

Abstract
V(D)J recombination assembles immunoglobulin (Ig) heavy or light chain (IgH or IgL) variable region exons in developing bone marrow B cells, while class switch recombination (CSR) exchanges IgH constant region exons in peripheral B cells. Both processes employ DNA double strand breaks (DSBs) repaired by non-homologous end-joining (NHEJ). Errors in either V(D)J recombination or CSR can initiate chromosomal translocations, including oncogenic IgH/c-myc translocations of peripheral B cell lymphomas. Collaboration between these processes also has been proposed to initiate translocations. However, occurrence of V(D)J recombination in peripheral B cells is controversial. Here, we report that activated NHEJ-deficient splenic B cells accumulate V(D)J recombination-associated IgL chromosomal breaks, as well as CSR-associated IgH breaks, often in the same cell. Moreover, IgL breaks frequently are joined to IgH breaks to form translocations, a phenomenon associated with specific IgH/IgL co-localization. IgH and c-myc also co-localize in these cells; correspondingly, introduction of frequent c-myc DSBs robustly promotes IgH/c-myc translocations. Our studies reveal peripheral B cells that attempt secondary V(D)J recombination and elucidate a role for mechanistic factors in promoting recurrent translocations in tumors.
Recombination activating gene 1/2 (RAG) endonuclease initiates V(D)J recombination by cleaving V, D and J segments, which are joined exclusively by NHEJ to form V(D)J exons1,2. V(D)J recombination in bone marrow (BM) pro-B cells first assembles IgH V(D)J exons leading to μ chain expression3. Subsequently, IgL VJ exons are assembled in pre-B cells, generating immature B cells that express μ plus IgL chains as surface IgM3. The two IgL families (Igκ and Igλ) are encoded in distinct loci, and primary Igκ V(D)J recombination usually precedes that of Igλ4. Individual B cells express either Igκ or Igλ, with about 95% of mouse IgM+ B cells being Igκ+ and the remainder Igλ+4. Newly generated BM B cells that express auto-reactive receptors can undergo tolerogenic secondary V(D)J recombination, termed receptor editing, in which they further rearrange or delete Igκ and may rearrange Igλ5-7 (See Suppl. Fig. 1 for schematic version of these processes).
Surface IgM+ B cells down-regulate RAG and migrate to peripheral lymphoid tissues (e.g. spleen) where they participate in antigen-dependent responses including CSR8. The various sets of germline IgH constant region exons (“CH genes”) are flanked by switch (S) regions9. Activation-induced cytidine deaminase (AID) initiates DSBs in Sμ and a downstream S region, which then are joined by NHEJ or, in its absence, by less efficient microhomology (MH)-mediated alternative end-joining (A-EJ)9,10. Thereby, Cμ is replaced with a downstream CH gene to effect CSR (Suppl. Fig. 1). Germinal center (GC) B cells have been argued to undergo antigen-dependent secondary V(D)J recombination, termed “receptor revision”, as a means of diversification11. Like receptor editing, receptor revision is proposed to target Igκ and Igλ, but to be distinct in location and activation mechanism11,12. However, whether or not V(D)J recombination occurs in the context of receptor revision in GC B cells has been debated11-14.
Human and mouse B lymphomas often harbor clonal translocations linking oncogenes, such as c-myc, to IgH, Igκ or Igλ15,16. Such recurrent oncogenic translocations are thought to represent highly selected, very low frequency events. Even so, aspects of c-myc, beyond coding sequences, may increase its translocation fequency17. In this regard, loci involved in recurrent oncogenic translocations often are spatially proximal within interphase nuclei18-23. RAG and AID have been implicated in collaboratively initiating oncogenic translocations in human BM-derived pro-B/pre-B lymphomas24,25. Many oncogenic translocations in mature B lymphomas occur during attempted CSR and involve AID-initiated breaks26-29; but others result from RAG-initiated DSBs15,30,31. Due to checkpoint defects, RAG-initiated IgH breaks in ATM-deficient BM pro-B cells persist and can be translocation substrates in IgM+ peripheral B cells32. Thus far, however, translocations have not been shown to result from RAG activity in peripheral B cells.
Xrcc4 is a critical NHEJ component2. In its absence, V(D)J recombination is abrogated33,34 and CSR impaired10,35. Conditional inactivation of LoxP-flanked Xrcc4 in p53-deficient peripheral B cells via a CD21-Cre transgene leads to recurrent “CXP” B cell lymphomas that harbor aberrant Igκ and Igλ V(D)J rearrangements, IgH CSR events, and Igλ and/or IgH/c-myc translocations36. We proposed CXP tumor progenitors to be peripheral B cells that undergo secondary V(D)J recombination and CSR36. To search for such putative CXP tumor progenitors, we now have analyzed splenic “CXc/−” B cells in which Xrcc4 was peripherally inactivated but p53 was left intact to obviate B cell lymphomas.
IgH Chromosomal Breaks in Xrcc4-Deficient Splenic B Cells are AID-dependent
CXc/− mice have normal IgM+ B cell numbers, as Xrcc4 is intact for primary V(D)J recombination in developing BM B cells, with inactivation starting in transitional stage peripheral B cells10,37. CXc/− splenic B cells activated for CSR have high levels of IgH breaks on chromosome 12 due to impaired NHEJ10. While Xrcc4 deficiency is not associated with known checkpoint defects34, we firmly tested AID-dependency of CXc/− B cell IgH breaks by breeding the CXc/− genotype onto an AID-deficient (A−/−) background38 to generate CXc/−A−/− mice. We stimulated CXc/−, CXc/−A−/− and control (Xc/c) splenic B cells with αCD40/IL4 for 4 days to promote IgG1 CSR and assayed metaphases for IgH breaks and translocations via fluorescence in situ hybridization (FISH) with 5′ and 3′ IgH probes. While general chromosomal breaks, as expected, were largely AID-independent in activated CXc/− splenic B cells (Suppl. Table 2), the vast majority of IgH breaks were AID-dependent (Fig. 1a and Suppl. Table 1).
Figure 1. Role of AID and RAG in Generating IgH, Igκ, and Igλ breaks in CXc/- Splenic B cells.
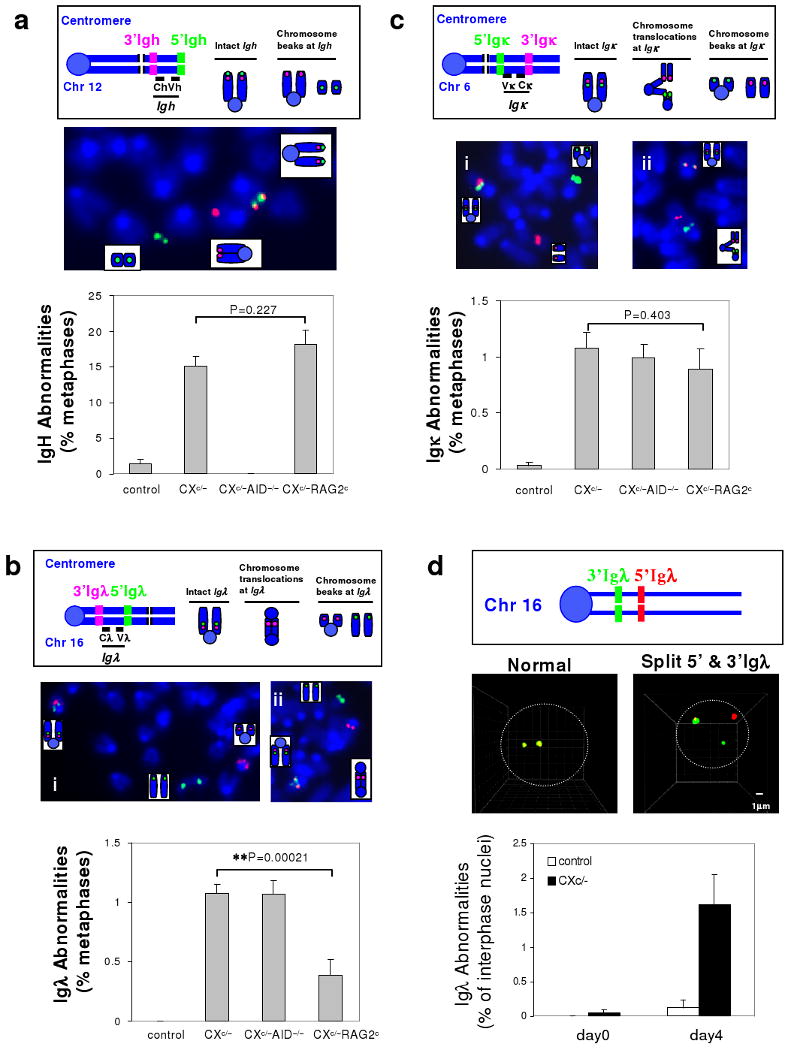
a, Upper: Diagram of IgH FISH probes. An intact IgH shows co-localized red and green signals while a broken locus appears as split red and green signals. Middle: Example of metaphase FISH showing IgH breaks. Lower: Quantification of IgH abnormalities in day4 αCD40/IL4-activated control (n=6), CXc/− (n=9), CXc/−A−/− (n=5) and CXc/−RAGc (n=8) splenic B cells (details in Suppl. Table 1). b, Upper: Diagram of Igλ FISH probes. Intact Igλ shows co-localized green and red signals, Igλ breaks appear as split green and red signals, either free or in translocations. Middle: Examples of metaphase FISH showing Igλ breaks (left) and an Igλ break and dicentric translocation (right). Lower: Quantification of Igλ abnormalities in day4 αCD40/IL4-activated control (n=11), CXc/− (n=11), CXc/−A−/− (n=3), CXc/−RAG2c (n=8) splenic B cells (details in Suppl. Table 3). c, Upper: Diagram of Igκ FISH probes. Igκ breaks are scored similarly as Igλ breaks. Middle: Examples of metaphase FISH showing an Igκ break (left) and Igκ break and translocations (right), involving both centromeric and telomeric portions of chromosome 6. Lower: Quantification of Igκ abnormalities in day4 αCD40/IL4-activated control (n=10), CXc/− (n=11), CXc/−A−/− (n=3), CXc/−RAGc (n=7) splenic B cells (details in Suppl. Table 4). d, Upper: Diagram of Igλ 3D interphase FISH Probes. Middle: Representative 3D interphase FISH showing intact Igλ (co-localization of green and red signals) and Igλ breaks (split green and red signals) (details in Suppl. Fig. 4). Lower: Quantification of Igλ abnormalities by 3D interphase FISH on day 0 (n=3) or day 4 (n=3) αCD40/IL4-activated splenic B cells. We could not do similar assays for Igκ due to the large size of this locus (greater than 3Mb). In all panels, data are presented as mean ± s.e.m. Statistical analyses were calculated by a Student's t-Test with two-tailed distribution.
RAG-dependent Igλ Breaks and Translocations in Xrcc4-deficient Splenic B Cells
We assayed activated CXc/− splenic B cells for Igλ breaks via metaphase FISH with 5′ and 3′ Igλ probes that flank the 200kb Igλ locus on chromosome 16 (Fig. 1b). After αCD40/IL4 stimulation for 4 days, we found Igλ breaks in over 1% of CXc/- B cells, with none in controls (Fig. 1b and Suppl. Table 3). Moreover, the Igλ breaks were frequently translocated (Fig. 1b; Suppl. Fig. 2). Metaphase FISH with BAC probes flanking Igκ revealed that 1% of activated CXc/- B cells also harbor Igκ breaks/translocations (Fig. 1c; Suppl. Fig. 2; Suppl. Table 4). In contrast, Xrcc4-deficient embryonic stem (ES) cells lacked Igκ or Igλ abnormalities (Suppl. Table 5). To elucidate when Igλ and Igκ breaks occurred, we assayed CXc/− splenic B cells at days 2, 3, and 4 of activation and observed both to accumulate during stimulation, with Igκ breaks kinetically preceding Igλ breaks (Suppl. Table 3, 4, and 6; Suppl. Fig. 3). We also assayed for Igλ breaks via 3D interphase FISH with 5′Igλ and 3′Igλ probes (Fig. 1d). Igλ breaks were rare in resting (day 0) CXc/− splenic B cells, but occurred in about 1.5% of day 4 activated CXc/- splenic B cells (Fig. 1d; Suppl. Fig 4, Table 7). We conclude that Igλ and Igκ breaks occur during expansion of activated CXc/- splenic B cells, a conclusion supported by our findings that p53 deficiency did not markedly enhance Igλ breaks (Suppl. Table 3) and that 50% of metaphases with Igλ breaks retained the acentric chromosome 16 fragment (Fig. 1b, data not shown).
To test AID involvement, we assayed for Igκ and Igλ breaks in day 4 αCD40/IL4-activated CXc/-A-/- B cells and found a comparable frequency as in CXc/− B cells (Fig. 1b,c, Suppl. Table 3 and 4, Suppl. Fig. 5). Similar to earlier studies32,39, we found only very low RAG expression in activated normal and CXc/− splenic B cells (data not shown). To further assess RAG involvement, we bred a LoxP-flanked Rag2 conditional allele (RAGc/c)40 into the CXc/− genotype to generate CXc/−RAGc/c or CXc/−RAGc/− (“CXc/−RAGc”) mice. Upon activation, the RAG conditional allele was largely deleted in day 3 and 4 activated CXc/−RAGc cells (Suppl. Fig. 6). While IgH break frequency was comparable between CXc/− and CXc/− RAGc B cells, Igλ break frequency was significantly reduced in CXc/−RAGc B cells (Fig. 1a,b, Suppl. Table 1 and 3). Thus, in activated CXc/− splenic B cells, IgH breaks are AID-dependent and RAG-independent; while Igλ breaks are AID-independent and mostly RAG-dependent. Igκ breaks were not significantly reduced in activated CXc/−RAGc B cells (Fig. 1c, Suppl. Table 4), suggesting they either are not initiated by AID or RAG or their earlier kinetic onset allows accumulation before RAG activity is eliminated.
RAG and AID Collaborate in Generating High Frequency IgH/Igλ Translocations
We employed sequential FISH to ask if IgH, Igκ or Igλ breaks occurred simultaneously in CXc/− B cell metaphases. Analyses of over 2000 day 4 αCD40/IL4-activated CXc/− B cell metaphases revealed none had both Igκ and Igλ breaks (Suppl. Fig. 7). However, analyses with a Jκ-Cκ probe showed that nearly 50% of metaphases with a broken Igλ had deleted Jκ-Cκ on one or both alleles (Suppl. Fig.8), similar to secondary V(D)J recombination events in CXP B lymphomas36. We found one Igκ/IgH translocation in over 2000 activated CXc/− B cell metaphases, consistent with a high frequency but at levels just below ready cytogenetic measurement (Suppl. Fig. 9a). Nearly 60% of metaphases with Igλ breaks also had IgH breaks and/or translocations and about 20% of these retained both centric and acentric portions of chromosome 12 and 16 (Suppl. Fig. 7; Suppl Fig. 9b,c; not shown), suggesting attempted V(D)J recombination and CSR in the same or successive cell cycles. In this regard, combined FISH with Igλ and IgH probes and chromosome paints revealed that 30% of Igλ translocations involved IgH (e.g. Fig. 2a; Suppl. Fig. 9b,c). As many IgH/Igλ translocations resulted in dicentrics with 3′Igλ and 3′ IgH probes juxtaposed (Suppl. Fig. 9b), we performed FISH with these probes simultaneously, which revealed AID-dependent IgH/Igλ translocations in about 0.2% of CXc/− B cells (Fig. 2a, Suppl. Table 8, Fig. 10). We conclude that unrepaired RAG-dependent Igλ breaks in activated CXc/− splenic B cells are frequently fused to AID-dependent IgH breaks in the same cell to form chromosome 12/16 translocations.
Figure 2. Frequent IgH/Igλ translocations in activated Xrcc4-deficient splenic B cells.
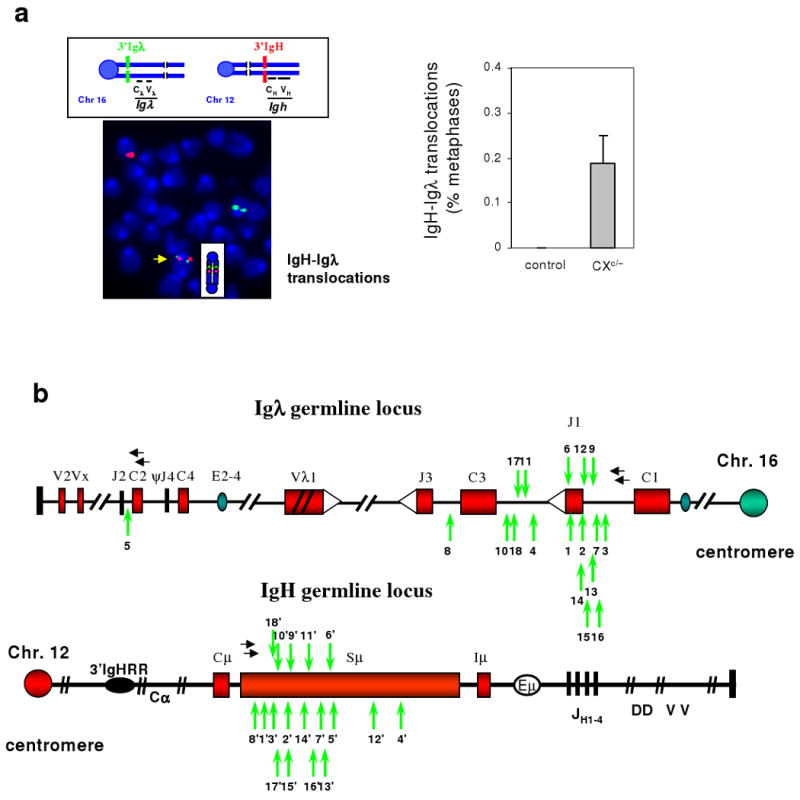
a, Top left: Diagram showing 3′Igλ probe (green) on chromosome 16 and 3′IgH probe (red) on chromosome 12. Bottom left: Representative Igλ/IgH translocation showing green and red signals juxtaposed on a dicentric chromosome (yellow arrow). Right: Quantification of IgH/Igλ translocations in day4 αCD40/IL4-activated control (n=2) or CXc/− (n=4) B cells analyzed by metaphase FISH (details in Suppl. Table 8). Data are presented as mean ± std. b, PCR-isolated Igh/Igλ translocation junctions from day4 activated CXc/− B cells (n=3) (primers indicated by horizontal black arrows). Junctional sequences are shown in Suppl. Fig. 11. A vertical green arrow indicates breakpoints. For a given translocation, the same number is used to indicate the corresponding IgH and Igλ breakpoints, with the IgH breakpoint denoted by a (') symbol.
We isolated IgH/Igλ translocation junctions from CXc/− B cells via PCR (Suppl. Fig. 10), and found most fused Sμ to sequences downstream of Jλ1/Jλ3 V(D)J recombination signal sequences (Fig.2b, Suppl. Fig.11). Consistent with AID-initiated IgH breaks joined to RAG-initiated Igλ breaks, point mutations and other alterations were observed in IgH- but not Igλ-derived junctional sequences (Suppl. Fig.11). Consistent with RAG-initiated breaks resolved in the absence of NHEJ, Igλ junctions were at variable distances downstream of Jλ1 and Jλ3. Finally, most IgH/Igλ junctions contained microhomologies indicative of A-EJ (Suppl. Fig. 11). We conclude that, in activated splenic CXc/− B cells, A-EJ joins RAG-induced Igλ breaks to AID-initiated IgH breaks at high frequency.
Cell-type Specific and Focal Co-localization of IgH and Igλ in B Cell Interphase Nuclei
3D interphase FISH with 3′IgH and 3′Igλ probes revealed co-localization of the loci (≤0.5μm apart) in about 14% of resting (day 0) and 7-8% of day 3.5 αC40/IL4-activated control and CXc/- splenic B cells (Fig. 3a-d; Suppl. Table 9,10). As there are no IgH or Igλ breaks in resting B cells (Figs. 1a, d), and AID-initiated breaks begin at day 241, we conclude IgH and Igλ co-localize before and after DSB induction and that Xrcc4 deficiency does not alter this association. To assess cell-type specificity, we assayed wt thymocyte and ES cell interphase nuclei and found only low-level IgH/Igλ co-localization (Fig. 3c, Suppl. Table 9). To examine specificity of the IgH/Igλ association within chromosome 16, we tested co-localization of IgH with two control loci (C2 and K10), which map, respectively, about 15Mb telomeric or centromeric to Igλ (Fig. 3b). IgH/C2 co-localization was at background levels in resting and activated B cells and thymocytes, while IgH/K10 co-localization occurred at substantially lower levels than IgH/Igλ co-localization (Fig. 3c, Suppl. Table 11). Therefore, IgH and Igλ co-localization is cell-type specific and focal on chromosome 16 with respect to Igλ. Notably, IgH and Igκ also specifically and focally, at least with respect to Igκ, co-localize in about 5% of splenic B cells (Suppl. Fig.12, Table 12).
Figure 3. Frequent cell-type and Igλ locus-specific IgH/Igλ co-localization.
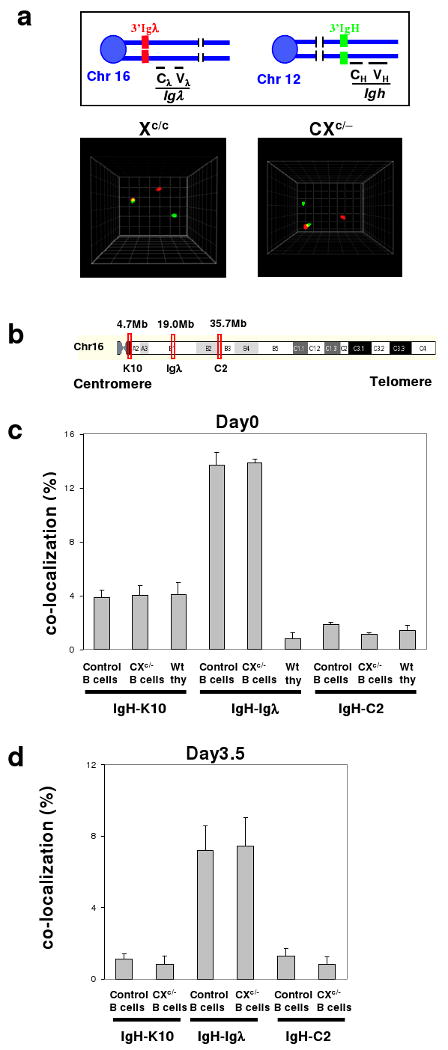
a, Top: Diagram showing 3′IgH (green) and 3′Igλ (red) probes used for 3D interphase FISH. Bottom: Representative co-localization of IgH/Igλ in day 0 control and CXc/− B cell interphase nuclei. b, Schematic map of Igλ, C2 and K10 BAC probes on chromosome 16. c, Quantification of co-localization of IgH-Igλ, IgH-C2, or IgH-K10 loci in nuclei of day 0 control and CXc/− splenic B cells and in nuclei of thymocytes (details in Suppl. Tables 9 and 11). d, Quantification of co-localization of IgH-Igλ, IgH-C2, or IgH-K10 loci in day3.5-activated control or CXc/− peripheral B cells (details in Suppl. Table 10). At least three mice were analyzed per data set; data are presented as mean ± s.e.m.
The c-myc DSB Frequency is Rate-limiting for IgH/c-myc Translocations
Given that CXP tumors routinely have IgH/c-myc translocations36, we tested for IgH/c-myc co-localization in B cell nuclei via 3D interphase FISH (Fig. 4b). Approximately, 4-6% of resting, 15′ activated and 3.5 day activated control or CXc/- B cell nuclei had co-localized IgH/c-myc signals (Fig. 4b,c; Suppl. Table 13), which were specific as IgH and c-myc did not co-localize in ES cells (Fig. 4c; Suppl. Table 13). While c-myc breaks and IgH/c-myc translocations were too infrequent to detect via FISH (Suppl. Table 14), PCR revealed an approximately 5-fold increase in IgH/c-myc translocations in activated CXc/− B cells over low (<1×10-6/cell) control levels (Fig. 4a; Suppl. Fig.13 and Table 15). Based on frequent IgH breaks and IgH/c-myc co-localization, we hypothesized c-myc breaks to be rate-limiting for IgH/c-myc translocations. To test this, we introduced 25 tandemly arrayed ISceI endonuclease target sites42 into the c-myc first intron to create the c-myc25IsceI allele (Fig. 4d; Suppl. Fig. 14). The array was used to increase ISceI cut frequency. Then, αCD40/IL4-activated peripheral B cells heterozygous for the c-myc25IsceI allele (c-myc25ISceI/wt) or wt control B cells (c-mycwt/wt) were infected with ISceI-expressing or control retrovirus43 and assayed for c-myc breaks via metaphase FISH. Strikingly, c-myc chromosomal breaks occurred in approximately 10% of c-myc25ISceI/wt B cells infected with the ISceI virus, but were absent in the various control B cells (Fig. 4e, Suppl. Table 16). PCR quantification demonstrated that IgH/c-myc translocations in ISceI virus-infected activated c-myc25ISceI/wt B cells were increased by at least 100 fold over control levels (Fig. 4a, Suppl. Fig. 15 and Table 16).
Figure 4. DSBs in c-myc are rate-limiting for IgH/c-myc translocations in activated splenic B cells.
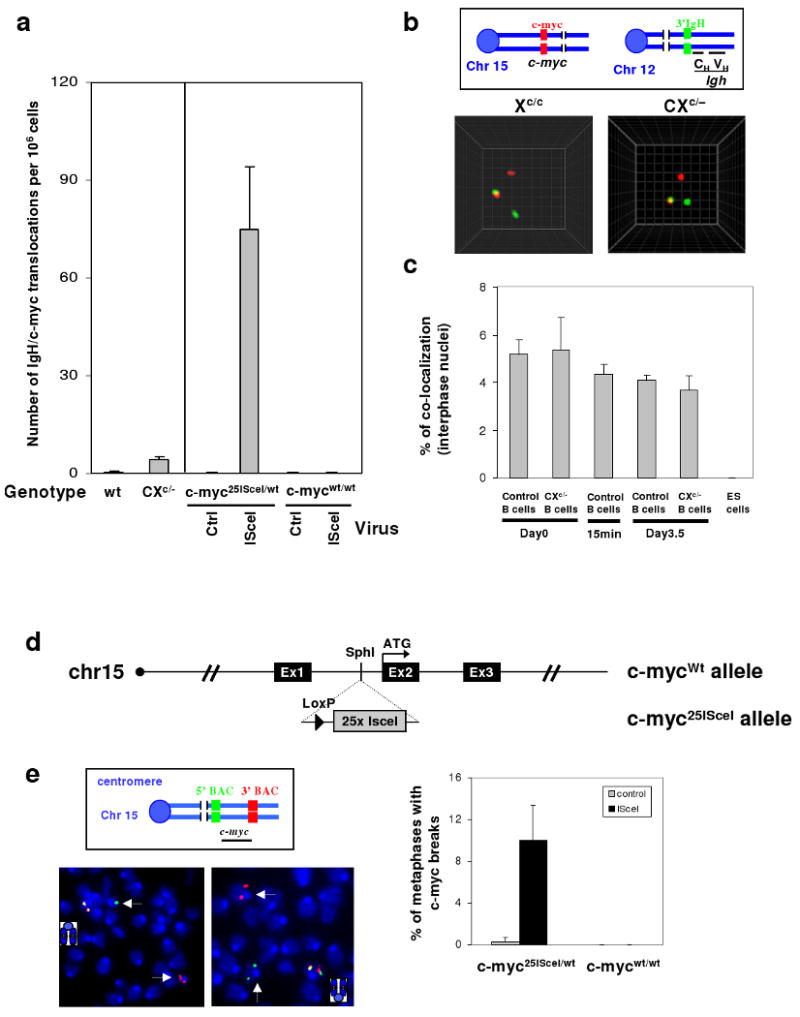
a, Frequency of IgH/c-myc translocations from day4 αCD40/IL4-activated wt (n=4) and CXc/− (n=4) splenic B cells, or B cells harboring c-myc25ISceI/wt (n=3) or c-mycwt/wt (n=1) infected with either control or ISceI-expressing retrovirus (details in Suppl. Fig. 13 and 15). b, Top: Schematic showing c-myc (red) probe on chromosome 15 and 3′IgH (green) probe on chromosome 12. Bottom: Representative images of IgH/c-myc co-localization in day 0 control and CXc/− B cell interphase nuclei. c, Quantification of IgH/c-myc association by 3D interphase FISH in control and CXc/− splenic B cells (n=3), and ES cells (n=3). Cells were analyzed at the indicated time points before or after stimulation. d, Schematic showing the wt c-myc allele (c-mycwt) and the modified c-myc allele containing 25 ISceI sites (c-myc25ISceI). e, Top left: Diagram of c-myc FISH probes. Bottom left: Representative c-myc abnormalities in αCD40/IL-4-activated c-myc25ISceI/wt B cells infected with IsceI-expressing retrovirus, appearing as green and red signals on separate chromosome fragments (white arrows). Bottom right: Quantification of c-myc breaks by metaphase FISH on day4 αCD40/IL4-activated B cells harboring either c-myc25ISceI/wt (n=4) or c-mycwt/wt (n=1) alleles after infection with control or ISceI-expressing retrovirus. Data are presented as mean ± std (details in Suppl. Table 16). High titer retrovirus infection appears to inhibit end joining allowing break visualization (see online methods).
Discussion
We show that some activated CXc/- splenic B cells harbor characteristics of postulated “editing and switching” CXP peripheral B cell lymphoma progenitors36, including Igκ deletions, aberrant Igλ V(D)J recombination, Igλ translocations, and aberrant IgH CSR associated with IgH translocations to c-myc or Igλ. Moreover, our studies clearly reveal V(D)J recombination-related events in CXc/- splenic B cells; because they leave telltale RAG-dependent Igλ breaks. We note that cultured splenic B cells do not represent GC B cells44 and CXP tumor progenitors do not appear of GC origin36. Therefore, we suggest that V(D)J recombination events in activated CXc/- splenic B cells and putative CXP lymphoma progenitors may represent peripheral “editing” mediated by low RAG expression, for example, as found in transitional B cells8,45,46. While potential physiological roles for such a process are unknown, it may be relevant for peripheral B cells subjected to chronic activation, such as those in gut-associated lymphoid tissues where CXP tumors arise36. In this context, we find RAG-dependent Igλ breaks in CXc/- mesenteric lymph node B cells taken directly from mice (unpublished data).
Our findings of RAG-initiated chromosomal breaks and translocations in Xrcc4-deficient peripheral B cells raises the possibility that translocations in some human peripheral B cell lymphomas, such as follicular lymphomas, might be initiated by V(D)J recombination in the periphery15,30. Our findings also demonstrate that AID and RAG can collaborate to generate frequent IgH/Igλ translocations in peripheral CXc/- B cells. It is particularly notable that these IgH/Igλ translocations offer no obvious cellular selective advantage. Therefore, their appearance as clonal translocations in CXP lymphomas simply may reflect the frequent occurrence of these translocations in tumor progenitors due to mechanistic factors that include the two loci being frequently broken and spatially proximal. In the latter context, our findings demonstrate that the co-localization of two loci on different chromosomes can be quite focal, implicating aspects of particular loci themselves, beyond broader chromosomal territories47, as important factors in determining spatial proximity and translocation frequency. Finally, analyses of oncogenic translocations in NHEJ-deficient pro-B and B cell lymphomas36,48 suggested A-EJ may be translocation prone relative to NHEJ49,50. The high frequency of specific translocations catalyzed by A-EJ in non-transformed CXc/- B cells supports this notion.
Methods Summary
Generation of mouse strains utilized
CXc/− mice were generated as previously described10 and crossed into AID-deficient mice38 to generate CXc/−A-/- or mice carrying floxed RAG2 alleles40 to generate CXc/−RAGc lines. We inserted a cassette containing 25 tandem ISceI target sites into the 1st intron of c-myc by gene targeting (details in online Methods). Mice were analyzed as outlined in the text at 8–16 weeks of age. The Institutional Animal Care and Use Committee of Children's Hospital (Boston, Massachusetts) approved all animal work.
Splenic B cell Purification, Activation in Culture, Retroviral Infection and CSR Assays
CD43− B cells were isolated from spleen, cultured, and assayed for CSR as previously described10,22. Cells were sampled on various days for DNA isolation, flow cytometry analyses and metaphase preparation. Retroviral infection was performed as previously described43 (details in online Methods).
Two-color FISH and telomere-FISH
Metaphase spreads from αCD40/IL4-activated B cell cultures were prepared and two-color FISH to detect IgH, Igκ, Igλ or c-myc chromosomal aberrations and telomere staining (T-FISH) to detect general aberrations were performed as previously described10. FISH probes are detailed in online Methods.
3D interphase FISH
3D FISH was performed as described previously32 (details in online Methods). Images of approximately 50 serial optical sections spaced by 0.2 microns were captured with Marianas spinning disk confocal microscope (63×) with a CCD detector (Intelligent Imaging Innovations) and analyzed with Slidebook software (Intelligent Imaging Innovations).
PCR assay to detect IgH/c-myc or IgH/Igλ translocations
IgH/c-myc translocation junctions were amplified by PCR from genomic DNA prepared from αCD40/IL4 activated splenic B cells using primers previously described26. PCR products were run on agarose gels and hybridized with an internal c-myc oligo. IgH/Igλ translocations were amplified using nested primers for Sμ and Jλ. PCR products were hybridized with Jλ and IgH probes, the bands positive for both probes were cloned into the pGEM-T vector (Promega), sequenced and analyzed using Lasergene software and the NCBI database. Primer sequences and PCR conditions are detailed in online Methods.
Supplementary Material
Suppl. Figure 1. Overview of V(D)J recombination, receptor editing, and IgH CSR in B cells.
a, Diagram of Primary and Secondary V(D)J recombination during B cell development. See text for details. b, Diagram of IgH V(D)J recombination and CSR. See text for details.
Suppl. Figure 2. Distribution of different types of Igλ and Igκ abnormalities in activated CXc/− or CXP splenic B cells.
The bar graphs show the distribution of the different types of Igκ and Igλ aberrations found in 4 day αCD40/IL4-activated CXc/− or CXP splenic B cells categorized as the proportion of metaphases with either breaks alone, breaks plus translocations, or translocations alone.
Suppl. Figure 3. Kinetics of Igκ and Igλ breaks in activated CXc/− splenic B cells.
Metaphases prepared from day 2, day 3 or day 4 αCD40/IL4-activated splenic B cells were analyzed via two-color FISH using Igκ or Igλ BAC probes as outlined in Figure 1. At least three mice were analyzed for each point (see Suppl. Table 6 for raw data). Data are presented as mean±s.e.m.
Suppl. Figure 4. Representative Igλ abnormalities visualized by 3D interphase FISH.
Interphase nuclei were isolated from day 4 αCD40/IL4-activated CXc/− splenic B cells and analyzed by 3D FISH using 5′ (red) and 3′ (green) Igλ BAC probes as outlined in Figure 1d. Representative images with isolated centromeric or telomeric signals are shown. Among 14 Igλ locus breaks scored, 3 had split signals (the distance between green and red signals greater than 1.5μm), 3 had isolated red signals only, and 8 had isolated green signals only.
Suppl. Figure 5. Igκ and Igλ abnormalities in activated CXc/− splenic B cells are not initiated by AID.
Metaphases from αCD40/IL4-activated splenic B cells (day 4) were analyzed by FISH for hybridization to Igκ or Igλ probes as outlined in Figure 1. Representative Igκ or Igλ breaks and translocations in CXc/−AID−/− splenic B cells are shown.
Suppl. Figure 6. Deletion of RAG conditional alleles in resting and activated splenic CXc/−RAGc B cells.
DNA samples were prepared from day 0, day 3, or day 4 αCD40/IL4-activated CXc/−RAGc/- B cells and from kidney (Kid) as a control. Genomic DNA was digested with StuI and hybridized with a 5′ RAG2 probe.
Suppl. Figure 7. Lack of Igκ and Igλ breaks or translocations in the same CXc/− metaphases.
Metaphases from αCD40/IL4-activated CXc/− B cells (day 4) were analyzed by sequential FISH hybridization first with 5′ and 3′ Igκ probes and then 5′ and 3′ Igλ probes as indicated. Upper: Representative metaphases with an intact Igκ locus and a broken/translocated Igλ locus in CXc/− splenic B cells are shown. Bottom: Quantification of these sequential FISH analyses.
Suppl. Figure 8. Increased Jκ-Cκ deletion in metaphases harboring Igλ abnormalities.
Metaphases prepared from day 4 aCD40/IL-4 activated wt (n=2) or CXc/− (n=3) splenic B cells were first analyzed via two color FISH to identify those with Igλ breaks and translocations. Subsequently the Igλ signals were stripped and the metaphases were assayed by two color FISH using a 5′ Igκ BAC probe (to identify chromosome 6 in the region of Igκ) and a Jκ-Cκ probe (indicated in the schematic map of Igκ locus) to assay for Igκ specific deletions. Upper: Bar graph showing that metaphases from wt B cells (n=207) have an intact Igκ locus in greater than 90% (open bars) while nearly 50% of metaphases from CXc/- B cells with Igλ abnormalities (n=21) (black bars) had deletions of the Cκ region on either one or both alleles (scored as absence of Jκ-Cκ probe signal on the chromosome carrying the 5′ Igκ signal). Bottom: Map of Igκ locus showing position of Jκ-Cκ probe and indicating potential mechanisms for Igκ deletions via rearrangements to 3′RS.
Suppl. Figure 9. Frequent IgH and Igλ abnormalities in activated CXc/− peripheral B cells.
a, Shown is an example of a metaphase with an Igκ break joined to an IgH break. Left: Igκ FISH shows a chromosomal translocation involving Igκ Sequential hybridizations of the same metaphase with chromosome 12 and chromosome 6 paints (center) followed by IgH FISH (right) revealed a translocation involving chromosome 12 (red) and chromosome 6 (green) with 3′IgH and 3′Igκ sequences at the breakpoint. b, Upper: Diagram of Igλ and IgH probes. Lower: Examples of metaphases with simultaneous Igλ and IgH breaks, as revealed by sequential Igλ FISH followed by IgH FISH. Metaphase 1: The broken Igλ locus (3′Igλ probe, red signal, left panel) is joined to a broken IgH locus (3′IgH probe, green signal, right panel) to form a dicentric chromosome, while the telomeric portion of chromosome 16 (5′Igλ probe, green signal, left panel) is fused with the telomeric portion of chromosome 12 (5′IgH probe, red signal, right panel) in an acentric fragment. The other chromosome 12 in the same metaphase is also involved in formation of a dicentric chromosome, which is only positive for the 3′IgH probe (green signal, right panel). Metaphase 2: Both the IgH and the Igλ loci are broken; and the chromosomal fragments containing the 5′Igλ probe (green, left panel) and the 5′IgH probe (red, right panel) are involved in translocations with different chromosomes. Metaphase 3: The Igλ locus is broken, with the 5′Igλ probe (green) joined to another chromosome to form a translocation (left panel), whereas the IgH locus is intact (right panel). c, Example is shown of a metaphase with an Igλ break joined to IgH break. Sequential hybridization of the same metaphase with Igλ probes, chromosome 12 and chromosome 16 paints and IgH probes revealed the telomeric portion of chromosome 16, containing the 5′Igλ probe (green, left panel) joined to the centromeric portion of chromosome 12, containing the 3′IgH probe (red, right panel).
Suppl. Figure 10. PCR assay to detect IgH/Igλ translocations in day 4 activated control and CXc/− splenic B cells.
a, Schematic representation of the PCR assay used for IgH/Igλ translocations. Primers used to detect chromosome 12/16 translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar. b, Representative Southern blots with the IgH or Jλ1 probe are shown for activated control, CXc/− and CXc/−A−/− peripheral B cells (day 4 of αCD40/IL4 activation). No IgH/Igλ translocation was detected in 1.5×106 cells from CXc/−A−/− mice. In each reaction DNA from ∼20000 cells was used.
Suppl. Figure 11. Sequence analyses of junctions of IgH/Igλ translocations.
IgH/Igλ translocations were cloned from 4 day αCD40/IL4-activated CXc/− (n=3) B cells by PCR. Sequences are aligned with genomic IgH (AJ851868) or Igλ (NG_004051) locus sequences. Mutations, insertions, or deletion were found in the Sμ regions (underlined). Microhomology at the junctions is underlined.
Suppl. Figure 12. Spatial proximity of IgH and Igκ.
The 3′ IgH probe and one of the three indicated Igκ region probes (F6, Igκ, I9) (diagrammed in lower panel) were used for 3D interphase FISH on control or CXc/− peripheral B cell nuclei. The bar graph in the upper panel shows the percentage of nuclei in which the 3′IgH was found co-localized (closer than 0.5μm) with each of the Igκ regions as indicated. At least three mice were analyzed for each point (see Suppl. Table 12). The percentage of co-localization is presented as mean ± s.e.m based on results of at least 3 different experiments per sample.
Suppl. Figure 13. PCR assay to detect IgH/c-myc translocations in day 4 activated control and CXc/− splenic B cells.
a, Schematic representation of the PCR assay used for IgH/c-myc translocations. Primers used to detect derivative chromosome 12 (der12) translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar (T13). b, Representative Southern blots with the T13 probe are shown for activated control and CXc/− peripheral B cells (day 4 of αCD40/IL4). In each reaction DNA from 100000 or 50000 cells was used (see Suppl. Table 15). c, Representative Southern blots with T13 probe are shown for activated control and CXc/−RAG2c splenic B cells at day 2 and day 3 of activation with αCD40/IL4. Each reaction used DNA from 50000 cells.
Suppl. Figure 14. Gene targeting strategy and Southern blot analyses of c-myc25ISceI allele.
Top Diagram: Schematic maps of the targeting strategy for insertion of 25 ISceI sites into the 1st intron of c-myc locus. The position of the 5′ and 3′ probes used for ES cell screening are shown in red. Bottom Panels: Southern blot analysis of a targeted clone before and after Neo cassette deletion.
Suppl. Figure 15. PCR assay to detect IgH/c-myc translocations in day 4-activated c-myc25IsceI/wt splenic B cells infected with control or ISceI virus.
a, Schematic representation of the PCR assay used for IgH/c-myc translocations. Primers used to detect derivative chromosome 12 (der12) translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar (T15). b, Representative Southern blots with the T15 probe are shown for activated c-myc25IsceI/wt and c-mycwt/wt peripheral B cells (day 4 of αCD40/IL4) infected with either control or ISceI virus. In each reaction, DNA from 50000 (control-infected samples) or 5000 (ISceI-infected samples) cells was used (see Suppl. Table 16).
Supplementary Table 1. Frequency of IgH breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 2. General Genomic Instability Measured by Telomere-FISH
Supplementary Table 3. Frequency of Igλ breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 4. Frequency of Igκ breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 5. Absence of Igκ and Igλ breaks in Xrcc4-deficient ES cells
Supplementary Table 6. Kinetics of Igλ and Igκ abnormalities in activated Xrcc4-deficient peripheral B cells
Supplemental Table 7. Igλ Abnormalities in CXc/- B cells measured by 3D Interphase FISH
Supplemental Table 8. IgH-Igλ, translocations measured by two color IgH-Igλ, FISH on control or CXc/− B cells activated with αCD40/IL4 for 4 days
Supplementary Table 9. Igλ-IgH co-localization in Xc/c and CXc/- B cells and other cell types measured by 3D interphase FISH
Supplementary Table 10. Igλ-IgH co-localization in activated control and CXc/- B cells measured by 3D interphase FISH
Supplementary Table 11. IgH-chr16 control loci co-localization in Xc/c and CXc/- B cells and other cell types by Interphase FISH
Supplementary Table 12. Igκ-IgH co-localization in control and CXc/- B cells and other cell types by 3D Interphase FISH
Supplementary Table 13. IgH-cmyc co-localization in B cells and other cell types by 3D Interphase FISH
Supplementary Table 14. No c-myc abnormalities detected in activated Xrcc4-deficient B cells by two color FISH
Supplementary Table 15. Kinetics of IgH-cmyc translocations in Xrcc4-deficient peripheral B cells
Supplementary Table 16. c-myc breaks and IgH/c-myc translocation in ISceI targeted B cells
Acknowledgments
We thank Alt lab members for discussions, and Y.L. Chen, J. M. Bianco and M. Moghimi for technical assistance. This work was supported by NIH grant 5P01CA92625 and a Leukemia and Lymphoma Society of America (LLS) SCORE grant (to F.W.A. and K.R.). M.G. is and J.H.W. was a Special Fellow of LLS. J.H.W. and D.R.W. are supported by an NIH training grant and C.T.Y. was supported by an NCI training grant. A.N. is supported by the Intramural Research program of the NIH, NCI, Center for Cancer Research. F.W.A. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author contributions F.W.A., J.H.W., M.G. and C.T.Y. planned studies and interpreted data. J.H.W. performed the majority of experiments, including mouse breeding, B cell studies, FISH, and IgH/Igλ PCR studies. C.T.Y. bred mice and performed B cell analyses. M.G. generated and analyzed c-myc25IsceI/wt mice and performed FISH and IgH/c-myc translocation studies. P.G., T.H., and E.H. provided technical assistance. S.D. and A.N. provided expertise in 3D interphase FISH. A.A.Z. generated the 25 IsceI array. D.R.W. performed RAG expression studies and mesenteric lymph node B cell analyses. K.R. provided RAG conditional knock-out mice and helped interpret data. F.W.A., J.H.W. and M.G. wrote the paper.
Author information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Jung D, Alt FW. Unraveling V(D)J recombination; insights into gene regulation. Cell. 2004;116:299–311. doi: 10.1016/s0092-8674(04)00039-x. [DOI] [PubMed] [Google Scholar]
- 2.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–31. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 3.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109(Suppl):S45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 4.Gorman JR, Alt FW. Regulation of immunoglobulin light chain isotype expression. Adv Immunol. 1998;69:113–81. doi: 10.1016/s0065-2776(08)60607-0. [DOI] [PubMed] [Google Scholar]
- 5.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–20. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol. 2006;6:728–40. doi: 10.1038/nri1939. [DOI] [PubMed] [Google Scholar]
- 8.Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annu Rev Immunol. 2004;22:485–501. doi: 10.1146/annurev.immunol.22.012703.104707. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhuri J, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 10.Yan CT, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–82. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 11.Nemazee D, Weigert M. Revising B cell receptors. J Exp Med. 2000;191:1813–7. doi: 10.1084/jem.191.11.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seagal J, Melamed D. Role of receptor revision in forming a B cell repertoire. Clin Immunol. 2002;105:1–8. doi: 10.1006/clim.2002.5290. [DOI] [PubMed] [Google Scholar]
- 13.Wilson PC, et al. Receptor revision of immunoglobulin heavy chain variable region genes in normal human B lymphocytes. J Exp Med. 2000;191:1881–94. doi: 10.1084/jem.191.11.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goossens T, Brauninger A, Klein U, Kuppers R, Rajewsky K. Receptor revision plays no major role in shaping the receptor repertoire of human memory B cells after the onset of somatic hypermutation. Eur J Immunol. 2001;31:3638–48. doi: 10.1002/1521-4141(200112)31:12<3638::aid-immu3638>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–94. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 16.Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst) 2006;5:1213–24. doi: 10.1016/j.dnarep.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Gostissa M, Ranganath S, Bianco JM, Alt FW. Chromosomal location targets different MYC family gene members for oncogenic translocations. Proc Natl Acad Sci U S A. 2009;106:2265–70. doi: 10.1073/pnas.0812763106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozubek S, et al. Distribution of ABL and BCR genes in cell nuclei of normal and irradiated lymphocytes. Blood. 1997;89:4537–45. [PubMed] [Google Scholar]
- 19.Neves H, Ramos C, da Silva MG, Parreira A, Parreira L. The nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood. 1999;93:1197–207. [PubMed] [Google Scholar]
- 20.Nikiforova MN, et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–41. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 21.Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet. 2003;34:287–91. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- 22.Osborne CS, et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meaburn KJ, Misteli T, Soutoglou E. Spatial genome organization in the formation of chromosomal translocations. Semin Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai AG, et al. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–42. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahowald GK, Baron JM, Sleckman BP. Collateral damage from antigen receptor gene diversification. Cell. 2008;135:1009–12. doi: 10.1016/j.cell.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 26.Ramiro AR, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–8. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Ramiro A, et al. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol. 2007;94:75–107. doi: 10.1016/S0065-2776(06)94003-6. [DOI] [PubMed] [Google Scholar]
- 28.Kovalchuk AL, et al. AID-deficient Bcl-xL transgenic mice develop delayed atypical plasma cell tumors with unusual Ig/Myc chromosomal rearrangements. J Exp Med. 2007;204:2989–3001. doi: 10.1084/jem.20070882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robbiani DF, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–38. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jager U, et al. Follicular lymphomas' BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood. 2000;95:3520–9. [PubMed] [Google Scholar]
- 31.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–45. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Callen E, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, et al. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–89. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 34.Gao Y, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 35.Soulas-Sprauel P, et al. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J Exp Med. 2007;204:1717–27. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JH, et al. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–90. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117:787–800. doi: 10.1016/j.cell.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 39.Gartner F, Alt FW, Monroe RJ, Seidl KJ. Antigen-independent appearance of recombination activating gene (RAG)-positive bone marrow B cells in the spleens of immunized mice. J Exp Med. 2000;192:1745–54. doi: 10.1084/jem.192.12.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao Z, Rajewsky K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J Exp Med. 2001;194:1151–64. doi: 10.1084/jem.194.8.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med. 2005;202:561–8. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130:451–60. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zarrin AA, et al. Antibody class switching mediated by yeast endonuclease-generated DNA breaks. Science. 2007;315:377–81. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- 44.Lahvis GP, Cerny J. Induction of germinal center B cell markers in vitro by activated CD4+ T lymphocytes: the role of CD40 ligand, soluble factors, and B cell antigen receptor cross-linking. J Immunol. 1997;159:1783–93. [PubMed] [Google Scholar]
- 45.Monroe RJ, et al. RAG2:GFP knockin mice reveal novel aspects of RAG2 expression in primary and peripheral lymphoid tissues. Immunity. 1999;11:201–12. doi: 10.1016/s1074-7613(00)80095-3. [DOI] [PubMed] [Google Scholar]
- 46.Yu W, et al. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature. 1999;400:682–7. doi: 10.1038/23287. [DOI] [PubMed] [Google Scholar]
- 47.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 48.Zhu C, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–21. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 49.Roth DB. Amplifying mechanisms of lymphomagenesis. Mol Cell. 2002;10:1–2. doi: 10.1016/s1097-2765(02)00573-7. [DOI] [PubMed] [Google Scholar]
- 50.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Figure 1. Overview of V(D)J recombination, receptor editing, and IgH CSR in B cells.
a, Diagram of Primary and Secondary V(D)J recombination during B cell development. See text for details. b, Diagram of IgH V(D)J recombination and CSR. See text for details.
Suppl. Figure 2. Distribution of different types of Igλ and Igκ abnormalities in activated CXc/− or CXP splenic B cells.
The bar graphs show the distribution of the different types of Igκ and Igλ aberrations found in 4 day αCD40/IL4-activated CXc/− or CXP splenic B cells categorized as the proportion of metaphases with either breaks alone, breaks plus translocations, or translocations alone.
Suppl. Figure 3. Kinetics of Igκ and Igλ breaks in activated CXc/− splenic B cells.
Metaphases prepared from day 2, day 3 or day 4 αCD40/IL4-activated splenic B cells were analyzed via two-color FISH using Igκ or Igλ BAC probes as outlined in Figure 1. At least three mice were analyzed for each point (see Suppl. Table 6 for raw data). Data are presented as mean±s.e.m.
Suppl. Figure 4. Representative Igλ abnormalities visualized by 3D interphase FISH.
Interphase nuclei were isolated from day 4 αCD40/IL4-activated CXc/− splenic B cells and analyzed by 3D FISH using 5′ (red) and 3′ (green) Igλ BAC probes as outlined in Figure 1d. Representative images with isolated centromeric or telomeric signals are shown. Among 14 Igλ locus breaks scored, 3 had split signals (the distance between green and red signals greater than 1.5μm), 3 had isolated red signals only, and 8 had isolated green signals only.
Suppl. Figure 5. Igκ and Igλ abnormalities in activated CXc/− splenic B cells are not initiated by AID.
Metaphases from αCD40/IL4-activated splenic B cells (day 4) were analyzed by FISH for hybridization to Igκ or Igλ probes as outlined in Figure 1. Representative Igκ or Igλ breaks and translocations in CXc/−AID−/− splenic B cells are shown.
Suppl. Figure 6. Deletion of RAG conditional alleles in resting and activated splenic CXc/−RAGc B cells.
DNA samples were prepared from day 0, day 3, or day 4 αCD40/IL4-activated CXc/−RAGc/- B cells and from kidney (Kid) as a control. Genomic DNA was digested with StuI and hybridized with a 5′ RAG2 probe.
Suppl. Figure 7. Lack of Igκ and Igλ breaks or translocations in the same CXc/− metaphases.
Metaphases from αCD40/IL4-activated CXc/− B cells (day 4) were analyzed by sequential FISH hybridization first with 5′ and 3′ Igκ probes and then 5′ and 3′ Igλ probes as indicated. Upper: Representative metaphases with an intact Igκ locus and a broken/translocated Igλ locus in CXc/− splenic B cells are shown. Bottom: Quantification of these sequential FISH analyses.
Suppl. Figure 8. Increased Jκ-Cκ deletion in metaphases harboring Igλ abnormalities.
Metaphases prepared from day 4 aCD40/IL-4 activated wt (n=2) or CXc/− (n=3) splenic B cells were first analyzed via two color FISH to identify those with Igλ breaks and translocations. Subsequently the Igλ signals were stripped and the metaphases were assayed by two color FISH using a 5′ Igκ BAC probe (to identify chromosome 6 in the region of Igκ) and a Jκ-Cκ probe (indicated in the schematic map of Igκ locus) to assay for Igκ specific deletions. Upper: Bar graph showing that metaphases from wt B cells (n=207) have an intact Igκ locus in greater than 90% (open bars) while nearly 50% of metaphases from CXc/- B cells with Igλ abnormalities (n=21) (black bars) had deletions of the Cκ region on either one or both alleles (scored as absence of Jκ-Cκ probe signal on the chromosome carrying the 5′ Igκ signal). Bottom: Map of Igκ locus showing position of Jκ-Cκ probe and indicating potential mechanisms for Igκ deletions via rearrangements to 3′RS.
Suppl. Figure 9. Frequent IgH and Igλ abnormalities in activated CXc/− peripheral B cells.
a, Shown is an example of a metaphase with an Igκ break joined to an IgH break. Left: Igκ FISH shows a chromosomal translocation involving Igκ Sequential hybridizations of the same metaphase with chromosome 12 and chromosome 6 paints (center) followed by IgH FISH (right) revealed a translocation involving chromosome 12 (red) and chromosome 6 (green) with 3′IgH and 3′Igκ sequences at the breakpoint. b, Upper: Diagram of Igλ and IgH probes. Lower: Examples of metaphases with simultaneous Igλ and IgH breaks, as revealed by sequential Igλ FISH followed by IgH FISH. Metaphase 1: The broken Igλ locus (3′Igλ probe, red signal, left panel) is joined to a broken IgH locus (3′IgH probe, green signal, right panel) to form a dicentric chromosome, while the telomeric portion of chromosome 16 (5′Igλ probe, green signal, left panel) is fused with the telomeric portion of chromosome 12 (5′IgH probe, red signal, right panel) in an acentric fragment. The other chromosome 12 in the same metaphase is also involved in formation of a dicentric chromosome, which is only positive for the 3′IgH probe (green signal, right panel). Metaphase 2: Both the IgH and the Igλ loci are broken; and the chromosomal fragments containing the 5′Igλ probe (green, left panel) and the 5′IgH probe (red, right panel) are involved in translocations with different chromosomes. Metaphase 3: The Igλ locus is broken, with the 5′Igλ probe (green) joined to another chromosome to form a translocation (left panel), whereas the IgH locus is intact (right panel). c, Example is shown of a metaphase with an Igλ break joined to IgH break. Sequential hybridization of the same metaphase with Igλ probes, chromosome 12 and chromosome 16 paints and IgH probes revealed the telomeric portion of chromosome 16, containing the 5′Igλ probe (green, left panel) joined to the centromeric portion of chromosome 12, containing the 3′IgH probe (red, right panel).
Suppl. Figure 10. PCR assay to detect IgH/Igλ translocations in day 4 activated control and CXc/− splenic B cells.
a, Schematic representation of the PCR assay used for IgH/Igλ translocations. Primers used to detect chromosome 12/16 translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar. b, Representative Southern blots with the IgH or Jλ1 probe are shown for activated control, CXc/− and CXc/−A−/− peripheral B cells (day 4 of αCD40/IL4 activation). No IgH/Igλ translocation was detected in 1.5×106 cells from CXc/−A−/− mice. In each reaction DNA from ∼20000 cells was used.
Suppl. Figure 11. Sequence analyses of junctions of IgH/Igλ translocations.
IgH/Igλ translocations were cloned from 4 day αCD40/IL4-activated CXc/− (n=3) B cells by PCR. Sequences are aligned with genomic IgH (AJ851868) or Igλ (NG_004051) locus sequences. Mutations, insertions, or deletion were found in the Sμ regions (underlined). Microhomology at the junctions is underlined.
Suppl. Figure 12. Spatial proximity of IgH and Igκ.
The 3′ IgH probe and one of the three indicated Igκ region probes (F6, Igκ, I9) (diagrammed in lower panel) were used for 3D interphase FISH on control or CXc/− peripheral B cell nuclei. The bar graph in the upper panel shows the percentage of nuclei in which the 3′IgH was found co-localized (closer than 0.5μm) with each of the Igκ regions as indicated. At least three mice were analyzed for each point (see Suppl. Table 12). The percentage of co-localization is presented as mean ± s.e.m based on results of at least 3 different experiments per sample.
Suppl. Figure 13. PCR assay to detect IgH/c-myc translocations in day 4 activated control and CXc/− splenic B cells.
a, Schematic representation of the PCR assay used for IgH/c-myc translocations. Primers used to detect derivative chromosome 12 (der12) translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar (T13). b, Representative Southern blots with the T13 probe are shown for activated control and CXc/− peripheral B cells (day 4 of αCD40/IL4). In each reaction DNA from 100000 or 50000 cells was used (see Suppl. Table 15). c, Representative Southern blots with T13 probe are shown for activated control and CXc/−RAG2c splenic B cells at day 2 and day 3 of activation with αCD40/IL4. Each reaction used DNA from 50000 cells.
Suppl. Figure 14. Gene targeting strategy and Southern blot analyses of c-myc25ISceI allele.
Top Diagram: Schematic maps of the targeting strategy for insertion of 25 ISceI sites into the 1st intron of c-myc locus. The position of the 5′ and 3′ probes used for ES cell screening are shown in red. Bottom Panels: Southern blot analysis of a targeted clone before and after Neo cassette deletion.
Suppl. Figure 15. PCR assay to detect IgH/c-myc translocations in day 4-activated c-myc25IsceI/wt splenic B cells infected with control or ISceI virus.
a, Schematic representation of the PCR assay used for IgH/c-myc translocations. Primers used to detect derivative chromosome 12 (der12) translocations are represented as horizontal black arrows. The internal oligonucleotide probe used in Southern blot experiments is shown as a horizontal black bar (T15). b, Representative Southern blots with the T15 probe are shown for activated c-myc25IsceI/wt and c-mycwt/wt peripheral B cells (day 4 of αCD40/IL4) infected with either control or ISceI virus. In each reaction, DNA from 50000 (control-infected samples) or 5000 (ISceI-infected samples) cells was used (see Suppl. Table 16).
Supplementary Table 1. Frequency of IgH breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 2. General Genomic Instability Measured by Telomere-FISH
Supplementary Table 3. Frequency of Igλ breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 4. Frequency of Igκ breaks in Xrcc4-deficient peripheral B cells activated with αCD40/IL4 for 4 days
Supplementary Table 5. Absence of Igκ and Igλ breaks in Xrcc4-deficient ES cells
Supplementary Table 6. Kinetics of Igλ and Igκ abnormalities in activated Xrcc4-deficient peripheral B cells
Supplemental Table 7. Igλ Abnormalities in CXc/- B cells measured by 3D Interphase FISH
Supplemental Table 8. IgH-Igλ, translocations measured by two color IgH-Igλ, FISH on control or CXc/− B cells activated with αCD40/IL4 for 4 days
Supplementary Table 9. Igλ-IgH co-localization in Xc/c and CXc/- B cells and other cell types measured by 3D interphase FISH
Supplementary Table 10. Igλ-IgH co-localization in activated control and CXc/- B cells measured by 3D interphase FISH
Supplementary Table 11. IgH-chr16 control loci co-localization in Xc/c and CXc/- B cells and other cell types by Interphase FISH
Supplementary Table 12. Igκ-IgH co-localization in control and CXc/- B cells and other cell types by 3D Interphase FISH
Supplementary Table 13. IgH-cmyc co-localization in B cells and other cell types by 3D Interphase FISH
Supplementary Table 14. No c-myc abnormalities detected in activated Xrcc4-deficient B cells by two color FISH
Supplementary Table 15. Kinetics of IgH-cmyc translocations in Xrcc4-deficient peripheral B cells
Supplementary Table 16. c-myc breaks and IgH/c-myc translocation in ISceI targeted B cells