

Abstract
Kir channels are important in setting the resting membrane potential and modulating membrane excitability. A common feature of Kir2 channels and several other ion channels that has emerged in recent years is that they are regulated by cholesterol, a major lipid component of the plasma membrane whose excess is associated with multiple pathological conditions. Yet, the mechanism by which cholesterol affects channel function is not clear. We have recently shown that the sensitivity of Kir2 channels to cholesterol depends on residues in the CD loop of the cytosolic domain of the channels with one of the mutations, L222I, abrogating cholesterol sensitivity of the channels completely. Here we show that in addition to Kir2 channels, members of other Kir subfamilies are also regulated by cholesterol. Interestingly, while similarly to Kir2 channels, several Kir channels, Kir1.1, Kir4.1 and Kir6.2Δ36 were suppressed by an increase in membrane cholesterol, the function of Kir3.4* and Kir7.1 was enhanced following cholesterol enrichment. Furthermore, we show that independent of the impact of cholesterol on channel function, mutating residues in the corresponding positions of the CD loop in Kir2.1 and Kir3.4*, inhibits cholesterol sensitivity of Kir channels, thus extending the critical role of the CD loop beyond Kir2 channels.
Cholesterol is one of the major lipid components of the plasma membrane in mammalian cells and it is essential for cell function and growth. The excess of cholesterol, however, is associated with multiple pathological conditions1–3 including the development of cardiovascular disease4–6. Multiple studies have shown that an increase in membrane cholesterol regulates the function of a variety of ion channels, including different types of K+ channels7–11, Ca+2 channels12–16, Na+ channels17,18 and Cl− channels19,20. Multiple types of ion channels were also shown to be associated with cholesterol-rich membrane domains21. It is also known that the impact of cholesterol on different types of ion channels is highly heterogeneous22. In most cases an increase in cholesterol levels leads to a decrease in channel function7,11,16,18,19 while in others, it leads to an increase in channel activity18,23. For example, cholesterol enrichment resulted in decreased activity of Ca2+ sensitive channels7, voltage-gated Na+ channels18, N-type Ca2+ channels16, volume-sensitive Cl− channels19 and the inwardly rectifying K+ channel, Kir2.111. On the other hand, epithelial Na+ channels (eNaC)23 and TrpC channels14 were inhibited by cholesterol depletion. However, little is still known about the mechanisms by which cholesterol regulates the function of ion channels. Our recent study provided the first structural insights into cholesterol sensitivity of ion channels by demonstrating that cholesterol sensitivity of Kir2 channels, a subfamily of inwardly rectifying K+ channels, critically depends on a specific loop in the C-terminus domain24. Here we extend our studies to test the effect of cholesterol on the representatives of all subfamilies of Kir channels. Our new observations show that effects of cholesterol are highly heterogenous even within the same family of ion channels but that corresponding structural domains may be important regardless of whether cholesterol has an inhibitory or a stimulatory effect.
Kir channels regulate multiple cellular functions in a variety of tissues including membrane excitability in neurons, cardiomyocytes and muscle cells, heart rate, vascular tone, insulin release and salt flow across epithelia. Fifteen members within the Kir channel family have been identified and classified in seven subfamilies (Kir1–7)25. We have previously shown that Kir2 channels are suppressed by the elevation of membrane cholesterol and enhanced by cholesterol depletion11,24,26. We have also shown that this effect is specific in terms of differential sensitivity of the channels to different cholesterol analogues and that most likely it should be attributed to a shift in channel conformation from active to silent26. The impact of cholesterol on other types of Kir channels, however, has not been systematically studied. Both Kir3 and Kir4 channels have been shown to partition into lipid rafts27,28. Yet, the implication of these observations in terms of cholesterol sensitivity of the channels is unclear. Kir3.1/3.2 fusion proteins were shown to partition into lipid rafts when expressed in CHO cells but their activity was not affected by blocking cholesterol synthesis with lovastatin27. Interestingly, however, regulation of Kir3.1/3.2 channels by Neural Cell Adhesion Molecule (NCAM) was compromised by lovastatin treatment suggesting that integrity of lipid rafts are important for the interactions of Kir3 channels with NCAM27. Kir4 channels were also found to partition into lipid rafts whose disruption resulted in loss of channel function28. Moreover, the role of cholesterol in regulation of Kir6 channels is quite controversial as well. While some studies have suggested that Kir6 channels are enhanced by plasma hypercholesterolemia29, other studies suggest that hypercholesterolemia blocks Kir6 channels30.
In this study, we first examine the effect of cholesterol enrichment on a representative homomeric member of each of the functional Kir subfamilies. Since Kir5.1 is not functional as a homomer, we concentrate on the remaining six subfamilies and include homomers of Kir1.1, Kir2.1, Kir3.4*, Kir4.1, Kir6.2Δ36 and Kir7.1. Kir3.4* refers to the homomerically active Kir3.4 pore mutant S143T31, and Kir6.2Δ36 refers to the C-terminal truncation mutant that renders these channels active as homomers in the absence of the SUR subunits32. We used the Xenopus oocyte heterologous expression system to test the effect of cholesterol on different Kir subfamily members.
Figure 1 depicts the effect of cholesterol enrichment on the activity of the representative Kir channels listed above. In agreement with data obtained for Kir2.1 expressed in endothelial cells11,26, and in CHO cells11,24, the function of Kir2.1 channels expressed in Xenopus oocytes was also significantly suppressed by cholesterol enrichment. As can be seen in Fig. 1A, similarly to Kir2.1, Kir1.1 and Kir6.2Δ36 showed a significant and comparable decrease in currents following cholesterol enrichment. In contrast, Kir3.4* currents were significantly enhanced by an increase in membrane cholesterol. The remaining two channels, Kir4.1 and Kir7.1, were also affected by cholesterol but to a lesser extent. While these two channels were less sensitive to cholesterol, the effect of cholesterol was systematic and consistent in different batches of oocytes. Specifically, comparison of four different batches of oocytes in each of these cases shows that Kir4.1 function is always suppressed by cholesterol enrichment while Kir7.1 currents are always enhanced by elevation of membrane cholesterol (see Fig. 1B).
Figure 1. Effect of cholesterol enrichment on Kir channels.

A. Whole cell basal currents of control and cholesterol-enriched Xenopus oocytes injected with Kir channels. Expression of channel proteins in oocytes was accomplished by injection of the following amounts of cRNA per oocyte: Kir1.1 and Kir7.1: 5ng; Kir2.1: 0.5ng; Kir3.4* and Kir6.2Δ36: 2ng; Kir4.1: 1ng. Two-electrode voltage clamp recordings were performed 1(for Kir2.1) −2 (for all other channels) days after injection. Oocytes injected with Kir6.2Δ36 were treated with 3mM NaAzide for 5min prior to recording. High-potassium solution was used to superfuse oocytes (in mM: 96 KCl, 1 NaCl, 1 MgCl2, 5 HEPES-KOH buffer, pH 7.4). Basal currents represent the difference of inward currents obtained at −80 mV in the presence of 3 mM (or 10mM for Kir7.1) BaCl2 in high-potassium solution from those in the absence of Ba2+. 2–6 batches of oocytes were tested for each normalized recording shown as detailed in the figure caption of Fig. 1B. For each construct, the total number of [control, enriched] oocytes tested was: Kir1.1 [19,20], Kir2.1 [64,52], Kir3.4* [45,33], Kir4.1 [28,26], Kir6.2Δ36 [15,10] and Kir7.1 [25,15]. Recordings from different batches of oocytes were normalized to the mean of whole-cell basal currents obtained from control untreated oocytes. Statistics (i.e., mean and standard error) of each construct were calculated from all of the normalized data recorded from different batches of oocytes. B. Average of normalized mean of whole-cell basal currents obtained from different batches of cholesterol enriched oocytes. The number of [batches] tested was: Kir1.1 [3], Kir2.1 [6], Kir3.4* [5], Kir4.1[4], Kir6.2Δ36 [2] and Kir7.1 [4]. The mean of each batch of control untreated oocytes was normalized to one, and therefore the average of the means of all batches of untreated oocytes was one, which is represented by the dashed line in the figure. The averages of all cholesterol enriched batches of oocytes were significantly different than one, as indicated by the asterisk (*p < 0.05).
These results demonstrate that even within the Kir family of channels itself, both the sensitivity and the impact of cholesterol on channel function vary. First, some Kir channels were more sensitive to cholesterol (Kir1.1, Kir2.1, Kir3.4* and Kir6.2Δ36) than others (Kir4.1, Kir7.1). Furthermore, the most striking observation is that while the function of most Kir channels was suppressed by elevation of cholesterol levels (Kir1.1, Kir3.1, Kir4.1 and Kir6.2Δ36), the function of two members of the Kir family, Kir3.4* and Kir7.1 was enhanced under the same conditions.
One possibility to explain the striking difference in cholesterol sensitivity between Kir2.1 and Kir3.4* is to suggest that cholesterol interacts with the two channels in completely different ways. An alternative possibility, however, is that the same structural domains of the two channels are responsible for their cholesterol sensitivity even though the functional effects are opposite. Comparison of the crystallographic structures of the cytosolic domains of Kir2.133, Kir3.133,34,36 and Kir3.235 shows that they share high structural similarity. This is evident, for example, in the structural alignment of the crystallized cytosolic domains of Kir2.1 and Kir3.1 depicted in Fig. 2A, which suggests that the topology of the cytosolic domain of Kir channels is conserved. It is expected, therefore, that Kir3.4* channels share the same structure of the cytosolic domain. To discriminate between the two possibilities described above, we examined whether cholesterol sensitivities of Kir2.1 and Kir3.4* depend on the same domain in the C-termini of the channels. Thus, since as we have recently shown24 mutations of residues in the CD loop (see Fig. 2A–C) of the cytosolic domain of Kir2.1 significantly decrease the sensitivity of Kir2.1 to cholesterol, we examined whether a similar effect would be observed for Kir3.4* channels. More specifically, since in Kir2.1, the L222I mutation resulted in the largest effect on cholesterol sensitivity in CHO cells24, we examined the equivalent position in Kir3.4* (Fig. 2B–C). Here we show that similarly to our previous study in CHO cells, L222I mutation significantly decreased cholesterol sensitivity of Kir2.1 channels in oocytes. Most importantly, the equivalent mutation in Kir3.4* channels, I229L, had the same effect on cholesterol sensitivity of Kir3.4* channels (Fig. 2D).
Figure 2. Effect of mutation in the CD loop of Kir2.1 and Kir3.4* on cholesterol sensitivity of the channels.
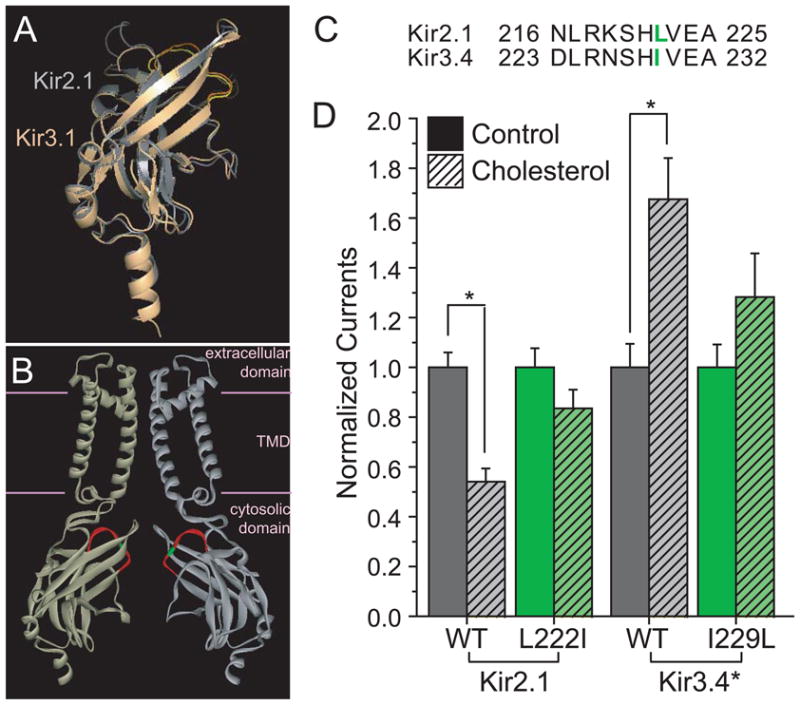
A. Structural alignment of the crystallographic structures of the cytosolic domains of Kir2.1 and Kir3.1. The Root Mean Square Deviation (RMSD) between the structures of the cytosolic domains of Kir2.1 (gray) (PDB accession no. 1U4F) and Kir3.1 (light orange) (PDB accession no. 1U4E) is only 1.1Å despite a sequence identity of only 54.2%. The CD loop is colored yellow in Kir2.1 and orange in Kir3.1. B. Homology model showing two opposite facing subunits of Kir2.1. The CD loop is colored red and the position of L222 is highlighted green. Due to the high structural similarity between the cytosolic domains of Kir2 and Kir3 channels as depicted in Fig. 2A, I229 in Kir3.4* would be located at approximately the same position. The structure shown is a homology model based on the cytosolic domain of Kir2.1 (PDB accession no. 1U4F) and the TMD of KirBac3.1 (PDB accession no. 1XL4). C. Alignment of the CD loop residues in Kir2.1 and Kir3.4. The residues included in the alignment correspond to the segment highlighted red in Fig. 2B, the CD loop. L222 in Kir2.1 and I229L in Kir3.4 are colored green and correspond to the residue highlighted in green in the homology model, Fig. 2B. D. Effect of CD-loop residue on the sensitivity to cholesterol of Kir2.1 and Kir3.4*. Whole cell basal currents of control and cholesterol-enriched Xenopus oocytes injected with Kir2.1, its L222I mutant, Kir3.4* and its I229L mutant. The recordings were obtained as described in the Figure caption of Fig. 1A. For each construct, the total number of [control, enriched] oocytes tested was: Kir2.1 [64,52], Kir2.1_L222I [35,32], Kir3.4* [45,33], Kir3.4*_I229L [17,12]. Number of [batches] tested: Kir2.1 [6], Kir2.1_L222I [4], Kir3.4* [5], Kir3.4*_I229L [3]. Significant difference is indicated by the asterisk (*p < 0.05).
Thus, although the impact of cholesterol enrichment on Kir2.1 and Kir3.4* function is opposite, cholesterol sensitivity in both channels is affected in a similar manner following mutations at the corresponding position in the CD loop of the cytosolic domain of the channel. This implies that the role of the CD loop in affecting cholesterol sensitivity extends beyond Kir2 channels to other Kir subfamilies, irrespective of the impact of cholesterol on the channel. Furthermore, these observations demonstrate that opposite effects of cholesterol on different ion channels may still be mediated by a common mechanism.
References
- 1.Kellner-Weibel G, Geng YJ, Rothblat GH. Cytotoxic cholesterol is generated by the hydrolysis of cytoplasmic cholesteryl ester and transported to the plasma membrane. Atherosclerosis. 1999;146:309–319. doi: 10.1016/s0021-9150(99)00155-0. [DOI] [PubMed] [Google Scholar]
- 2.Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 3.Yeagle PL. Modulation of membrane function by cholesterol. Biochimie. 1991;73:1303–1310. doi: 10.1016/0300-9084(91)90093-g. [DOI] [PubMed] [Google Scholar]
- 4.Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med. 2001;1:633–653. doi: 10.2174/1566524013363212. [DOI] [PubMed] [Google Scholar]
- 5.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- 7.Bolotina V, Omelyanenko V, Heyes B, Ryan U, Bregestovski P. Variations of membrane cholesterol alter the kinetics of Ca2+-dependent K+ channels and membrane fluidity in vascular smooth muscle cells. Pflugers Archives. 1989;415:262–268. doi: 10.1007/BF00370875. [DOI] [PubMed] [Google Scholar]
- 8.Heaps CL, Tharp DL, Bowles DK. Hypercholesterolemia abolishes voltage-dependent K+ channel contribution to adenosine-mediated relaxation in porcine coronary arterioles. Am J Physiol Heart Circ Physiol. 2005;288:H568–576. doi: 10.1152/ajpheart.00157.2004. [DOI] [PubMed] [Google Scholar]
- 9.Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM. Differential Targeting of Shaker-like Potassium Channels to Lipid Rafts. J Biol Chem. 2000;275:7443–7446. doi: 10.1074/jbc.275.11.7443. [DOI] [PubMed] [Google Scholar]
- 10.Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific Localization of Voltage-gated K+ Channels to Distinct Lipid Raft Populations. Targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- 11.Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–3861. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambudkar IS. Cellular Domains That Contribute to Ca2+ Entry Events. Sci STKE. 2004:pe32. doi: 10.1126/stke.2432004pe32. [DOI] [PubMed] [Google Scholar]
- 13.Bowles DK, Heaps CL, Turk JR, Maddali KK, Price EM. Hypercholesterolemia inhibits L-type calcium current in coronary macro-, not microcirculation. J Appl Physiol. 2004;96:2240–2248. doi: 10.1152/japplphysiol.01229.2003. [DOI] [PubMed] [Google Scholar]
- 14.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 15.Lundbaek JA, Birn P, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 16.Toselli M, Biella G, Taglietti V, Cazzaniga E, Parenti M. Caveolin-1 expression and membrane cholesterol content modulate N-type calcium channel activity in NG108–15 cells. Biophys J. 2005;89:2443–2457. doi: 10.1529/biophysj.105.065623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundbaek JA, Birn P, Hansen AJ, Sogaard R, Nielsen C, Girshman J, Bruno MJ, Tape SE, Egebjerg J, Greathouse DV, Mattice GL, Koeppe RE, II, Andersen OS. Regulation of sodium channel function by bilayer elasticity: The importance of hydrophobic coupling. Effects of micelle-forming amphiphiles and cholesterol. J Gen Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu CC, Su MJ, Chi JF, Chen WJ, Hsu HC, Lee YT. The effect of hypercholesterolemia on the sodium inward currents in cardiac myocyte. J Mol Cell Cardiol. 1995;27:1263–1269. doi: 10.1016/s0022-2828(05)82388-0. [DOI] [PubMed] [Google Scholar]
- 19.Levitan I, Christian AE, Tulenko TN, Rothblat GH. Membrane cholesterol content modulates activation of volume-regulated anion current (VRAC) in bovine endothelial cells. J Gen Physiol. 2000;115:405–416. doi: 10.1085/jgp.115.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romanenko VG, Rothblat GH, Levitan I. Sensitivity of volume-regulated anion current to cholesterol structural analogues. J Gen Physiol. 2004;123:77–88. doi: 10.1085/jgp.200308882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807. doi: 10.1016/j.cardiores.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Levitan I. Cholesterol and Kir channels. IUBMB Life. 2009;61:781–790. doi: 10.1002/iub.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlyonsky VG, Mies F, Sariban-Sohraby S. Epithelial sodium channel activity in detergent-resistant membrane microdomains. Am J Physiol Renal Physiol. 2003;284:F182–188. doi: 10.1152/ajprenal.00216.2002. [DOI] [PubMed] [Google Scholar]
- 24.Epshtein Y, Chopra AP, Rosenhouse-Dantsker A, Kowalsky GB, Logothetis DE, Levitan I. Identification of a C-terminus domain critical for the sensitivity of Kir2.1 to cholesterol. Proc Natl Acad Sci USA. 2009;106:8055–60. doi: 10.1073/pnas.0809847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reimann F, Ashcroft FM. Inwardly rectifying potassium channels. Curr Opin Cell Biol. 1999;11:503–8. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- 26.Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–22. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delling M, Wischmeyer E, Dityatev A, Sytnyk V, Veh RW, Karschin A, Schachner M. The Neural Cell Adhesion Molecule Regulates Cell-Surface Delivery of G-Protein-Activated Inwardly Rectifying Potassium Channels Via Lipid Rafts. J Neurosci. 2002;22:7154–7164. doi: 10.1523/JNEUROSCI.22-16-07154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hibino H, Kurachi Y. Distinct detergent-resistant membrane microdomains (lipid rafts) respectively harvest K+ and water transport systems in brain astroglia. Eur J Neurosci. 2007;26:2539–2555. doi: 10.1111/j.1460-9568.2007.05876.x. [DOI] [PubMed] [Google Scholar]
- 29.Mathew V, Lerman A. Altered effects of potassium channel modulation in the coronary circulation in experimental hypercholesterolemia. Atherosclerosis. 2001;154:329–335. doi: 10.1016/s0021-9150(00)00493-7. [DOI] [PubMed] [Google Scholar]
- 30.Genda S, Miura T, Miki T, Ichikawa Y, Shimamoto K. KATP channel opening is an endogenous mechanism of protection against the no-reflow phenomenon but its function is compromised by hypercholesterolemia. J Am Coll Cardiol. 2002;40:1339–1346. doi: 10.1016/s0735-1097(02)02156-3. [DOI] [PubMed] [Google Scholar]
- 31.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J Biol Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 32.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 33.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 34.Nishida M, MacKinnon R. Structural Basis of Inward Rectification: Cytoplasmic Pore of the G Protein-Gated Inward Rectifier GIRK1 at 1.8A Resolution. Cell. 2002;111:957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 35.Inanobe A, Matsuura T, Nakagawa A, Kurachi Y. Structural diversity in the cytoplasmic region of G protein-gated inward rectifier K+ channels. Channels. 2007;1:39–45. [PubMed] [Google Scholar]
- 36.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–15. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]