

Abstract
An unusual reaction between 1,3-dithiane anions and by 5-(alkylsulfanyl)-1-phenyltetrazoles has been discovered in which the dithiane anion formally displaces the 1-phenyltetrazole ring from sulfur to provide a sulfanylated dithiane.
Keywords: Tetrafibricin; Total synthesis; 1,3-Dithianes; 5-(Alkylsulfanyl)-1-phenyltetrazoles; Nucleophilic additions; Sulfanylations; Sulfur chemistry
The natural product tetrafibricin 1 was isolated in 1993 by Kamayama and coworkers, and they assigned its two-dimensional constitution by a battery of spectroscopic methods1. The compound has a linear carbon backbone featuring 11 stereocenters (10 with hydroxy groups and one with a methyl group), is a tetraene, with three isolated trans double bonds, and assorted other functional groups. In 2003, Kishi and coworkers assigned the three-dimensional structure (configuration) of 1 without resorting to derivatization or degradation by comparing NMR spectra of the natural product in both chiral and achiral solvents to an NMR database of fragment spectra2. Roush3 and Cossy4 have described syntheses of fragments of 1.
We have advanced a retrosynthetic strategy in Fig. 1 of the disassembled tetrafibricin 1 into six main fragments 2–7. Though the synthesis has not yet been completed, all of the fragments have been made, and assorted couplings have been established5. Along the way to making the C21–C30 fragment 4, we encountered an unusual reaction between a dithiane anion6 and a 5-(alkylsulfanyl)-1-phenyltetrazoles. Herein we describe this reaction, which, in the context of the tetrafibricin synthesis, proved to be a temporary roadblock. We also describe a straightforward detour around this roadblock to complete the synthesis of a modified C21–C30 fragment.
Fig.1.

Structure of tetrafibricin 1 and high level retrosynthetic analysis
RESULTS AND DISCUSSION
The two fragments 13 and 17 needed to make 4 were synthesized as shown in Scheme 1. Oxidation7 of commercially available dioxolane alcohol (S)-8 to aldehyde 9 was followed by the Wittig reaction and non-selective epoxidation of the resulting alkene 10 to give a mixture of epoxide isomers. Hydrolytic kinetic resolution with the (S,S)-Jacobsen catalyst then gave 11 in 45% yield8. Dithiane opening of the epoxide 11 gave alcohol 12, which was protected with tert-butyldimethylsilyltriflate (TBSOTf) to give 13. Synthesis of 17 started from the enantiomer (R)-8. The alcohol was converted to a phenylthiotetrazole 14, then the acetal was cleaved to give diol 15. Standard mono-tosylation and base treatments of 16 provided epoxide 17.
Scheme 1.

Synthesis of the precursors 13 and 17 for dithiane alkylation
Treatment of dithiane 13 with t-BuLi in THF at −30 to −20 °C to generate the anion was followed by addition of epoxide 17 (Scheme 2). After warming to 0 °C and stirring for 3 h, TLC analysis showed that the starting materials were mostly consumed and a single new spot was evident. After standard workup and flash chromatography, the new product was isolated in 56% yield. It was clear from the initial NMR spectrum that the two fragments were united. However, it was also clear that the target product 18 was not formed because the resonances for the epoxide protons remained. A complete characterization by 1D and 2D NMR spectroscopy5 and mass spectrometry showed that the product was 19, resulting from formal sulfanylation of the dithiane anion by 17. The epoxide survives 17, but the 1-phenyltetrazole is apparently displaced!
Scheme 2.

Sulfanylation of dithiane 13 by S-alkyl-N-phenylthioalkyltetrazole 17
Central to the structure assignment was the high resolution MS spectrum of 19, which showed the presence of three sulfur atoms (HRMS EI, 479.1780 corresponding to [M+ – Me] for C21H39O4S3Si). The connection of the sulfur of the starting phenylthiotetrazole to the carbon of the 1,3-dithiane was indicated by the MS fragmentation pattern as well as by features in the NMR spectra including the absence of a dithiane proton in the 1H NMR spectrum and the absence of cross-peaks in HMBC spectrum between right and left sides of the molecule (due to insulation by the chain sulfur atom).
The unusual transformation of 13 and 17 to 19 presumably requires only a dithiane anion and a 5-(alkylsulfanyl)-1-phenyltetrazole. Accordingly, we generated the anion from 2-phenylethyldithiane 20 as usual and then added 5-(ethylsulfanyl)-1-phenyltetrazole (21) (Scheme 3). Standard workup and purification provided the now expected product 22 in 61% yield. The same product 22 was also prepared by a standard sulfanylation of the anion of dithiane 20 with diethyldisulfide. These two samples of 22 were identical.
Scheme 3.
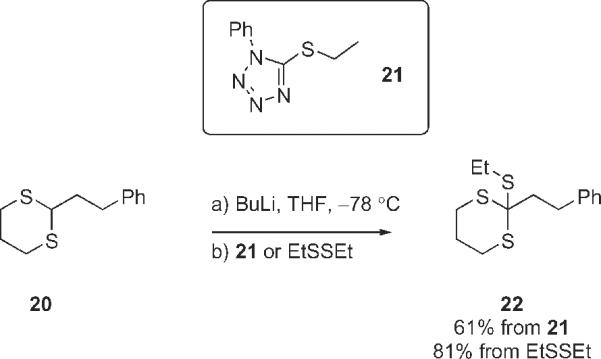
Simple model sulfanylations
The mechanism for this formal displacement of the phenyltetrazole ring by the dithiane anion is not clear; however, the economical SN2 displacement mechanism at sulfur does not seem reasonable since tetrazole is not a leaving group and since the epoxide survives. We postulate that the transformation may be initiated by addition of the dithiane anion to the tetrazole ring. This could ultimately result in a role reversal with the thiolate anion emerging as a nucleophile and the dithiane attached to a leaving group. A highly speculative series of steps for this process is outlined in Fig. 2.
Fig.2.

A speculative mechanism for the sulfanylation
Though interesting, the observed reaction is a minor roadblock for the synthesis of the C21–C30 fragment of tetrafibricin. A detour was quickly put into place as shown in Scheme 4. Reaction of dithiane anion derived from 13 with 4-methoxybenzyl-protected epoxide 23 provided alkylated dithiane 24. Cleavage of the dithiane, 1,3-anti-reduction of the resulting hydroxy ketone 25 and finally TBS protection of 26 provided protected pentaol 27 as a single isomer. The hydroxy groups of this pentaol are suitably differentiated for further fragment coupling on either end of the molecule, and significant progress along these lines has been made5.
Scheme 4.

Synthesis of alternative C21–C30 fragment 27
CONCLUSIONS
An unusual reaction between dithiane anions and 5-(alkylsulfanyl)-1-phenyltetrazoles has been discovered. The dithiane anion formally displaces the tetrazole ring to provide a sulfanylated 1,3-dithiane. The mechanism of the process is not clear, though direct displacement at sulfur does not seem probable. Instead, addition of the dithiane anion to the tetrazole ring may initiate the transformation. While there are simpler ways to sulfanylate 1,3-dithianes, the observed chemoselectivity of the new transformation (terminal epoxide survives unscathed) may render it interesting in some settings.
EXPERIMENTAL
General
Optical rotations were measured on a Perkin-Elmer 241 polarimeter at the Na D-line (λ = 589 nm) using a 1-dm cell at 20 °C. [α]D values are given in deg cm2 g−1. IR spectra were recorded on a Mattson Genesis Series FTIR using thin film deposition on NaCl plates (wave-numbers in cm−1). 1H NMR spectra were measured on a Bruker Avance DPX 300 spectrometer (at 300 MHz) or on an Avance DRX 500 spectrometer (at 500 MHz) in CDCl3. 13C NMR spectra were measured on a Bruker Avance DPX 300 spectrometer (at 75 MHz), or on an Avance DRX 500 spectrometer (at 125 MHz) in CDCl3. Chemical shifts are given in ppm (δ-scale) and were determined relative to the residual proton signal for CHCl3 (7.27) and the carbon signal for CDCl3 (77.09). Coupling constants (J) are given in Hz. High resolution mass spectra were obtained on a VG Autospec double focusing instrument and are reported in units of m/z. For column chromatography 230–400 mesh silica gel was used (Sorbent Technologies).
2-[(4S)-2,2-Dimethyl-1,3-dioxolan-4-yl]acetaldehyde (9)
Aldehyde 9 was obtained by oxidation of the commercially available 2-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]ethan-1-ol ((S)-8) using the Parikh–Doering protocol7a (pyr·SO3, DMSO) in 86% yield.
(4S)-4-Allyl-2,2-dimethyl-1,3-dioxolane (10)
To a solution of CH3PPh3Br (15.8 g, 44.2 mmol) in THF (500 ml) at 0 °C, n-BuLi (1.6 M in hexane, 27.6 ml, 44.2 mmol) was added. The reaction mixture was stirred for 20 min followed by cooling to −78 °C. A solution of aldehyde 9 (4.9 g, 34 mmol) in THF (5 ml) was slowly added. After 30 min, the reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was poured into saturated aqueous NH4Cl (300 ml) and the aqueous layer was extracted with Et2O (2 × 300 ml). The combined organic layers were dried over anhydrous MgSO4 and concentrated. Purification of the crude product by flash column chromatography (silica gel, 20% ethyl acetate in hexanes) afforded 10 (4.0 g, 83%) as a volatile oil. 1H NMR (300 MHz, CDCl3): 1.37 (s, 3 H), 1.43 (s, 3 H), 2.25–2.34 (m, 1 H), 2.37–2.48 (m, 1 H), 3.59 (dd, J = 8.2, 7.1, 1 H), 4.03 (dd, J = 8.2, 6.0, 1 H), 4.13–4.21 (m, 1 H), 5.07–5.17 (m, 2 H), 5.81 (ddt, J = 17.0, 10.4, 7.1, 1 H). 13C NMR (75 MHz, CDCl3): 25.7, 26.9, 38.1, 69.0, 75.2, 109.0, 117.7, 133.7.
(4S)-2,2-Dimethyl-[(2S)-epoxyprop-1-yl]-1,3-dioxolane (11)
MCPBA (5.82 g, 33.74 mmol) was added to a solution of the alkene 10 (4.0 g, 28.12 mmol) in dichloromethane (200 ml). The resulting mixture was stirred at room temperature for 1.5 h. The reaction was quenched by addition of saturated aqueous NaHCO3 solution. The layers were separated and the aqueous layer was extracted twice with dichloromethane. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under vacuum. Purification by column chromatography (30% EtOAc in hexanes) gave 4.35 g (98%) of epoxide as a ~1:1 mixture of epimers. This mixture was used directly in the next step.
Acetic acid (62 μl, 1.09 mmol) and (S,S)-Jacobson catalyst (164.7 mg, 0.27 mmol) were added to a solution of the above epoxide (4.30 g, 27.18 mmol) in THF (0.26 ml). The reaction mixture was cooled to 0 °C and water (0.27 ml, 14.95 mmol) was added in one portion. The mixture was warmed up to room temperature and stirred for 16 h. The volatile components were removed by vacuum transfer into a cooled (−78 °C) receiving flask. The recovered epoxide was further purified by flash column chromatography (30% EtOAc in hexanes) to obtain 1.95 g (45%) of the pure epoxide 11 as a colorless oil. 1H NMR (500 MHz, CDCl3): 1.31 (s, 3 H), 1.36 (s, 3 H), 1.49 (ddd, J = 13.8, 7.3, 5.5, 1 H), 1.91 (ddd, J = 14.2, 7.8, 4.1, 1 H), 2.45 (dd, J = 4.6, 2.3, 1 H), 2.75 (t, J = 4.6, 1 H), 2.97–3.00 (m, 1 H), 3.53 (t, J = 7.3, 1 H), 4.05 (dd, J = 7.8, 5.6, 1 H), 4.22–4.27 (m, 1 H). 13C NMR (126 MHz, CDCl3): 25.6, 26.9, 37.1, 47.1, 49.3, 69.3, 73.6, 108.9. IR (neat): 2987, 2942, 2872, 1454, 1371, 1060. HRMS for C7H11O3 (M – CH3)+: calculated 143.0708, found 143.0706.
(2S)-1-[(4S)-2,2-Dimethyl-1,3-dioxolan-4-yl]-3-(1,3-dithian-2-yl)propan-2-ol (12)
t-BuLi (1.7 M in pentane, 5.3 ml, 9.0 mmol) was added to a solution of 1,3-dithiane (1.10 g, 9.04 mmol) in THF/HMPA (5 ml/0.3 ml) at −78 °C. After 30 min, a solution of 11 (1.3 g, 8.2 mmol) in THF (3 ml) and HMPA (1 ml) was added to the above reaction mixture. After 1 h at −78 °C, the reaction mixture was allowed to warm to 0 °C, followed by addition of saturated aqueous NH4Cl solution. The aqueous layer was extracted with ethyl acetate, the combined organic layers were dried over MgSO4 and concentrated. The crude product was purified by flash column chromatography (silica gel, 25% ethyl acetate in hexanes) to yield 12 (1.9 g, 83%) as an oil, [α]D +6.75 (c 0.80 CHCl3). 1H NMR (500 MHz, CDCl3): 1.36 (s, 3 H), 1.42 (s, 3 H), 1.69–1.79 (m, 2 H), 1.84–1.99 (m, 3 H), 2.10–2.16 (m, 1 H), 2.67 (d, J = 5.0, 1 H), 2.82–2.95 (m, 4 H), 3.59 (t, J = 7.8, 1 H), 4.09 (dd, J = 8.2, 6.0, 1 H), 4.14–4.20 (m, 1 H), 4.27 (dd, J = 8.9, 5.3, 1 H), 4.31–4.36 (m, 1 H). 13C NMR (126 MHz, CDCl3): 25.6, 25.9, 26.9, 30.1, 30.3, 39.8, 42.8, 44.2, 66.1, 69.4, 73.4, 108.9. IR (neat): 3435, 2983, 2935, 2899, 1456, 1423, 1370. EIMS: (M+) 278. HRMS for C12H22O3S2 (M+): calculated 278.1010, found 278.1006.
2-{(2S)-2-[(tert-Butyldimethylsilyl)oxy]-3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]propyl}-1,3-dithiane (13)
To a solution of alcohol 12 (320 mg, 1.15 mmol) and 2,6-lutidine in CH2Cl2 at −78 °C, TBSOTf was added. The reaction mixture was stirred for 1.5 h, then poured into water (25 ml). The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and concentrated. The crude product was purified by flash column chromatography to yield 13 (410 mg, 91%) as an oil, [α]D −1.65 (c 4.79 CHCl3). 1H NMR (500 MHz, CDCl3): 0.07 (s, 3 H), 0.09 (s, 3 H), 0.87 (s, 9 H), 1.30 (s, 3 H), 1.36 (s, 3 H), 1.59 (ddd, J = 13.7, 8.2, 5.0, 1 H), 1.77 (ddd, J = 13.7, 7.8, 4.6, 1 H), 1.79–1.88 (m, 3 H), 2.05–2.11 (m, 1 H), 2.75–2.87 (m, 4 H), 3.44 (t, J = 7.8, 1 H), 3.99–4.09 (m, 3 H), 4.11–4.16 (m, 1 H), 4.13 (dtd, J = 10.5, 7.8, 5.0, 1 H). 13C NMR (126 MHz, CDCl3): −4.6, −4.4, 25.8, 25.9, 27.1, 30.3, 30.5, 41.4, 43.6, 43.9, 66.6, 69.9, 72.8, 77.3, 108.5.
5-({2-[(4R)-2,2-Dimethyl-1,3-dioxolan-4-yl]ethyl}sufanyl)-1-phenyl-1H-tetrazole (14)
Diisopropyl azodicarboxylate (2.80 g, 14.0 mmol) was added to a solution of (R)-8 (1.2 g, 8.2 mmol), 1-phenyl-1H-tetrazole-5-thiol (2.60 g, 14.8 mmol) and triphenylphosphine (3.00 g, 11.5 mmol) in THF (20 ml) at 0 °C. The mixture was stirred at room temperature for 3 h, water (20 ml) was added. The aqueous layer was extracted with ethyl acetate and the combined organic layers were dried over anhydrous MgSO4 and concentrated. Purification of the crude product by flash column chromatography (silica gel, 25% ethyl acetate in hexanes) provided 14 (2.23 g, 7.29 mmol, 89%) as an oil, [α]D +12.3 (c 1.19 CHCl3). 1H NMR (500 MHz, CDCl3): 1.90–2.09 (m, 2 H), 2.55 (br s, 1 H), 3.42–3.55 (m, 1 H), 3.56–3.68 (m, 2 H), 3.68–3.81 (m, 1 H), 3.85–4.13 (m, 2 H), 7.58 (br s, 5 H). 13C NMR (126 MHz, CDCl3): 29.8, 33.7, 66.4, 69.5, 124.0, 129.9, 130.4, 133.5, 155.1. IR (neat): 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280. EIMS: (M + H)+ 267. HRMS for C10H11N4O1S (M – CH3O)+: calculated 235.0653, found 235.0658.
(2R)-4-[(1-Phenyl-1H-tetrazol-5-yl)sulfanyl]butane-1,2-diol (15)
To a solution of 14 (600 mg, 1.96 mmol) in methanol (10 ml), a drop of acetyl chloride (estimated amount, 20 mg) was added. After 30 min, the reaction mixture was concentrated and the crude product was purified by flash column chromatography (80% ethyl acetate in hexanes) to provide the title compound (510 mg, 86 %) as a viscous oil, [α]D +11 (c 0.78 CHCl3). 1H NMR (500 MHz, CDCl3): 1.90–2.09 (m, 2 H), 2.55 (br s, 1 H), 3.42–3.55 (m, 1 H), 3.56–3.68 (m, 2 H), 3.68–3.81 (m, 1 H), 3.85–4.13 (m, 2 H), 7.58 (br s, 5 H). 13C NMR (126 MHz, CDCl3): 29.8, 33.7, 66.4, 69.5, 124.0, 129.9, 130.4, 133.5, 155.1. IR (neat): 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280. EIMS: (M + H)+ 267. HRMS for C10H11N4O1S (M – CH3O)+: calculated 235.0653, found 235.0658.
(2R)-2-Hydroxy-4-[(1-phenyl-1H-tetrazol-5-yl)sufanyl]butyl 4-Methylbenzene-1-sulfonate (16)
To a solution of 15 (290 mg, 1.28 mmol) in CH2Cl2 (10 ml), Bu2SnO (64 mg, 0.26 mmol), tosyl chloride (244 mg, 1.28 mmol) and triethylamine (130 mg, 1.28 mmol) were added. The reaction mixture was stirred for 3 h followed by dilution with water (10 ml). The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and concentrated. The crude product was purified by flash column chromatography (silica gel, ethyl acetate-hexanes 1:1) to yield the title compound (400 mg, 74%), [α]D +10.0 (c 0.74 CHCl3). 1H NMR (500 MHz, CDCl3): 1.87–2.03 (m, 2 H), 2.39 (s, 3 H), 3.43–3.50 (m, 2 H), 3.92–4.03 (m, 4 H), 7.30 (d, J = 8.2, 2 H), 7.53 (s, 5 H), 7.75 (d, J = 8.2, 2 H). 13C NMR (126 MHz, CDCl3): 21.4, 29.2, 32.7, 66.9, 73.1, 123.6, 127.7, 129.7, 129.8, 130.1, 132.3, 133.3, 144.9, 154.4. IR (neat): 3400, 3060, 2946, 1597, 1499, 1387, 1357, 1243. HRMS for C18H20N4O4S2: calculated 443.0824, found 443.0804.
5-({2-[(2R)-Oxiran-2-yl]ethyl}sulfanyl)-1-phenyl-1H-tetrazole (17)
To a solution of 16 (440 mg, 1.05 mmol) in CH3OH–dichloromethane (9:1, 10 ml), K2CO3 (173 mg, 1.25 mmol) was added. The reaction mixture was stirred at room temperature for 1 h, concentrated, then diluted with dichloromethane (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic extracts were dried over anhydrous MgSO4 and concentrated to provide 17 (211 mg, 81%) as an oil, [α]D +15.0 (c 1.13 CHCl3). 1H NMR (500 MHz, CDCl3): 1.92 (td, J = 14.2, 6.9, 1 H), 2.21–2.29 (t, J = 4.6, 1 H), 2.52 (dd, J = 4.6, 2.7, 1 H), 2.74–2.78 (m, 1 H), 3.01–3.06 (m, 1 H), 3.45–3.54 (m, 2 H), 7.49–7.56 (m, 5 H). 13C NMR (126 MHz, CDCl3): 29.8, 32.0, 46.8, 50.6, 123.7, 129.8, 130.1, 133.5, 153.9. IR (neat): 3056, 2991, 2924, 1596, 1500, 1461. HRMS (EI) for C11H12N4O1S: calculated 248.0732, found 248.0721.
2-{(2S)-2-[(tert-Butyldimethylsilyl)oxy]-3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]propyl}-2-({2-[(2R)-oxiran-2-yl]ethyl}sulfanyl)-1,3-dithiane (19)
t-BuLi (1.7 M in pentane, 0.14 ml, 0.24 mmol) was added to a solution of dithiane 13 in THF (0.8 ml) at −30 °C. After 30 min, epoxide 17 (58.9 mg, 0.284 mmol) in THF (0.2 ml) was added to the reaction mixture and stirred at −30 °C for additional 30 min. Then the mixture was warmed to 0 °C and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were dried over MgSO4 and concentrated. The crude product was purified by flash column chromatography (silica gel, 25% ethyl acetate in hexanes) to yield 19 (66 mg, 56%) as a colorless oil, [α]D −21.2 (c 1.72 CHCl3). 1H NMR (500 MHz, CDCl3): 0.12 (s, 3 H), 0.16 (s, 3 H), 0.89 (s, 9 H), 1.33 (s, 3 H), 1.41 (s, 3 H), 1.61 (ddd, J = 13.7, 8.3, 4.6, 1 H), 1.81–1.87 (m, 3 H), 2.04–2.09 (m, 1 H), 2.23–2.29 (m, 3 H), 2.53 (dd, J = 5.0, 2.8, 1 H), 2.58–2.71 (m, 4 H), 2.78–2.80 (m, 1 H), 2.99–3.04 (m, 1 H), 3.14–3.35 (m, 2 H), 3.51–3.54 (m, 1 H), 4.06 (dd, J = 7.8, 5.5, 1 H), 4.17–4.23 (m, 1 H), 4.26–4.31 (m, 1 H). 13C NMR (126 MHz, CDCl3): −4.2, −3.8, 18.1, 24.6, 25.9, 26.1, 27.1, 27.5, 27.8, 29.6, 31.9, 43.3, 47.2, 51.4, 52.0, 60.4, 67.2, 70.1, 73.2, 108.6. IR (neat): 2983, 2953, 2855, 1472, 1252. HRMS (EI) for C21H39O4S3Si (M – CH3)+: calculated 479.1779, found 479.1780.
2-(Ethylsulfanyl)-2-phenethyl-1,3-dithiane (22)
BuLi (0.5 ml, 0.8 mmol) was added dropwise to a solution of dithiane9 20 (179.2 mg, 0.8 mmol) in THF (10 ml) at −30 °C. The reaction mixture was stirred at this temperature for 1 h followed by addition of a solution of tetrazole10 21 (164.8 mg, 0.8 mmol) in THF (2 ml). The reaction mixture was further stirred at −30 °C for 3 h and then quenched by the addition of saturated NH4Cl solution. The mixture was diluted with ether and water, the layers were separated, and the aqueous layer was extracted with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and concentrated under vacuum. Purification by flash column chromatography (silica gel, 5% EtOAc in hexanes) gave 205.4 mg of 22. 1H NMR (300 MHz, CDCl3): 7.32–7.27 (m, 2 H), 7.23–7.18 (m, 3 H), 3.41–3.31 (m, 2 H), 3.00 (dd, J = 4.86, 12.48, 1 H), 3.00 (dd, J = 8.49, 8.59, 1 H), 2.70 (dd, J = 3.19, 4.49, 1 H), 2.66 (d, J = 3.70, 4.49, 1 H), 2.59 (q, J = 7.49, 2 H), 2.30 (dd, J = 4.62, 12.50, 1 H), 2.30 (dd, J = 8.38, 8.60, 1 H), 2.20–2.10 (m, 1 H), 1.95–1.80 (m, 1 H), 1.28 (t, J = 7.49, 3 H). 13C NMR (75 MHz, CDCl3): 141.4, 128.5, 128.4, 126.0, 62.1, 44.7, 30.6, 27.4, 26.8, 25.0, 13.9. IR (neat): 2952, 2926, 1729, 1601, 1495, 1452, 1421, 1276. EIMS: (M – SC2H5)+ 223. HRMS for C12H15S2 (M – SC2H5)+ calculated 223.0615, found 223.0552.
Preparation of an Authentic Sample of 22
BuLi (0.55 ml, 0.89 mmol) was added dropwise to a solution of dithiane 20 (200 mg, 0.89) in THF (5 ml) at −30 °C. The resulting mixture was stirred at this temperature for 1 h followed by the addition of diethyl disulfide (0.20 ml, 0.89 mmol). The reaction mixture was stirred at −30 °C for 3 h and then quenched by the addition of saturated NH4Cl solution. The reaction mixture was diluted with ether, the layers were separated, and the aqueous layer was extracted with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, concentrated under vacuum, and purified by column chromatography (5% EtOAc in hexanes) to afford 205.4 mg (81%) of pure 22. The spectroscopic data of this sample were identical to those of the product obtained by the reaction of dithiane 20 with tetrazole 21.
(2R)-1-(2-{(2S)-2-[(tert-Butyldimethylsilyl)oxy]-3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-propyl}-1,3-dithian-2-yl)-4-[(4-methoxybenzyl)oxy]butan-2-ol (24)
To a solution of dithiane 13 (0.33 g, 0.84 mmol) in THF (1.2 ml) at 0 °C, n-BuLi (1.6 M in hexane, 0.6 ml) was added. The reaction mixture was stirred at 0 °C for 20 min, followed by cooling to −20 °C. Then a solution of epoxide 2311 (0.18 g, 0.88 mmol) in THF (0.5 ml) was added. After 3 h at −10 °C, the reaction was quenched with saturated aqueous NH4Cl and the layers were separated. The aqueous layer was extracted with Et2O and the combined organic layers were dried over anhydrous MgSO4 and concentrated. The crude product was purified by flash column chromatography (silica gel, 25% ethyl acetate in hexanes) to yield 24 (340 mg, 67%) as a clear oil. 1H NMR (300 MHz, CDCl3): 0.06 (s, 3 H), 0.12 (s, 3 H), 0.88 (s, 9 H), 1.34 (s, 3 H), 1.41 (s, 3 H), 1.52–1.80 (m, 3 H), 1.88–2.19 (m, 5 H), 2.20–2.39 (m, 2 H), 2.75–2.99 (m, 4 H), 3.51 (t, J = 7.9, 1 H), 3.57–3.63 (m, 3 H), 3.80 (s, 3 H), 4.06 (dd, J = 13.5, 7.7, 1 H), 4.12–4.23 (m, 3 H), 4.45 (s, 2 H). 13C NMR (75 MHz, CDCl3): −4.2, −3.7, 14.3, 18.0, 24.7, 25.9, 26.0, 26.2, 26.7, 27.1, 37.7, 43.0, 46.7, 48.2, 51.2, 55.3, 60.4, 67.0, 67.5, 68.0, 70.1, 72.9, 73.3, 108.6, 113.8, 129.4, 130.4, 159.2. IR (neat): 3497, 2974, 2932, 2856, 1613, 1514, 1249, 1093. HRMS (EI) for C30H52O6S2Si (M)+: calculated 600.2975, found 600.2955.
[(2S,6R)-2-(tert-Butyldimethylsilyl)oxy]-3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-6-hydroxy-8-[(4-methoxybenzyl)oxy]octan-4-one (25)
To a solution of 24 (135 mg, 0.224 mmol) in acetonitrile and water (6:1, 3 ml), MeI (80 mg, 0.56 mmol) and K2CO3 (34 mg, 0.35 mmol) were added. The reaction mixture was stirred at 45 °C for 6 h. The reaction mixture was diluted with Et2O (5 ml), dried over anhydrous MgSO4, concentrated and purified by flash column chromatography (silica gel, 25% ethyl acetate in hexanes) to yield 25 (93 mg, 81%) as an oil. 1H NMR (500 MHz, CDCl3): 0.09 (s, 3 H), 0.13 (s, 3 H), 0.91 (s, 9 H), 1.36 (s, 3 H), 1.42 (s, 3 H), 1.64 (ddd, J = 13.8, 8.3, 6.7, 1 H), 1.71–1.86 (m, 3 H), 2.60–2.68 (m, 4 H), 3.41 (d, J = 3.2, 1 H), 3.49 (t, J = 7.8, 1 H), 3.61–3.70 (m, 2 H), 3.83 (s, 3 H), 4.06 (dd, J = 7.8, 6.0, 1 H), 4.13–4.20 (m, 1 H), 4.25–4.31 (m, 1 H), 4.33–4.38 (m, 1 H), 4.47 (s, 2 H), 6.91 (d, J = 8.7, 2 H), 7.28 (d, J = 8.7, 2 H). 13C NMR (125 MHz, CDCl3): −4.7, −4.5, 18.0, 25.9, 27.1, 36.2, 41.6, 50.9, 52.0, 55.3, 66.2, 66.4, 67.6, 69.8, 72.7, 72.9, 108.8, 113.9, 129.3, 130.2, 159.3, 209.5. IR (neat): 3466, 2952, 2931, 1708, 1514, 1302, 1090, 777. HRMS (ES) for C27H46NaO7Si (M + Na)+: calculated 533.2911, found 533.2886.
(3S,5S,7R)-5,7-Bis[(tert-butyldimethylsilyl)oxy]-8-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-1-[(4-methoxybenzyl)oxy]octan-3-ol (26)
To a solution of 25 (88 mg, 0.17 mmol) in acetonitrile (0.54 ml) at −25 °C, a solution of (CH3)4NBH(OAc)3 (68 mg, 0.26 mmol) in acetic acid (0.1 ml) was added. The reaction mixture was stirred at that temperature for 48 h, quenched with aqueous sodium potassium tartarate (1.0 M, 0.8 ml), diluted with ethyl acetate and neutralized with sodium hydrogen carbonate. The aqueous layer was extracted with ethyl acetate; the combined organic layers were dried over anhydrous MgSO4 and concentrated to yield the title compound (76 mg, 86%) as an oil. Crude 26 was taken to the next step without further purification. 1H NMR (500 MHz, CDCl3): 0.12 (s, 3 H), 0.14 (s, 3 H), 0.90 (s, 9 H), 1.34 (s, 3 H), 1.38 (s, 3 H), 1.50–1.62 (m, 3 H), 1.66–1.75 (m, 2 H), 1.78–1.92 (m, 3 H), 3.48 (t, J = 8.0, 1 H), 3.57 (brs, 1 H), 3.60–3.71 (m, 2 H), 3.80 (s, 3 H), 3.89 (brs, 1 H), 4.05 (dd, J = 7.8, 6.0, 1 H), 4.09–4.24 (m, 3 H), 4.26–4.34 (m, 1 H), 4.45 (s, 2 H), 6.87 (d, J = 8.7, 2 H), 7.24 (d, J = 8.7, 2 H). 13C NMR (76 MHz, CDCl3): −4.8, −4.4, 18.0, 25.9, 27.1, 36.9, 40.4, 43.1, 43.9, 55.3, 66.0, 68.4, 68.8, 69.3, 69.9, 70.1, 72.8, 73.0, 108.9, 113.9, 129.3, 130.3, 159.4. IR (neat): 3449, 2984, 2934, 2857, 1613, 1514, 1249. HRMS (EI) for C27H48NaO7Si (M + Na)+: calculated 535.3067, found 535.3072.
(4S)-2,2-Dimethyl-4-{(2S,4S,6R)-2,4,6-tris[(tert-butyldimethylsilyl)oxy]-8-[(4-methoxybenzyl)oxy]octyl}-1,3-dioxolane (27)
To a solution of 26 (44.0 mg, 0.0858 mmol) in dichloromethane (5 ml) at 0 °C, TBSOTf (46.5 mg, 0.176 mmol) and 2,6-lutidine (54.3 mg, 0.506 mmol) were added. The reaction mixture was stirred at that temperature for 1 h followed by quenching with water. The aqueous layer was extracted with dichloromethane and the combined organic layers were dried over MgSO4 and concentrated. Purification of the crude reaction mixture by flash column chromatography (silica gel, 10% ethyl acetate in hexanes) gave 27 (50 mg, 80%) as a clear oil. 1H NMR (300 MHz, CDCl3): 0.07 (s, 18 H), 0.88 (s, 18 H), 0.89 (s, 9 H), 1.33 (s, 3 H), 1.38 (s, 3 H), 1.48–1.87 (m, 8 H), 3.42–3.52 (m, 3 H), 3.75–3.84 (m, 4 H), 3.85–3.94 (m, 2 H), 4.00–4.04 (m, 1 H), 4.13–4.26 (m, 1 H), 4.41 (s, 2 H), 6.87 (d, J = 8.8, 2 H), 7.25 (d, J = 7.7, 2 H). 13C NMR (75 MHz, CDCl3): −4.4, −4.2, −4.1, −3.7, −3.5, 18.1, 26.0, 27.2, 37.9, 41.3, 46.3, 47.3, 55.3, 66.7, 67.4, 67.5, 67.7, 70.0, 72.7, 108.6, 113.8, 129.3, 130.8, 159.1. IR (neat): 2953, 2857, 1511, 1250. HRMS (EI) for C39H76NaO7Si3 (M + Na)+: calculated 129.0552, found 129.0550.
Footnotes
Dedicated to Dr. Alfred Bader on the occasion of his 85th birthday.
REFERENCES
- 1.a) Kamiyama T, Itezono Y, Umino T, Satoh T, Nakayama N, Yokose K. J. Antibiot. 1993;46:1047. doi: 10.7164/antibiotics.46.1047. [DOI] [PubMed] [Google Scholar]; b) Kamiyama T, Umino T, Fujisaki N, Fujimori K, Satoh T, Yamashita Y, Ohshima S, Watanabe J, Yokose K. J. Antibiot. 1993;46:1039. doi: 10.7164/antibiotics.46.1039. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi Y, Czechtizky W, Kishi Y. Org. Lett. 2003;5:93. doi: 10.1021/ol0272895. [DOI] [PubMed] [Google Scholar]
- 3.Lira R, Roush WR. Org. Lett. 2007;9:533. doi: 10.1021/ol0629869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.BouzBouz S, Cossy J. Org. Lett. 2004;6:3469. doi: 10.1021/ol048862i. [DOI] [PubMed] [Google Scholar]
- 5.Gudipati V. Ph.D. Thesis. University of Pittsburgh; Pittsburgh: 2008. http://etd.library.pitt.edu/ETD/available/etd-04212008-115104/. The thesis contains copies of key spectra of 19 and other compounds. [Google Scholar]
- 6.a) Seebach D. Angew. Chem., Int. Ed. Engl. 1969;8:639. [Google Scholar]; b) Yus M, Najera C, Foubelo F. Tetrahedron. 2003;59:6147. [Google Scholar]; c) Smith AB, Adams CM. Acc. Chem. Res. 2004;37:365. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]
- 7.a) Parikh JR, Doering WE. J. Am. Chem. Soc. 1967;89:5505. [Google Scholar]; b) Dineen TA, Roush WR. Org. Lett. 2004;6:2043. doi: 10.1021/ol049331x. [DOI] [PubMed] [Google Scholar]
- 8.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 9.Hon Y-S, Lee C-F, Chen R-J, Huang Y-F. J. Org. Chem. 2008;73:2018. [Google Scholar]
- 10.Fuchs E, Keller M, Breit B. Chem. Eur. J. 2006;12:6930. doi: 10.1002/chem.200600180. [DOI] [PubMed] [Google Scholar]
- 11.Marshall JA, Schaaf G, Nolting A. Org. Lett. 2005;7:5331. doi: 10.1021/ol0523493. [DOI] [PubMed] [Google Scholar]