

Abstract
Elevation of nitric oxide (NO˙) can either promote or inhibit ultraviolet B light (UVB)-induced apoptosis. In this study, we determined real-time concentration of NO˙ and peroxynitrite (ONOO−) and their role in regulation of membrane integrity and apoptosis. Nanosensors (diameter 300–500 nm) were used for direct in situ simultaneous measurements of NO˙ and ONOO− generated by UVB in cultured keratinocytes and mice epidermis. An exposure of keratinocytes to UVB immediately generated ONOO− at maximal concentration of 190 nm followed by NO release with a maximal concentration of 91 nm. The kinetics of UVB-induced NO˙/ONOO− was in contrast to cNOS agonist stimulated NO˙/ONOO− from keratinocytes. After stimulating cNOS by calcium ionophore (CaI), NO˙ release from keratinocytes was followed by ONOO− production. The [NO˙] to [ONOO−] ratio generated by UVB decreased below 0.5 indicating a serious imbalance between cytoprotective NO˙ and cytotoxic ONOO−—a main component of nitroxidative stress. The NO˙/ONOO− imbalance increased membrane damage and cell apoptosis was partially reversed in the presence of free radical scavenger. The results suggest that UVB-induced and cNOS-produced NO˙ is rapidly scavenged by photolytically and enzymatically generated superoxide (O2˙−) to produce high levels of ONOO−, which enhances oxidative injury and apoptosis of the irradiated cells.
Introduction
UVB induces the production of nitric oxide (NO˙), which plays a role in regulation of apoptosis in skin cells (1–5). NO˙ is produced from l-arginine and oxygen in reaction catalyzed by nitric oxide synthase (NOS). The family of NO˙ synthases consists of constitutive enzymes (cNOS), including neuronal (nNOS) and endothelial NOS (eNOS), and inducible NO˙ synthase (iNOS). The activation of cNOS could be immediately triggered by elevation of the intracellular calcium level, which induces the binding of calmodulin to the inactive cNOS (6–8). The induction of iNOS is regulated through multiple signaling pathways and it could take hours to increase the expression of iNOS (9).
Most cell types residing in the skin have been reported to produce NO˙ in response to appropriate stimulation. Keratinocytes (10), Langerhans cells (11), dermal fibroblasts (12), melanocytes (13) and melanoma (14) cells express iNOS upon stimulation with inflammatory cytokines. Among these cells, keratinocytes account for 90–95% of total cells in the epidermis. Keratinocytes contain cNOS, mainly neuronal NOS, which is activated by UV-induced calcium flux (15–20). UV also induces iNOS expression with a maximized mRNA level at 24 h postirradiation in human skin (9,21). UV-induced NO˙ production was shown to protect cultured keratinocytes and skin from apoptotic death (2,3,5,22,23). However, all the reported studies were based on indirect observation that the inhibition of NOS or supplementation of NO˙ donor during a period of 18–24 h protected keratinocytes from apoptotic death. An elevation of NO˙ was detected immediately after UV irradiation in endothelial and epithelial cells (18,24). The impact of the early release of NO˙ on UV-induced apoptosis has not been studied yet. In this report, we used a direct method (electrochemical nanosensors) to measure simultaneously at real time, in situ, the NO˙ and ONOO− release from cultured keratinocytes and skin tissue of mice after irradiation of UVB. We demonstrated that UVB generated high level of ONOO− shifts unfavorable NO˙/ONOO− balance and has a pro-apoptotic effect on both cultured cells and living skin.
Materials and Methods
Animals
Adult BALB/c mice were housed in a pathogen-free barrier facility in accordance with the standards of Ohio University. Mice were kept in groups of two per cage in a 12 h light/12 h dark cycle and housed at 25°C and 50% relative humidity.
Cell culture
The immortalized human keratinocyte cell line HaCaT was kindly provided by Dr. Hongtao Yu (Jackson State University, MS). The cells were cultured as monolayer in DMEM (Cellgro) with 10% FBS (Cellgro) at 37°C with 5% CO2.
Nanosensors for continuous measurement of NO˙ and ONOO−
Concurrent measurements of NO˙ and ONOO− were performed with electrochemical nanosensors (300–500 nm diameter). The designs are based on previously developed and well-characterized chemically modified carbon-fiber technology (25–29). Each of the sensors was made by depositing a sensing material on the tip of the carbon fiber. We used a conductive film of polymeric nickel (II) tetrakis (3-methoxy-4hydroxy-phenyl) porphyrinic for the NO˙ sensor (25,28,29) and a polymeric film of Mn (III)–paracyclophanyl-porphyrin for the ONOO− sensor (26,27). Amperometry was used to measure changes in NO˙ and ONOO− concentrations from its basal level with time (detection limit of 1 nm and resolution time <50 ms for each sensor). Linear calibration curves were constructed for each sensor from 5 nm to 3 μm before and after measurements with aliquots of NO˙ and ONOO− standard solutions, respectively.
Determination of UVB-induced NO˙, and ONOO− production in a single cell
Cells were seeded at 103 cells cm−2 and cultured for 12 h in complete medium. The sensors were positioned near the surface (5 ± 2 μm) of a selected cell with the help of a computer-controlled micromanipulator. The background signals of NO˙ and ONOO− were stabilized and the cells were exposed to UVB irradiation at a power of 0.5 mW cm−2. The signals generated by the nanosensors were recorded continuously for 80 s.
Determination of UVB-induced NO˙ and ONOO− production in living mouse skin
The back of mice was shaved with an electric clipper 1 day prior to the experiment. Under anesthesia (ketamine 50 mg kg−1 + xylazine 5 mg kg−1), the module nanosensors (total diameter 3.0 ± 0.5 μm) were inserted into the epidermal and dermal layers. L-shaped carbon fibers with the tip sharpened in microwave plasma were used in this study. Computer-controlled micromanipulators with x,y,z resolution ±2 μm were employed to implant the sensors in the skin. The z coordinate (depth) was calibrated using piezoelectric currents recorded at zero distance from the skin (electrode touching the skin). The productions of NO˙ and ONOO− were continuously recorded. Once the background signals of NO˙ and ONOO− were stabilized, the mice were UVB-irradiated at a power of 0.5 mW cm−2. The productions of NO˙ and ONOO− were continuously recorded for 40 s.
Inhibition of early release of NO˙ and ONOO− in cultured cells and skin epidermis
An N-substituted l-arginine analog NG-methyl-l-arginine (l-NMMA; Sigma) was used to inhibit NOS activity; the glutathione (GSH) synthesis precursor N-acetyl-l-cysteine (l-NAC; Sigma) was used to scavenge free radicals and superoxide dismutase attached to polyethylene glycol (PEG-SOD) was used to dismutate superoxide (O2˙−). The cells were pretreated with l-NMMA (100 μm) or l-NAC (25 mm) for 2 h and then UVB-irradiated (50 mJ cm−2). Immediately after irradiation, the cells were cultured in fresh medium without the inhibitors until further analysis. The mice were administered an intraperitoneal injection of l-NMMA (10 mg kg−1) or l-NAC (500 mg kg−1) 1 h before UVB irradiation.
Determination of cell apoptosis and death by flow cytometry
At 24 h postirradiation, the cells were digested with 0.01% trypsin and combined with the cells floating in the medium. An Annexin V: FITC Apoptosis Detection Kit II (BD Pharmingen) was used following the manufacturer's protocol. The annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) stained cells were analyzed by using a FACSort Flow Cytometer (Becton Dickinson) equipped with Cell-Quest software (Becton Dickinson). The parameter of the measurement was set at SSC 350, FL1 700 and FL2 700, and a total 10 000 cells were counted.
Cell injury assay using calcium green-1 acetoxymethylester (CAG-AM) and PI fluorescence staining
At 1 h postirradiation, the cells were incubated with CAG-AM (2.5 μm; Invitrogen) and PI (50 μg mL−1) for 30 min at room temperature. After washing three times with PBS, fluorescence images were acquired by a camera connected to a confocal microscope (LSM 510; Carl Zeiss) at excitation and emission wavelengths of 506/531 nm for calcium green-1 and 535/617 nm for PI, respectively.
Analysis of membrane damage in mice epidermis
At 24 h before UVB exposure, mice were shaved with electric clippers. Non-UVB-treated controls were also shaved to maintain a constant protocol. The mice were anesthetized with Avertin and treated with the chemical and/or UVB (100 mJ cm−2). Immediately after irradiation, the mice were injected subcutaneously with 0.1 mL of PI (100 μg mL−1; Sigma). Thirty minutes after PI injection, the mice were euthanized by decapitation and skin tissue was harvested and sliced with a cryostat section in 40 nm thickness. The fluorescent images were acquired by a camera connected to a Nikon fluorescent microscope at excitation and emission wavelengths of 535 and 617 nm, respectively. The intensity of the PI staining was analyzed using ImageJ (v1.42k; NIH).
Western blotting
HaCaT cells were treated with l-NMMA and l-NAC for 2 h before UVB irradiation. At 24 h post-UVB, the cells were lysed at 4°C in NP-40 lysis buffer (2% NP-40, 80 mm NaCl, 100 mm Tris-HCl, 0.1% SDS) containing a Proteinase Inhibitor Cocktail (Complete™; Roche Molecular Biochemicals). The protein samples were then added to five-fold Laemmli buffer (0.32 m Tris-HCl, pH 6.8, 0.5 m glycine, 10% SDS, 50% glycerol and 0.03% bromophenol blue) and boiled. These samples were separated on an SDS-PAGE gel and then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% (wt/vol) skim milk in TBST (20 mm Tris [pH 7.5], 150 mm NaCl, 0.1% Tween 20) for 1 h and then incubated with anti-poly(ADP-ribose) polymerase (anti-PARP) antibodies (obtained from Santa Cruz Biotechnology) at 4°C overnight. After washing with TBST, the membrane was incubated with HRP-conjugated anti-rabbit antibody for 1 h at room temperature. The membrane was then washed three times in TBST and two times in TBS, and developed in West Pico Supersignal chemiluminescent substrate (Pierce).
UVB irradiation
UVB was generated from an 8 or 15 W UVB lamp (UVP). The intensity of UVB was standardized by an UVB-meter (UVP). The media was replaced with PBS during the irradiation. After UVB irradiation, fresh medium added to each plate was used in the experiments.
Statistical analysis
Student's t-test was used to analyze the significance of data. P < 0.05 was considered significant.
Results
UVB induces release of NO˙ and ONOO− in cultured human keratinocytes
NO˙ can rapidly react with O2˙− to form ONOO− (30,31), a highly oxidative molecule (32). To determine the biological property of NO˙ after UV irradiation, we measured the realtime production of NO˙ and ONOO− in UVB-treated HaCaT by using a NO˙ or ONOO− nanosensor. After positioning the nanosensors near the surface of keratinocytes (5 ± 2 μm), the productions of NO˙ or ONOO− were continuously monitored. Our data showed that 4 s after the exposure of the keratinocytes to UVB, a rapid release of ONOO− was observed (Fig. 1A). A maximal [ONOO−] of 190 ± 20 was reached after 15 ± 2 s postradiation (Fig. 1A,B). NO˙ release was recorded after 20 ± 2 s and reached a maximal level of 91 ± 8 nm at 40 ± 5 s postradiation (Fig. 1A,B). These results suggest that the cells were under high oxidative stress when NO˙ was just released after UVB irradiation.
Figure 1.
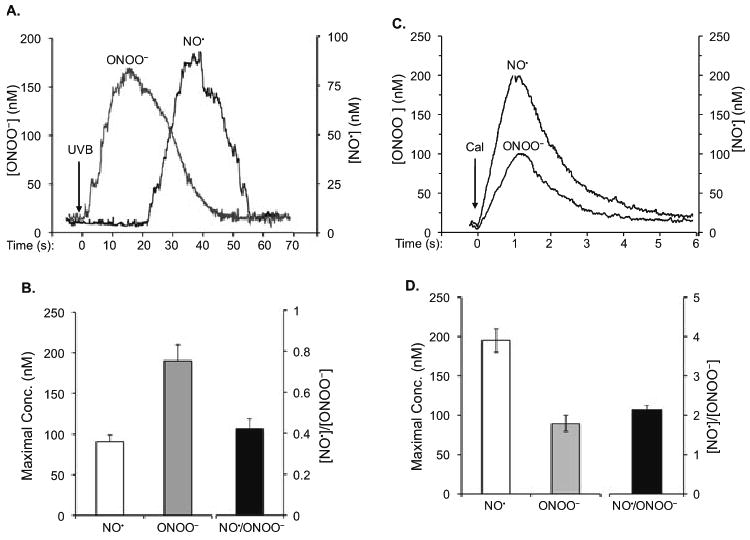
NO˙ and ONOO− amperograms (current calibrated as concentration vs time) and maximal [NO˙], [ONOO−] and a ratio of [NO˙]/[ONOO−] measured in a keratinocyte after UVB irradiation. (A) Amperograms of NO˙ and ONOO− release from the cells irradiated with UVB for 1 min at 0.5 mW cm−2. (B) Maximal [NO˙], [ONOO−] and a ratio of maximal [NO˙]/[ONOO−] produced by a keratinocyte after treating with UVB. (C) Amperograms of NO˙ and ONOO− release from the cells stimulated by CaI (1 μm) in 6 s. (D) Maximal [NO˙], [ONOO−] and a ratio of maximal [NO˙]/[ONOO−] produced by a keratinocyte after treating with CaI. The data in (B) and (D) represent the average of three sets of independent measurements.
To determine maximal concentrations of NO˙ and ONOO− in the keratinocytes, we measured NO˙/ONOO− after stimulation of cNOS with the calcium-independent agonist, CaI. The kinetics of CaI-stimulated ONOO− and NO˙ release was distinctively different from that observed after UVB stimulation (Fig. 1C vs 1A). A rapid increase in [NO˙] was observed after less than 0.1 s after treatment of CaI. The maximal [NO˙] (195 ± 15 nm) was reached about 1.0 ± 0.1 s postinjection of CaI. The release of NO˙ was followed by the release of ONOO− (0.3 ± 0.1 s after treatment of CaI), which reached a maximum of 90 ± 10 nm after 1.2 ± 0.1 s. A ratio of [NO˙]/[ONOO−] was used to quantify a level of oxidative/nitroxidative stress and NO˙/ONOO− imbalance in keratinocytes after stimulation with UVB or CaI (Fig. 1B,D). High [NO˙]/[ONOO−] ratio indicates high concentration of bioavailable, cytoprotective NO˙ and/or low levels of cytotoxic ONOO−. The ratio of [NO˙]/[ONOO−] was 0.42 ± 0.05 after stimulation of keratinocytes with UVB radiation and 2.15 ± 0.10 after stimulation with CaI. These results suggest that there is a rapid increase in O2˙−, which reacts with NO˙ to form ONOO− after UVB irradiation.
To confirm that the UVB-induced elevation of O2˙− leads to the imbalance of [NO˙]/[ONOO−], we analyzed the production of [NO˙] and [ONOO−] in the presence of the cNOS inhibitor l-NAME and a membrane-permeable superoxide dismutase PEG-SOD. In the presence of l-NAME, both [NO˙] and [ONOO−] decreased significantly (Fig. 2). However, the decrease in NO˙ was more pronounced (about 85%) than the decrease in ONOO− (about 70%). An incubation of keratinocytes with PEG-SOD increased the NO˙ level to 165 ± 15 nm (80% increase vs control) while ONOO− level decreased by about 30% compared to control. [NO˙]/[ONOO−] ratio did not change significantly in the presence of l-NAME and increased significantly in the presence of PEG-SOD (Fig. 2B). These results demonstrate that UVB-induced production of NO˙ was accompanied by an elevation of O2˙−, most likely due to photolytic reaction of oxygen and/or O2˙− production by uncoupled cNOS.
Figure 2.
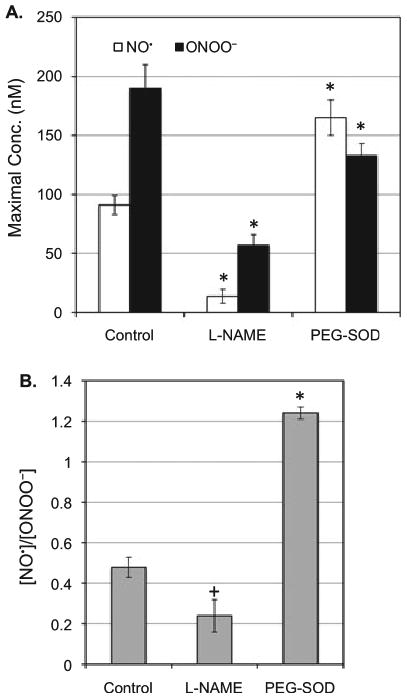
NO˙ and ONOO− release from the cells irradiated with UVB (0.5 mW cm−2) for 1 min in the presence or absence of l-NAME (2 μm) or PEG-SOD (100 U). (A) Maximal [NO˙] and [ONOO−] produced by a keratinocyte after UVB treatment. (B) A ratio of maximal [NO˙]/[ONOO−] produced by a keratinocyte after UVB treatment. The data represent the average of three sets of independent measurements. *P < 0.001 vs control; + P < 0.1 vs control.
UVB induces NO˙ and ONOO− production in skin epidermis in vivo
After quantitatively analyzing NO˙ and ONOO− in cultured cells, we determined the UVB-induced release of NO˙ and ONOO− in living mouse skin. The module nanosensors (total diameter 3.0 ± 0.5 μm) were inserted underneath the skin surface of the anesthetized mouse (Fig. 3A) and the productions of NO˙ or ONOO− were continuously recorded (Fig. 3B) before and after the irradiation. Our data show that the exposure of the living skin to UVB causes a rapid release of NO˙ and ONOO− at 4 s postirradiation. Interestingly, while the release of ONOO− rapidly reaches a maximum of 270 ± 20 nm at 15 s postirradiation and slowly reduced, the release of NO˙ remains raising to 150 ± 8 nm at the end of the measurement (Fig. 3B,C). Our results demonstrate that the patterns for the release of NO˙ and ONOO− in response to UVB irradiation are distinctive in cultured cells and living skin (Fig. 3B vs 1A).
Figure 3.
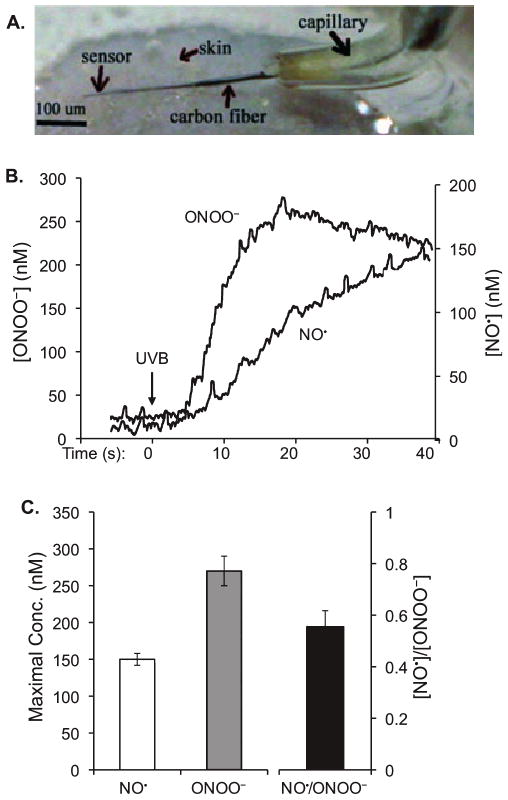
In vivo measurement of NO˙ and ONOO− in UVB-irradiated mouse skin using a nanosensor. (A) A micrograph of L-shaped sharpened carbon fibers with a module of NO˙ and ONOO− sensors deposited at the tip of the carbon fibers. The module was implanted in the skin of a mouse. (B) Maximal [NO˙], [ONOO−] and a ratio of maximal [NO˙]/[ONOO−] produced by the irradiated skin. (C) Maximal [NO˙], [ONOO−] and a ratio of maximal [NO˙]/[ONOO−] produced by a keratinocyte after UVB treatment. The data represent the average of three sets of independent measurements.
Early elevation of NO˙ and oxidative stress mediates UVB-induced death of keratinocytes
As an elevation of NO˙ could be pro- or anti-apoptotic (33), we determined whether this early release of NO˙ in combination with ONOO− inhibits or promotes apoptosis of cultured HaCaT cells upon UVB irradiation. The cell death was analyzed using annexin V and PI double-staining to determine the loss of membrane phospholipid symmetry and membrane integrity (34,35). At 24 h postirradiation, the UVB-induced apoptotic cell death was reduced from 17.6 ± 1.7% to 14.4 ± 1.5% or 10.8 ± 1.0%, respectively, upon l-NMMA or l-NAC treatment (Fig. 4A). The effects of l-NMMA and l-NAC on UVB-induced cell death were also analyzed by determination of cleavage of poly (ADP-ribose) polymerase (PARP). Our data showed that UVB induced a cleavage of 116 kDa PARP to an 89-kDa fragment (Fig. 4B, lane 2 vs 1), which is the marker for cell apoptosis. Treating the cells with l-NMMA and l-NAC partially protected PARP cleavage upon UVB irradiation (Fig. 4B, lanes 3 and 4 vs lane 2 [correction added after online publication Jan 13 2010: lane 4 changed to lane 2]). Interestingly, treating the cells with l-NAC did not only reduce the PARP cleavage but also reduced the PARP expression after UVB irradiation (Fig. 4B, lane 4 vs lanes 1–3). The data agreed with our previous reports, which indicated that l-NAC protects eIF2α from UV-induced phosphorylation (36) and elimination of eIF2α phosphorylation reduces PARP expression after UV treatment (37).
Figure 4.
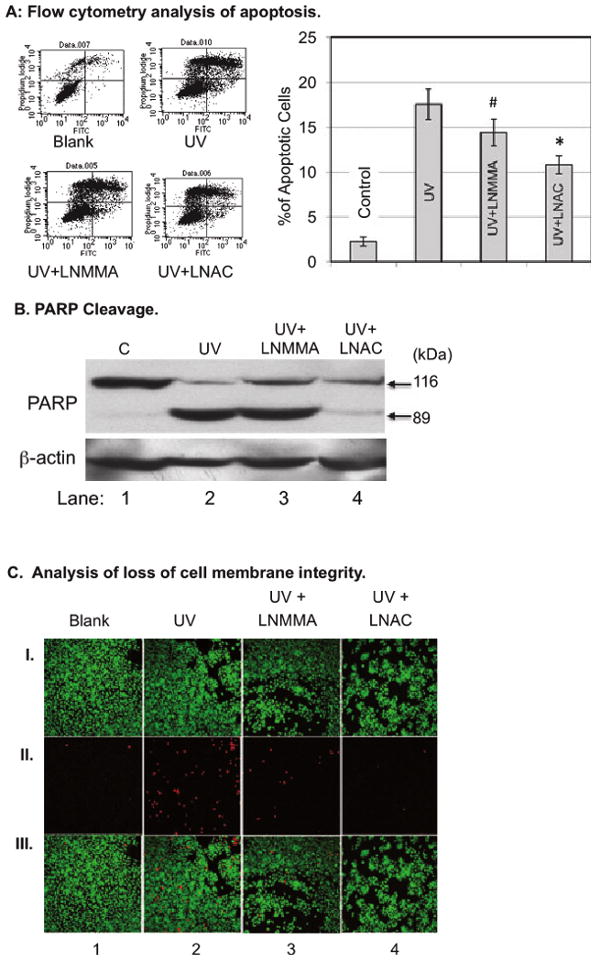
HaCaT cells were treated with l-NMMA (100 μm) or l-NAC (25 mm) for 2 h and then irradiated with UVB (50 mJ cm−2). (A) The cells were double-stained with Annexin V/PI at 24 h postirradiation. The percentages of apoptotic cells were determined by flow cytometric analysis. The data represent the average of three sets of independent measurements. #P < 0.01 vs UVB alone; *P < 0.005 vs UVB alone. (B) The cells were lyzed at 24 h postirradiation and Western blot analysis was used to determine the PARP cleavage. (C) The cells were double-stained with CAG/PI at 1 h postirradiation. The images were captured by fluorescence confocal microscopy. (I) Living cells stained by CAG-AM (2.5 μm). (II) Injured cells that lost membrane integrity were stained with PI (50 μg mL−1). (III) Pictures (I) and (II) overlaid on top of each other.
In addition to apoptosis, we also determined whether the early release of NO˙ initiates necrotic death of UVB-treated HaCaT cells. Necrosis is initiated due to injury to the cells, which could be characterized by the loss of membrane integrity (38). We analyzed membrane integrity using the PI staining method. The living cells were stained and visualized by using CAG-AM staining. Our data show that UVB irradiation induces a loss of membrane integrity in the cells within 1 h of treatment (Fig. 4C, column 2 vs 1). The inhibition of NOS or free radicals significantly reduces the amount of injured cells (Fig. 5B, columns 3 and 4 vs 2). Our results demonstrate that the early release of NO˙ and ONOO− mediates UVB-induced apoptotic and necrotic death of cells.
Figure 5.
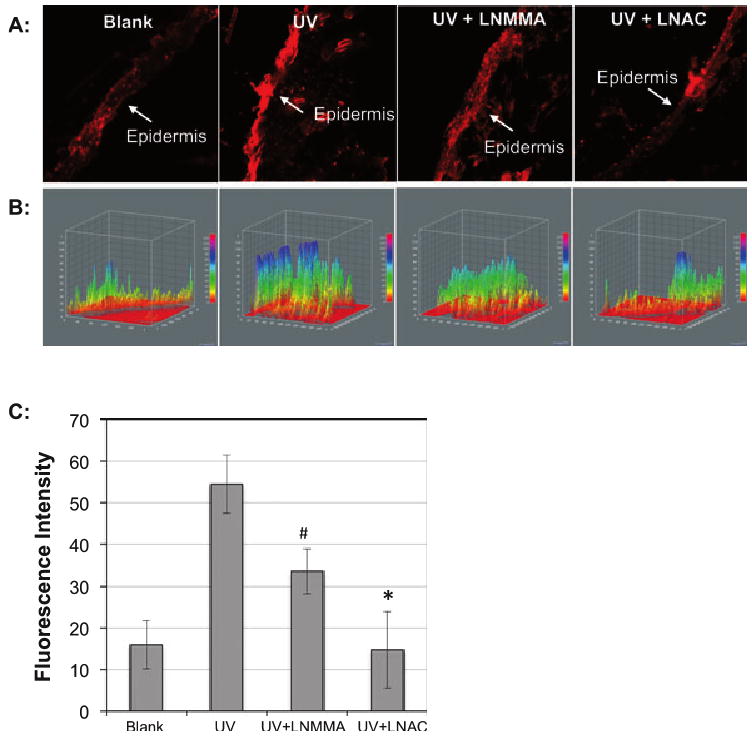
PI staining was used to determine the UVB-induced injury of skin tissue of mice. The mice were injected intraperitoneally with l-NMMA (10 mg kg−1) or l-NAC (500 mg kg−1) at 1 h before UVB irradiation. Immediately after irradiation, the mice were injected subcutaneously with 0.1 mL PI (100 μg mL−1) and the skin tissues were collected at 30 min postirradiation. The images of the skin sections were captured by fluorescence microscope. (A) Images of the PI-stained skin slices. (B) A 3-D analysis of the fluorescence intensity of the PI staining skin tissue using ImageJ (v1.34k; NIH). The fluorescence intensity is analyzed against distance of the slice in two dimensions. (C) The average fluorescence intensity of three measurements. #P < 0.01 vs UVB alone; *P < 0.02 vs UVB alone.
UVB-induced elevation of NO˙ and oxidative stress leads to tissue damage
To assess the role of NOS and oxidative stress in UVB-induced skin injury, we analyzed the effect of l-NMMA and l-NAC on UVB-induced loss of membrane integrity in living mice skin tissue. Compared to the samples from the UVB-irradiated mice, the skin tissue harvested from the mice with l-NMMA showed a significant decrease in fluorescence intensity in the epidermis (Fig. 5), which consists of 90–95% of keratinocytes. The treatment of l-NAC almost totally inhibited UVB-induced tissue injury (Fig. 5). These results demonstrated that UVB-induced rapid activation of NOS in combination with oxidative stress led to the loss of membrane integrity and skin injury.
Discussion
UVB induces a rapid release of NO˙ and O2˙− in cultured keratinocytes (24). However, the formation of the more reactive ONOO− has never been directly measured after UVB irradiation. Furthermore, the effect of UVB radiation on NO˙ production in the skin has been assessed only with the use of indirect methods (39). Indirect measurements cannot separate an effect of NO˙ in biological milieu from the effect of ONOO−. In this study we used highly sensitive, selective, millisecond response-time electrochemical nanosensors (40) to directly measure the concentration of UVB- and CaI-stimulated NO˙ and ONOO− release in cultured keratinocytes and in the epidermis of mice.
Our results demonstrate that a short-time exposure of cultured keratinocytes or the epidermis of mice to UVB irradiation stimulated NO˙ release as well as ONOO− production (Figs. 1 and 3). The study of NOS activity showed dependence upon calcium, indicating the involvement of the cNOS rather than the iNOS in NO˙/ONOO− production (Fig. 2). As the crucial role of NO˙ in the physiology of vasculature has become well established, the question arises whether NO˙ directly or indirectly, through the formation of more reactive oxidative species such as ONOO−, averts its deleterious biological effects. It is interesting to note that the UVB-induced ONOO− release in cultured keratinocytes precedes the production of bioavailable (diffusible) NO˙ (Fig. 1). This is in contrast to calcium-stimulated NO˙ release where the generation of ONOO− follows the production of NO˙ (Fig. 2). To explain the differences in the effect of UVB on NO˙ production vs ONOO− production, it may help to realize that the NO˙ sensor used in this study detects the net concentration of NO˙ (i.e. NO˙ that is not consumed in fast chemical reactions and can freely diffuse to a target cell and trigger cGMP production). This net concentration depends not only on the activity of eNOS but also on the production of O2˙−. NO˙ rapidly reacts with O2˙− in diffusion-controlled reactions (k = 6 × 1010 nmol−1s−1) to form ONOO−. There are several potential sources of O2˙− in cells including NAD(P)H, cNOS, mitochondria and others. It is also well established that UVB induces O2˙− production (24,41). UVB exposure generates O2˙− immediately after the exposure, and the reactive oxygen species thus produced remains for 100–1200 s. The data presented here indicate that initially produced ONOO− is most likely a result of the reaction between O2˙− generated by photolytic reduction of oxygen and NO˙ generated by uncoupled cNOS. ONOO− is formed when NO˙ and O2˙− react in a fast (k = 2 × 1010 mol−1s−1) reaction (30,31). Formation of ONOO− is favored by the overproduction of O2˙− and/or NO˙.
After measuring the kinetics of NO˙ and ONOO− productions, we determined the role of the early cNOS activation and NO˙/ONOO− imbalance in UVB-induced apoptosis by analyzing Annexin V-FITC/PI double-stained cells or PARP cleavage. l-NAC, a commonly used ONOO− reducer (36,42,43), was used to increase the ratio of NO˙/ONOO−. Our data showed that preincubation of the cells with either an NOS inhibitor or an antioxidant reduced apoptotic death of the UVB-treated cells (Fig. 4). These results suggest that the early NO˙ release enhances oxidative stress-induced apoptosis upon UVB irradiation. The UVB-induced apoptosis is likely to be trigged by the oxidative damage to the cell membrane at the early stage of irradiation. NO˙ is an unstable molecule, which rapidly reacts with O2˙− to form ONOO− and its protonated form (ONOOH). ONOO− is a potent inducer of apoptosis (44,45). While [O2˙−] is low, ONOO− can isomerize to harmless NO3−. However, at high [O2˙−], ONOO− undergoes a hemolytic or heterolytic cleavage to form strong oxidants including HO˙−, NO2− and NO2+. These species initiate a cascade of events leading to an increase in cytotoxicity and trigger cellular damage. In UVB-treated cells, high levels of O2˙− can be generated by reduction of O2, by uncoupled eNOS or by other sources like NADP(H) oxidase. However, the most effective generator of O2˙− is uncoupled cNOS (46). Our data showed that the early activation of cNOS led to an injury of the cultured cells and skin tissue within 1 h postirradiation (Figs. 4C and 5). The UVB-induced membrane damage of cells or skin tissue could be reduced or prevented by pretreatment of l-NMMA or l-NAC (Figs. 4C and 5). Furthermore, the early membrane damage correlated with the late apoptosis of the irradiated cells. These results suggest that UVB-induced apoptosis could be trigged by an early-induced NO˙ release in combination with high production of O2˙−. Based on our results, we propose a novel model (Fig. 6) that UVB induced immediate activation of cNOS and production of NO˙, which rapidly reacts with O2˙− to form ONOO−, which induces oxidative membrane damage and apoptosis of the irradiated cells.
Figure 6.
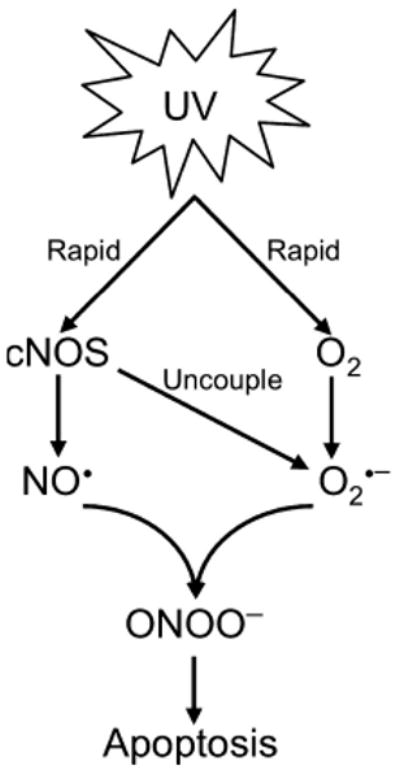
Model of UVB-induced and NO˙-enhanced apoptotic signaling pathways.
Acknowledgments
This work is partially supported by R56 CA86928 (to S.W.) and RO1 CA086928 (to S.W.).
References
- 1.Oplander C, Cortese MM, Korth HG, Kirsch M, Mahotka C, Wetzel W, Pallua N, Suschek CV. The impact of nitrite and antioxidants on ultraviolet-A-induced cell death of human skin fibroblasts. Free Radic Biol Med. 2007;43:818–829. doi: 10.1016/j.freeradbiomed.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 2.Suschek CV, Briviba K, Bruch-Gerharz D, Sies H, Kroncke KD, Kolb-Bachofen V. Even after UVA-exposure will nitric oxide protect cells from reactive oxygen intermediate-mediated apoptosis and necrosis. Cell Death Differ. 2001;8:515–527. doi: 10.1038/sj.cdd.4400839. [DOI] [PubMed] [Google Scholar]
- 3.Suschek CV, Krischel V, Bruch-Gerharz D, Berendji D, Krutmann J, Kronckei KD, Kolb-Bachofen V. Nitric oxide fully protects against UVA-induced apoptosis in tight correlation with Bcl-2 up-regulation. J Biol Chem. 1999;274:6130–6137. doi: 10.1074/jbc.274.10.6130. [DOI] [PubMed] [Google Scholar]
- 4.Weller R, Billiar T, Vodovotz Y. Pro- and anti-apoptotic effects of nitric oxide in irradiated keratinocytes: The role of superoxide. Skin Pharmacol Appl Skin Physiol. 2002;15:348–352. doi: 10.1159/000064540. [DOI] [PubMed] [Google Scholar]
- 5.Yamaoka J, Kawana S, Miyachi Y. Nitric oxide inhibits ultraviolet B-induced murine keratinocyte apoptosis by regulating apoptotic signaling cascades. Free Radic Res. 2004;38:943–950. doi: 10.1080/10715760412331284807. [DOI] [PubMed] [Google Scholar]
- 6.Madajka M, Korda M, White J, Malinski T. Effect of aspirin on constitutive nitric oxide synthase and the biovailability of NO. Thromb Res. 2003;110:317–321. doi: 10.1016/s0049-3848(03)00419-5. [DOI] [PubMed] [Google Scholar]
- 7.Newman E, Spratt DE, Mosher J, Cheyne B, Montgomery HJ, Wilson DL, Weinberg JB, Smith SM, Salerno JC, Ghosh DK, Guillemette JG. Differential activation of nitric-oxide synthase isozymes by calmodulin-troponin C chimeras. J Biol Chem. 2004;279:33547–33557. doi: 10.1074/jbc.M403892200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZG, Chopp M, Bailey F, Malinski T. Nitric oxide changes in the rat brain after transient middle cerebral artery occlusion. J Neurol Sci. 1995;128:22–27. doi: 10.1016/0022-510x(94)00216-b. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn A, Fehsel K, Lehmann P, Krutmann J, Ruzicka T, Kolb-Bachofen V. Aberrant timing in epidermal expression of inducible nitric oxide synthase after UV irradiation in cutaneous lupus erythematosus. J Invest Dermatol. 1998;111:149–153. doi: 10.1046/j.1523-1747.1998.00253.x. [DOI] [PubMed] [Google Scholar]
- 10.Arany I, Brysk MM, Brysk H, Tyring SK. Regulation of inducible nitric oxide synthase mRNA levels by differentiation and cytokines in human keratinocytes. Biochem Biophys Res Commun. 1996;220:618–622. doi: 10.1006/bbrc.1996.0452. [DOI] [PubMed] [Google Scholar]
- 11.Qureshi AA, Hosoi J, Xu S, Takashima A, Granstein RD, Lerner EA. Langerhans cells express inducible nitric oxide synthase and produce nitric oxide. J Invest Dermatol. 1996;107:815–821. doi: 10.1111/1523-1747.ep12330572. [DOI] [PubMed] [Google Scholar]
- 12.Wang R, Ghahary A, Shen YJ, Scott PG, Tredget EE. Human dermal fibroblasts produce nitric oxide and express both constitutive and inducible nitric oxide synthase isoforms. J Invest Dermatol. 1996;106:419–427. doi: 10.1111/1523-1747.ep12343428. [DOI] [PubMed] [Google Scholar]
- 13.Rocha IM, Guillo LA. Lipopolysaccharide and cytokines induce nitric oxide synthase and produce nitric oxide in cultured normal human melanocytes. Arch Dermatol Res. 2001;293:245–248. doi: 10.1007/s004030100211. [DOI] [PubMed] [Google Scholar]
- 14.Tsatmali M, Manning P, McNeil CJ, Thody AJ. alpha-MSH inhibits lipopolysaccharide induced nitric oxide production in B16 mouse melanoma cells. Ann N Y Acad Sci. 1999;885:474–476. doi: 10.1111/j.1749-6632.1999.tb08716.x. [DOI] [PubMed] [Google Scholar]
- 15.Deliconstantinos G, Villiotou V, Stavrides JC. Nitric oxide and peroxynitrite released by ultraviolet B-irradiated human endothelial cells are possibly involved in skin erythema and inflammation. Exp Physiol. 1996;81:1021–1033. doi: 10.1113/expphysiol.1996.sp003986. [DOI] [PubMed] [Google Scholar]
- 16.Deliconstantinos G, Villiotou V, Stravrides JC. Release by ultraviolet B (u.v.B) radiation of nitric oxide (NO) from human keratinocytes: A potential role for nitric oxide in erythema production. Br J Pharmacol. 1995;114:1257–1265. doi: 10.1111/j.1476-5381.1995.tb13341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubaszewski E, Peters A, McClain S, Bohr D, Malinski T. Light-activated release of nitric oxide from vascular smooth muscle of normotensive and hypertensive rats. Biochem Biophys Res Commun. 1994;200:213–218. doi: 10.1006/bbrc.1994.1436. [DOI] [PubMed] [Google Scholar]
- 19.Marletta MA. Nitric oxide: Biosynthesis and biological significance. Trends Biochem Sci. 1989;14:488–492. doi: 10.1016/0968-0004(89)90181-3. [DOI] [PubMed] [Google Scholar]
- 20.Nathan C. Inducible nitric oxide synthase: What difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meeran SM, Katiyar N, Singh T, Katiyar SK. Loss of endogenous interleukin-12 activates survival signals in ultraviolet-exposed mouse skin and skin tumors. Neoplasia. 2009;11:846–855. doi: 10.1593/neo.09528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukunaga-Takenaka R, Fukunaga K, Tatemichi M, Ohshima H. Nitric oxide prevents UV-induced phosphorylation of the p53 tumor-suppressor protein at serine 46: A possible role in inhibition of apoptosis. Biochem Biophys Res Commun. 2003;308:966–974. doi: 10.1016/s0006-291x(03)01516-x. [DOI] [PubMed] [Google Scholar]
- 23.Schneiderhan N, Budde A, Zhang Y, Brune B. Nitric oxide induces phosphorylation of p53 and impairs nuclear export. Oncogene. 2003;22:2857–2868. doi: 10.1038/sj.onc.1206431. [DOI] [PubMed] [Google Scholar]
- 24.Aitken GR, Henderson JR, Chang SC, McNeil CJ, Birch-Machin MA. Direct monitoring of UV-induced free radical generation in HaCaT keratinocytes. Clin Exp Dermatol. 2007;32:722–727. doi: 10.1111/j.1365-2230.2007.02474.x. [DOI] [PubMed] [Google Scholar]
- 25.Brovkovych V, Patton S, Brovkovych S, Kiechle F, Huk I, Malinski T. In situ measurement of nitric oxide, superoxide and peroxynitrite during endotoxemia. J Physiol Pharmacol. 1997;48:633–644. [PubMed] [Google Scholar]
- 26.Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, Jankowski M, Martyniec L, Angielski S, Malinski T. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: A novel mechanism for antihypertensive action. Circulation. 2003;107:2747–2752. doi: 10.1161/01.CIR.0000066912.58385.DE. [DOI] [PubMed] [Google Scholar]
- 27.Kalinowski L, Malinski T. Endothelial NADH/-NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim Pol. 2004;51:459–469. [PubMed] [Google Scholar]
- 28.Malinski T, Radomski MW, Taha Z, Moncada S. Direct electrochemical measurement of nitric oxide released from human platelets. Biochem Biophys Res Commun. 1993;194:960–965. doi: 10.1006/bbrc.1993.1914. [DOI] [PubMed] [Google Scholar]
- 29.Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–678. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- 30.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 31.Groves JT. Peroxynitrite: Reactive, invasive and enigmatic. Curr Opin Chem Biol. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 32.Pataer A, Vorburger SA, Barber GN, Chada S, Mhashilkar AM, Zou-Yang H, Stewart AL, Balachandran S, Roth JA, Hunt KK, Swisher SG. Adenoviral transfer of the melanoma differentiation-associated gene 7 (mda7) induces apoptosis of lung cancer cells via up-regulation of the double-stranded RNA-dependent protein kinase (PKR) Cancer Res. 2002;62:2239–2243. [PubMed] [Google Scholar]
- 33.Snyder SH. Janus faces of nitric oxide. Nature. 1993;364:577. doi: 10.1038/364577a0. [DOI] [PubMed] [Google Scholar]
- 34.Schindl A, Klosner G, Honigsmann H, Jori G, Calzavara-Pinton PC, Trautinger F. Flow cytometric quantification of UV-induced cell death in a human squamous cell carcinoma-derived cell line: Dose and kinetic studies. J Photochem Photobiol B, Biol. 1998;44:97–106. doi: 10.1016/s1011-1344(98)00127-4. [DOI] [PubMed] [Google Scholar]
- 35.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 36.Lu W, Laszlo CF, Miao Z, Chen H, Wu S. The role of nitric oxide synthase in regulation of ultraviolet light-induced phosphorylation of the alpha-subunit of eukaryotic initiation factor 2. J Biol Chem. 2009;284(36):24281–24288. doi: 10.1074/jbc.M109.008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker SH, Parker TA, George KS, Wu S. The roles of translation initiation regulation in ultraviolet light-induced apoptosis. Mol Cell Biochem. 2006;293:173–181. doi: 10.1007/s11010-006-9239-y. [DOI] [PubMed] [Google Scholar]
- 38.Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: Analysis of apoptosis and accidental cell death (necrosis) Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- 39.Virag L, Szabo E, Bakondi E, Bai P, Gergely P, Hunyadi J, Szabo C. Nitric oxide-peroxynitrite-poly(ADP-ribose) polymerase pathway in the skin. Exp Dermatol. 2002;11:189–202. doi: 10.1034/j.1600-0625.2002.110301.x. [DOI] [PubMed] [Google Scholar]
- 40.Heeba G, Moselhy M, Hassan M, Khalifa M, Gryglewski R, Malinski T. Anti-atherogenic effect of statins: Role of nitric oxide, peroxynitrite and haem oxygenase-1. Br J Pharmacol. 2009;156(8):1256–1266. doi: 10.1111/j.1476-5381.2009.00125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hakozaki T, Date A, Yoshii T, Toyokuni S, Yasui H, Sakurai H. Visualization and characterization of UVB-induced reactive oxygen species in a human skin equivalent model. Arch Dermatol Res. 2008;300(Suppl 1):S51–S56. doi: 10.1007/s00403-007-0804-3. [DOI] [PubMed] [Google Scholar]
- 42.Failli P, Palmieri L, D'Alfonso C, Giovannelli L, Generini S, Rosso AD, Pignone A, Stanflin N, Orsi S, Zilletti L, Matucci-Cerinic M. Effect of N-acetyl-l-cysteine on peroxynitrite and superoxide anion production of lung alveolar macrophages in systemic sclerosis. Nitric Oxide. 2002;7:277–282. doi: 10.1016/s1089-8603(02)00120-9. [DOI] [PubMed] [Google Scholar]
- 43.Lin KT, Xue JY, Sun FF, Wong PY. Reactive oxygen species participate in peroxynitrite-induced apoptosis in HL-60 cells. Biochem Biophys Res Commun. 1997;230:115–119. doi: 10.1006/bbrc.1996.5897. [DOI] [PubMed] [Google Scholar]
- 44.Brune B, von Knethen A, Sandau KB. Nitric oxide (NO): An effector of apoptosis. Cell Death Differ. 1999;6:969–975. doi: 10.1038/sj.cdd.4400582. [DOI] [PubMed] [Google Scholar]
- 45.Dimmeler S, Zeiher AM. Nitric oxide and apoptosis: Another paradigm for the double-edged role of nitric oxide. Nitric Oxide. 1997;1:275–281. doi: 10.1006/niox.1997.0133. [DOI] [PubMed] [Google Scholar]
- 46.Huk I, Nanobashvili J, Neumayer C, Punz A, Mueller M, Afkhampour K, Mittlboeck M, Losert U, Polterauer P, Roth E, Patton S, Malinski T. L-arginine treatment alters the kinetics of nitric oxide and superoxide release and reduces ischemia / reperfusion injury in skeletal muscle. Circulation. 1997;96:667–675. doi: 10.1161/01.cir.96.2.667. [DOI] [PubMed] [Google Scholar]