows:
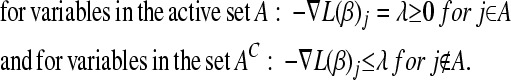 |
(B2) |
These conditions hold true by design of the algorithm. We describe now explicitly the calculations implied by 2b of the algorithm in the least-squares case. For calculating
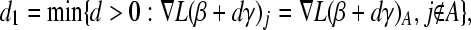 |
we must find for each 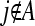 a value of d such that
a value of d such that
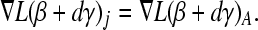 |
(B3) |
The left-hand side of (B3) is
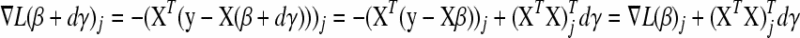 |
and the right-hand side of (B3) is simply
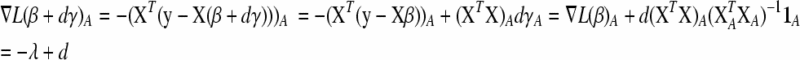 |
as 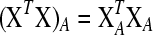 . Solving of (B3) for d gives
. Solving of (B3) for d gives
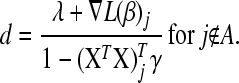 |
(B4) |
Variables for which 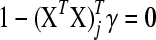 are assigned
are assigned 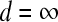 ; such variables do not need to be included in the active set A, as they satisfy condition (B2) for all new λ − d ≥ 0. The solution for d1 is the minimum positive value of so calculated d's. In these formulas
; such variables do not need to be included in the active set A, as they satisfy condition (B2) for all new λ − d ≥ 0. The solution for d1 is the minimum positive value of so calculated d's. In these formulas 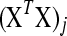 is the jth column of the XTX matrix.
is the jth column of the XTX matrix.
Calculating
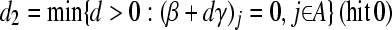 |
amounts to calculating
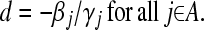 |
The solution for d2 is the minimum positive value of so calculated d's.
References
- Besnier, F., and Ö. Carlborg, 2007. A general and efficient method for estimating continuous IBD functions for use in genome scans for QTL. BMC Bioinformatics 8 e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bink, M., and T. H. E. Meuwissen, 2004. Fine mapping of quantitative trait loci using linkage disequilibrium in inbred plant populations. Euphytica 137 95–99. [Google Scholar]
- Bink, M., M. Boer, C. ter Braak, J. Jansen, R. Voorrips et al., 2008. a Bayesian analysis of complex traits in pedigreed plant populations. Euphytica 161 85–96. [Google Scholar]
- Bink, M. C. A. M., A. D. Anderson, W. E. van de Weg and E. A. Thompson, 2008. b Comparison of marker-based pairwise relatedness estimators on a pedigreed plant population. Theor. Appl. Genet. 117 843–855. [DOI] [PubMed] [Google Scholar]
- Bink, M. C. A. M., C. J. F. ter Braak, O. S. Smith and L. R. Totir, 2010. Statistical approach for optimal use of genetic information collected on historical pedigrees, genotyped with dense marker maps, into routine pedigree analysis of active maize breeding populations, U.S. Patent Application Publication US2010/0095394.
- Chapman, N. H., and E. A. Thompson, 2003. A model for the length of tracts of identity by descent in finite random mating populations. Theor. Popul. Biol. 64 141–150. [DOI] [PubMed] [Google Scholar]
- Efron, B., T. Hastie, I. Johnstone and R. Tibshirani, 2004. Least angle regression. Ann. Stat. 32 407–499. [Google Scholar]
- Fernando, R. L., and M. Grossman, 1989. Marker assisted selection using best linear unbiased prediction. Genet. Sel. Evol. 21 467–477. [Google Scholar]
- George, A. W., P. M. Visscher and C. S. Haley, 2000. Mapping quantitative trait loci in complex pedigrees: a two-step variance component approach. Genetics 156 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, R. A., J. W. Belmont, P. Hardenbol, T. D. Willis, F. L. Yu et al., 2003. The international HapMap project. Nature 426 789–796. [DOI] [PubMed] [Google Scholar]
- Gill, P. E., W. Murray and M. H. Wright, 1981. Practical Optimization. Academic Press, London.
- Gourlay, A. R., and G. A. Watson, 1973. Computational Methods for Matrix Eigenproblems. Wiley, New York.
- Heath, S. C., 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, M. O., 1973. Diversity and evenness: a unifying notation and its consequences. Ecology 54 427–432. [Google Scholar]
- Kang, H. M., N. A. Zaitlen, C. M. Wade, A. Kirby, D. Heckerman et al., 2008. Efficient control of population structure in model organism association mapping. Genetics 178 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, C. L., and R. J. Hanson, 1974. Solving Least Squares Problems. Prentice-Hall, Englewood Cliffs, NJ.
- Leutenegger, A. L., B. Prum, E. Genin, C. Verny, A. Lemainque et al., 2003. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 73 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malosetti, M., R. G. F. Visser, C. Celis-Gamboa and F. A. van Eeuwijk, 2006. QTL methodology for response curves on the basis of non-linear mixed models, with an illustration to senescence in potato. Theor. Appl. Genet. 113 288–300. [DOI] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2000. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 155 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2001. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 33 605–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, M. E. J., 2006. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E 74 036104. [DOI] [PubMed] [Google Scholar]
- Noy-Meir, I., 1973. Data transformation in ecological ordination. I. Some advantages of non-centering. J. Ecol. 61 329–341. [Google Scholar]
- Patterson, N., A. L. Price and D. Reich, 2006. Population structure and eigenanalysis. PLoS Genet. 2 2074–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong-Wong, R., A. W. George, J. A. Woolliams and C. S. Haley, 2001. A simple and rapid method for calculating identity-by-descent matrices using multiple markers. Genet. Sel. Evol. 33 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 2002. Numerical Recipes in C++. The Art of Scientific Computing, Ed. 2. Cambridge University Press, Cambridge, UK.
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. A. Rosenberg, 1999. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 65 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosset, S., and J. Zhu, 2007. Piecewise linear regularized solution paths. Ann. Stat. 35 1012–1030. [Google Scholar]
- Sato, M., and Y. Sato, 1994. An additive fuzzy clustering model. Jpn. J. Fuzzy Theory Syst. 6 185–204. [Google Scholar]
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, E. H., 1949. Measurement of diversity. Nature 163 688. [Google Scholar]
- Sneath, P. H. A., and R. R. Sokal, 1973. Numerical Taxonomy. Freeman, San Francisco.
- ter Braak, C. J. F., Y. A. I. Kourmpetis, H. A. L. Kiers and M. C. A. M. Bink, 2009. Approximating a similarity matrix by a latent class model: a reappraisal of additive fuzzy clustering. Comp. Stat. Data Anal. 53 3183–3193. [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. B 58 267–288. [Google Scholar]
- Uimari, P., and M. J. Sillanpaa, 2001. Bayesian oligogenic analysis of quantitative and qualitative traits in general pedigrees. Genet. Epidemiol. 21 224–242. [DOI] [PubMed] [Google Scholar]
- Van De Casteele, T., P. Galbusera and E. Matthysen, 2001. A comparison of microsatellite-based pairwise relatedness estimators. Mol. Ecol. 10 1539–1549. [DOI] [PubMed] [Google Scholar]
- van Eeuwijk, F., M. Boer, L. R. Totir, M. Bink, D. Wright et al., 2010. Mixed model approaches for the identification of QTLs within a maize hybrid breeding program. Theor. Appl. Genet. 120 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T., R. L. Fernando, S. van der Beek, M. Grossman and J. A. M. van Arendonk, 1995. Covariance between relatives for a marked quantitative trait locus. Genet. Sel. Evol. 27 251–274. [Google Scholar]
- Zhu, C., and J. Yu, 2009. Nonmetric multidimensional scaling corrects for population structure in association mapping with different sample types. Genetics 82 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]