

Abstract
Attacks of migraine with aura represent a phenomenon in which abnormal neuronal activity in the cortex produces sensory disturbances (aura) some 20–40 min before the onset of headache. The purpose of this study was to determine whether cortical spreading depression (CSD)—an event believed to underlie visual aura—can give rise to activation of nociceptors that innervate the meninges—an event believed to set off migraine headache. CSD was induced in anesthetized male rats by stimulation of the visual cortex with electrical pulses, pin prick, or KCl; single-unit activity of meningeal nociceptors was monitored in vivo in the rat before and after CSD. Regardless of the method of cortical stimulation, induction of CSD was recorded in 64 trials. In 31 of those trials, CSD induced a twofold increase in meningeal nociceptor firing rate that persisted for 37.0 ± 4.6 min in trials in which activity returned to baseline, or >68 min in trials in which activity remained heightened at the time recording was interrupted. In two-thirds of the trials, onset of long-lasting neuronal activation began ∼14 min after the wave of CSD. The findings demonstrates for the first time that induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of nociceptors that innervate the meninges. We suggest that migraine with aura is initiated by waves of CSD that lead up to delayed activation of the trigeminovascular pathway.
Introduction
Migraine is a heterogeneous, episodic neurovascular disorder affecting ∼11% of the population worldwide (Stovner et al., 2007). In patients experiencing migraine with aura, the impending headache is heralded some 20–40 min earlier by sensory disturbances (aura) lasting up to 1 h (Olesen et al., 2005). Such sequence of events has stimulated extensive research into the premise that cortical mechanisms underlying aura can give rise to activation of cranial meningeal nociceptors, which constitute the first-order neurons of the trigeminovascular pain pathway of migraine.
Based on his animal studies and personal experience of spreading scintillations during migraine, Leão (1944, 1986) proposed that visual aura is a manifestation of cortical spreading depression (CSD), a phenomenon where waves of depression of spontaneous electrical activity propagate slowly through the occipital lobe of the cortex. Human studies have shown that the aura phase of migraine is associated with biphasic changes in cortical blood flow (initial hyperemia followed by sustained oligemia) that propagate throughout the surface of the visual cortex (Lauritzen and Olesen, 1984; Lauritzen, 1994; Cao et al., 1999; Hadjikhani et al., 2001). The going theory is that the hyperemic phase of CSD is associated with local release of ATP and glutamate molecules and potassium and hydrogen ions by neurons, glia, or vascular cells (Brinley et al., 1960; Rapoport and Marshall, 1964; Csiba et al., 1985; Marrannes et al., 1988; D'Andrea et al., 1991; Lauritzen and Hansen, 1992; Fabricius et al., 1993; Mayevsky et al., 1996; James et al., 2001; Brennan et al., 2007; Schock et al., 2007; Charles and Brennan, 2009). Regardless of the origin of their release, these mediators are thought to diffuse outward into the leptomeninges overlaying the affected cortical region, resulting in activation of pial nociceptors, local neurogenic inflammation, and the persistent activation of dural nociceptors, which triggers the migraine headache (Moskowitz, 1993). This hypothesis has been supported by indirect evidence for a CSD-induced increase in cellular c-fos expression in areas of the trigeminal nucleus caudalis known to receive input from the cranial meninges (Moskowitz et al., 1993; Bolay et al., 2002) and evidence that trigeminal denervation abolishes CSD-evoked increases in dural blood flow (Moskowitz et al., 1993; Bolay et al., 2002). Whether CSD can trigger activation of meningeal nociceptors has not been tested directly hitherto. The present study tested the hypothesis that induction of CSD by focal stimulation of the rat visual cortex can lead to activation of meningeal nociceptors in the trigeminal ganglion.
Materials and Methods
Surgical preparation.
Experiments were approved by the standing committee on animals of Harvard Medical School and Beth Israel Deaconess Medical Center, in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague Dawley rats weighing 250–350 g were deeply anesthetized with urethane (1.8 g/kg, i.p.) and mounted on a stereotaxic frame. Core temperature was kept at 37°C using a heating blanket. End-tidal CO2 was continuously monitored and kept within a physiological range of 3.5–4.5%. The dura over the left hemisphere was exposed using an opening in the roof of the skull between the bregma and 2 mm caudal to lambda. The exposed dura was kept moist throughout the experiment, using modified synthetic interstitial fluid (SIF, pH 7.2) containing 135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 5 mm CaCl2, 10 mm glucose, and 10 mm HEPES.
Excision of the sphenopalatine ganglion.
In 10 experiments, the left sphenopalatine ganglion (SPG) was surgically exposed and removed using the approach described by Spencer et al. (1990). Briefly, an incision was made along the left supraorbital ridge above the eye, ∼1.5 cm in length. The skin was retracted outward and the underlying tissue was loosened away from the frontal bone. The eyeball and ocular muscles were gently retracted laterally to expose the sphenopalatine fossa, the site between the frontal bone and the maxillary nerve where the SPG is embedded and the nasociliary nerve enters the anterior ethmoidal foramen. The SPG was carefully dissected away from its rich vascular plexus; sections collected from the excised tissue were processed for vasoactive intestinal peptide immunohistochemistry to confirm the chemical identity of the ganglion.
Recording and identification of meningeal nociceptors.
Single-unit activity of meningeal nociceptors (1 unit/rat) was recorded in the trigeminal ganglion as described in detail before (Strassman et al., 1996; Levy et al., 2005). A platinum-coated tungsten microelectrode (impedance 50 kΩ; FHC) was lowered toward the left trigeminal ganglion through an incision made in the dura, ∼2 mm caudal to the bregma suture and 2 mm lateral to the midline. Included in the study were meningeal nociceptors that exhibited discrete bursts of activity in response to mechanical stimulation of the dura overlying the ipsilateral transverse sinus (indentation with von Frey monofilaments), plus consistent response latencies to repeated electrical stimulation of the dura (0.5 ms pulse, 5 mA, 0.5 Hz). The tip of the stimulating electrode was moved to different sites within the dural receptive field to find a point at which electrical pulses yielded the shortest neuronal response latency. This response latency was divided by the average distance between the dural receptive field and the trigeminal ganglion (12.5 mm), yielding the conduction velocity (CV) of the neuron under study. Slow-responding neurons (CV ≤ 1.5 m/s) were classified as C-units; faster responders (CV > 1.5 m/s) were identified as Aδ-units. Real-time waveform discriminator was used to create and store a template for the action potential evoked in the neuron under study by electrical pulses on the dura; spikes of activity matching the template waveform were acquired and analyzed online and offline using Spike 2 software (CED).
Induction and recording of CSD.
Single waves of CSD were induced using mechanical, electrical, and chemical stimulation of the visual cortex, ∼6 mm away from the dural receptive field of the neuron under study (Fig. 1). Changes in steady potential were recorded on the surface of the cortex using a glass micropipette filled with 150 mm NaCl (impedance 70–120 kΩ), approximately halfway between the dural receptive field and the site of cortical stimulation (Fig. 1). At a propagation rate of 3–5 mm/min (Lauritzen, 1994), a wave of CSD was expected to be registered by the recording electrode and enter the neuronal receptive field within 1–2 min of cortical stimulation. Mechanical stimulation (pin prick) was delivered by inserting a glass micropipette (diameter 25 μm) ∼1 mm into the visual cortex for 10 s. Electrical stimulation (cathodal pulses) was delivered through a concentric bipolar electrode at incremental magnitude every 90 min until a wave of CSD was registered at the recording site (current—0.5 or 1.0 mA; duration—1 or 3 ms; frequency—between 0.2 and 50 Hz). In many trials, the overall magnitude of electrical simulation was insufficient to induce CSD, thus controlling for nonspecific neuronal activation. Chemical stimulation was delivered by placing a granule of crystalline KCl on the surface of the cortex; the granule was washed away with SIF at the end of the CSD wave.
Figure 1.

Experimental setup and induction of CSD. Extracellular recording electrode was lowered into the trigeminal ganglion (1) in search for a single meningeal nociceptor (red). The target unit was identified using electrical stimulation of the dura overlying the transverse sinus and visual cortex (2), and the receptive field of the unit was mapped using mechanical stimulation of the dura (3). Stimulation of the cortex with electrical pulses, pin prick, or KCl was applied ∼6 mm rostral to the neuronal receptive field (4). Waves of CSD were recorded through the tip of a glass micropipette touching the surface of the cortex midway between the site of cortical stimulation and the neuronal receptive field (5). TG, Trigeminal ganglion; SpV, spinal trigeminal nucleus. Midsagittal and transverse sinuses are marked in blue.
Experimental paradigm.
Dural receptive field of the nociceptor under study was mapped using calibrated von Frey monofilaments. Small incisions were made in the dura overlaying the visual cortex at the site designated for cortical stimulation and CSD recording. Ongoing activity of the nociceptor was recorded over 30 min before dural incision, and for another 30 min afterward. After confirming that ongoing neuronal firing rate remained unchanged between the two intervals, the cortex was stimulated, and ongoing activity was monitored for a period of 30–120 min after registering a wave of CSD. A neuron was considered to be activated if its firing rate after cortical stimulation increased ≥25% compared to baseline and remained elevated for a period longer than 10 min.
Data analysis.
Data were analyzed by nonparametric statistics, using two-tailed level of significance set at α = 0.05. Neurons were divided into subgroups according to the pattern of activity recorded before and after the induction of CSD (see Results). Latency to onset of neuronal activation and the duration of neuronal activation were compared across three subgroups of neurons using Kruskal–Wallis one-way ANOVA. Mean neuronal firing rate (spikes/s) before cortical stimulation (baseline) was calculated in each trial over a period of 30 min. Mean firing rate after induction of CSD was calculated in each trial over the entire period during which neuronal activity was ≥25% higher than baseline activity. Mean firing rates before and after the induction of CSD were compared using Wilcoxon matched-pairs signed-ranks test.
Results
The temporal relationship between the initiation of CSD and the activation of meningeal nociceptors was studied using simultaneous recordings of shifts in direct current in the cortex and single-unit activity (1 unit/rat) in the trigeminal ganglion (Fig. 1). Cortical stimulation was applied in a total of 83 trials performed in 53 rats.
In 64 trials, waves of CSD lasting 64.0 ± 2.1 s (mean ± SEM) were induced within 56.3 ± 6.1 s of cortical stimulation, most effectively with crystalline KCl and pin prick (Table 1). In 33 of those trials, neuronal firing rate recorded after the induction of CSD was similar to the baseline activity recorded before CSD (1.09 ± 0.21 vs 1.18 ± 0.21 spikes/s, respectively; p > 0.05). However, the remaining 31 trials resulted in long-lasting (≥11 min) increase in neuronal firing rate (≥25% compared to baseline) that began either immediately after cortical stimulation (Fig. 2) or between 3 and 32 (14.4 ± 1.7) min after the end of the CSD wave (Figs. 3, 4). Sustained neuronal activation was only seen in 1 of the 19 trials (5%) in which cortical stimulation failed to produce CSD.
Table 1.
Induction of CSD and sustained activation of meningeal nociceptors by stimulation of the visual cortex
| Cortical stimulation | CSD per trials | Sustained neuronal activation per CSD |
|---|---|---|
| Electrical | 13/30 (43%) a | 6/13 (46%) |
| Pin prick | 22/24 (92%) | 13/22 (59%) |
| Crystalline KCl | 29/29 (100%) | 12/29 (41%) |
| All trials | 64/83 (77%) | 31/64 (52%) |
aBy design, the parameters of electrical pulses were suboptimal in many trials for inducing CSD in order to control for nonspecific neuronal activation.
Figure 2.

Immediate onset of sustained activation of meningeal nociceptors. A, An example of a CSD wave induced by electrical stimulation of the cortex (upper trace) that was associated with immediate, long-lasting (>93 min) activation of a C-unit (bottom trace). Note that neuronal activity (numbers in parentheses) after cortical stimulation (pink) was approximately twofold higher than baseline activity (green). Bin width = 10 s. B, Temporal relationship between waves of CSD (yellow) and onset of neuronal activation induced by stimulation of the cortex with electrical pulses (blue), pin prick (red) or KCl (green). Left, Latencies to onset of CSD and neuronal activation during the first 5 min. Right, Duration of neuronal activation. Numbers at end of each bar indicate duration (in minutes) of neuronal activation; numbers in parentheses indicate trials in which neuronal activity remained heightened at the time recording was stopped.
Figure 3.

Delayed onset of sustained activation of meningeal nociceptors. A, An example of a CSD wave induced by mechanical stimulation of the cortex (upper trace) that was associated with delayed onset (gray area) of sustained activation of an Aδ-unit that lasted 38 min (bottom trace). Note that neuronal (numbers in parentheses) activity after CSD (pink) was approximately sevenfold higher than baseline activity (green). Bin width = 10 s. B, Temporal relationship between waves of CSD and onset of neuronal activation. Note that neuronal activation did not occur during the first 5 min in all but two trials. See Figure 2 A,B for further details.
Figure 4.

Delayed onset of sustained activation of meningeal nociceptors, which was preceded by a short-lived surge of activity. A, An example of a CSD wave induced by chemical stimulation of the cortex (upper trace) that was associated an initial burst of neuronal firing, followed by delayed onset (gray area) of long-lasting (>51 min) activation of an Aδ-unit (bottom trace). Note that neuronal activity (numbers in parentheses) after CSD (pink) was approximately sixfold higher than baseline activity (green). Bin width = 10 s. B, Temporal relationship between waves of CSD and onset of neuronal activation. Note that delayed neuronal activation did not occur during the first 5 min. See Figure 2 A,B for further details.
Immediate onset of sustained neuronal activation was manifested in 10 trials by two Aδ- and seven C-units as a twofold increase in the rate of firing (1.37 ± 0.44 spikes/s vs 0.73 ± 0.29 at baseline; p = 0.0051) that began either before (three trials) or during the wave of CSD (seven trials). In six trials, neuronal activity returned to baseline after 41.2 ± 13.3 min; in the remaining four trials, neuronal activity remained heightened for 33, 87, 93, and 107 min, at which time the recording was stopped (Fig. 2).
Delayed onset of sustained neuronal activation was manifested in 13 trials by six Aδ- and six C-units as a twofold increase in the rate of firing (1.59 ± 0.46 spikes/s vs 0.73 ± 0.26 at baseline; p = 0.0015) that began 11.9 ± 1.9 min after the end of the CSD wave. In seven trials, neuronal activity returned to baseline after 36.9 ± 7.0 min; in the remaining six trials, neuronal activity remained heightened for 14, 22, 68, 75, 80, or 80 min, at which time the recording was stopped (Fig. 3).
Another set of eight trials performed on seven Aδ-units and one C-unit yielded delayed onset of sustained neuronal activation, which was preceded by a short-lived surge of activity that began either before (six trials) or during (two trials) the CSD wave and lasted for 60.0 ± 14.7 s. As in the previous set of trials, sustained activation was manifested as a twofold increase of the firing rate (1.05 ± 0.29 spikes/s vs 0.49 ± 0.18 at baseline; p = 0.012) that began 18.4 ± 2.8 min after the end of the CSD wave. Neuronal activity returned to baseline after 33.7 ± 3.8 min in seven trials, and was still heightened 79 min after CSD in one trial, at which time the recording was stopped (Fig. 4).
In a set of 10 trials performed on one Aδ-unit and eight C-units, CSD was only associated with an immediate, short-lived surge of activity that began either before the onset of CSD (seven trials) or during the CSD wave (three trials) and lasted for 77.8 ± 19.0 s (Fig. 5). Short-lived neuronal activity in this group and the previous group occurred in 28% of all trials with CSD, and in 26% of the trials in which cortical stimulation failed to produce CSD.
Figure 5.

Immediate, short-lived burst of firing by meningeal nociceptors. A, An example of a CSD wave induced by chemical stimulation of the cortex (upper trace) that was associated with a brief burst of neuronal firing by a C-unit (bottom trace). Note that neuronal firing (numbers in parentheses) rate returned to baseline level (green) at the end of the initial burst of activity (pink). B, Temporal relationship between waves of CSD and the acute burst of neuronal firing. See Figure 2 A,B for further details.
Regardless of the method of cortical stimulation (electrical pulses, pin prick, or KCl), long-lasting neuronal activation was manifested as a significant twofold increase in neuronal firing rate (Fig. 6 A). In 19 cases, long-lasting activation started with an overall delay of 14.4 ± 1.7 min after the end of the CSD wave, with no significant effect of the method of cortical stimulation (Fig. 6 B). Independent of the method of cortical stimulation, sustained neuronal activation persisted for 37.0 ± 4.6 min in 20 trials where firing rate returned to baseline, and >68 min in 8/11 trials in which firing rate remained heightened at the time recording was interrupted (Fig. 6 C).
Figure 6.
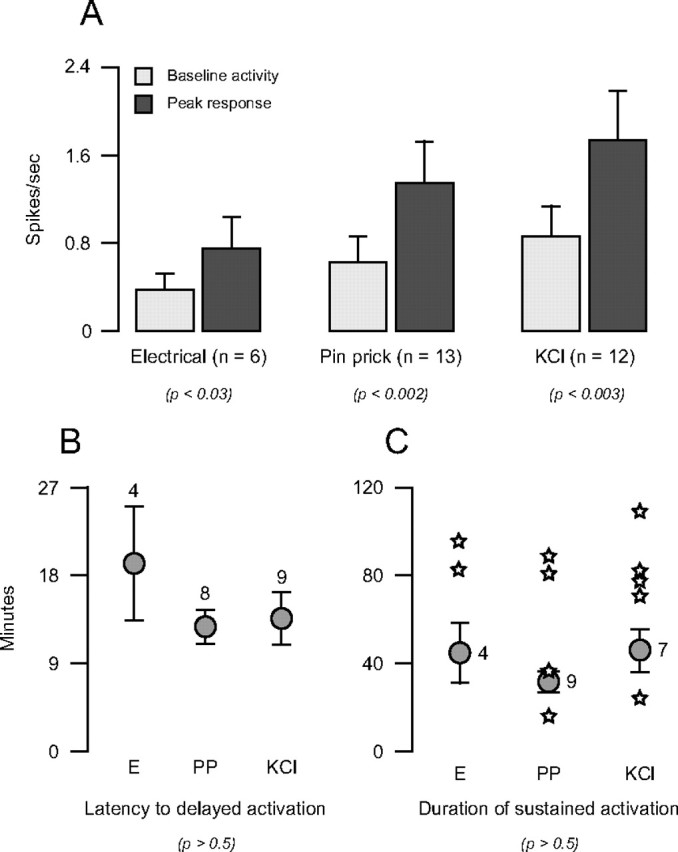
Characterization of sustained neuronal activation induced by stimulation of the cortex with electrical pulses (E), pin prick (PP), or KCl. A, Rate of neuronal firing before and after cortical stimulation. B, Latency to onset of delayed neuronal activation. C, Duration of sustained neuronal activation. The means of 20 trials in which activity returned to baseline are indicated by circles with error bars. Individual trials in which firing rate remained heightened at the time recording was interrupted are indicated by stars. Note that neuronal response parameters were consistently similar, regardless of the type of stimulus used to induce CSD. Numbers indicate sample size. Probability values derive from Wilcoxon test (A) and Kruskal–Wallis test (B, C).
No apparent differences in the pattern of neuronal activation were observed between Aδ- and C-units. Ongoing neuronal firing increased approximately twofold from 0.30 ± 0.10 at baseline to 0.84 ± 0.18 after CSD in Aδ-units, and from 1.01 ± 0.25 to 1.89 ± 0.41 in C-units. Delayed neuronal activation began with similar latencies in Aδ- and C-units (15.7 ± 2.0 vs 12.1 ± 3.0 min) and lasted for similar durations in trials where firing rates returned back to baseline (36.5 ± 6.2 vs 37.5 ± 7.3 min).
The relative contribution of parasympathetic innervation of the dura to long-lasting activation of meningeal nociceptors was assessed by stimulating the cortex in the absence of the ipsilateral SPG (Fig. 7 A–D). Waves of CSD were induced in 25 trials that were performed in 10 rats with excised SPG, using electrical stimulation (2 trials), pin prick (14 trials), or a KCl granule (9 trials). In nine of those trials (performed on 2 Aδ- and 5 C-units), neuronal firing rate increased significantly from 0.87 ± 0.32 spikes/s at baseline to 1.35 ± 0.43 after CSD (p < 0.008, Wilcoxon test). This pattern of neuronal activation began immediately upon cortical stimulation (three trials), or 20.3 ± 4.8 min after the end of CSD (six trials), lasting 23.7 ± 5.7 and 48.5 ± 6.8 min, respectively (Fig. 7 E). An immediate, short-lived surge of activity that preceded or coincided with the wave of CSD was recorded in three of the six trials with delayed long-lasting activation, and in five trials in which CSD was not associated with sustained neuronal activation.
Figure 7.
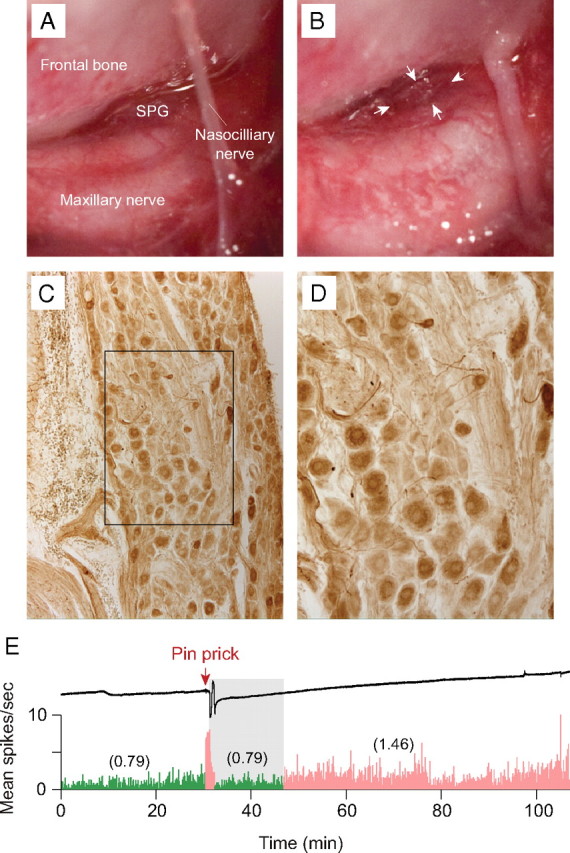
Effects of ipsilateral SPG ablation on sustained neuronal activation. A, Surgical preparation exposing the rat SPG and adjacent structures. B, The same preparation after removal of the SPG. C, D, Immunostaining for vasoactive intestinal peptide in the isolated SPG (D shows higher-power detail of the boxed area in C). E, An example of a CSD wave induced by mechanical stimulation of the cortex (upper trace) that was associated an initial burst of neuronal firing, followed by delayed onset (gray area) of sustained activation (bottom trace). Note that neuronal activity (numbers in parentheses) after CSD (pink) was approximately twofold higher than baseline activity (green). Bin width = 10 s.
Discussion
The notion that waves of CSD can precipitate migraine headache by activating meningeal nociceptors was tested in vivo in anesthetized rats. This is the first study to show that induction of CSD by focal stimulation of the rat visual cortex can indeed lead to activation of meningeal nociceptors. The most significant finding of this study was that neuronal activation lasted over a long period of time and that it was tightly correlated with the occurrence of the CSD wave. This conclusion supports the view that CSD—the presumed mechanism of aura—can activate the trigeminovascular pathway underlying the headache phase (Moskowitz, 1993).
In 22% of the trials, long-lasting neuronal activation coincided with the wave of CSD. It has been suggested that the cascade of events leading to migraine headache involves immediate activation of pial branches of meningeal nociceptors due to release of potassium ions, hydrogen ions, and glutamate molecules in the wake of the CSD wave (Bolay et al., 2002). The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache, a condition recognized by the International Headache Classification Committee (Olesen et al., 1990; Headache Classification Subcommittee of the International Headache Society, 2004).
In 68% of the trials, onset of long-lasting neuronal activation was unexpectedly delayed, occurring some 14 min after CSD. Delayed neuronal activation may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown. Other investigators observed that cortical oligemia occurred with a similar delay after a wave of CSD (Fabricius and Lauritzen, 1993; Busija et al., 2008). It remains to be determined whether cortical oligemia is associated with delayed release of excitatory molecules from the parenchyma or cerebral vasculature, and whether such events could promote persistent activation of meningeal nociceptors. We suggest that units exhibiting delayed activation were not activated earlier during the registration of the CSD wave because their receptive fields did not extend to the pia mater.
The duration of neuronal activation lasted 37 min on average in most trials, and beyond 1 h in other trials. We propose two scenarios for sustained activation of meningeal nociceptors. According to one scenario, short-lasting release of algesic molecules during CSD promotes acute activation that gives rise to ongoing sensitization of the nociceptor, which typically outlasts the stimulus by 30–60 min (Strassman et al., 1996). According to a second scenario, CSD induces ongoing release of algesic molecules, which may last up to 1 h. The duration of sustained activation of meningeal nociceptors, however, may not be sufficient, in and of itself, to explain the duration of the headache phase of migraine, which typically lasts 4–72 h. Assuming that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway, we propose that the duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours (Burstein and Jakubowski, 2004; Burstein et al., 2004). Previous studies, which failed to demonstrate activation of central trigeminovascular neurons by CSD, derived from electrophysiological recording of neurons located in spinal white matter or in laminae III and IV, or V of the dorsal horn (Lambert et al., 1999; Ebersberger et al., 2001). These studies, however, did not sample trigeminovascular neurons in laminae I and II, where most neuronal c-fos expression was induced by CSD (Moskowitz et al., 1993).
Short-lasting neuronal activity occurred promptly upon cortical stimulation in 28% of the trials with CSD (mostly before the onset of CSD), and in 26% of the trials without CSD, suggesting it was unrelated to the induction of CSD. Indeed, meningeal nociceptors exhibit only immediate, short-lasting bursts of action potentials (1–5 s) in response to direct stimulation at their dural receptive field with electrical pulses, mechanical indentation, or KCl (Levy and Strassman, 2002; Strassman and Levy, 2006). Similar acute responses were reported for a variety of somatic and visceral nociceptors elsewhere in the body (Bessou and Perl, 1969; LaMotte and Campbell, 1978; Belmonte and Giraldez, 1981; Mense and Stahnke, 1983; Gallar et al., 1993; Cervero, 1996). Considering that we stimulated the cortex outside the dural receptive field of the nociceptors, we postulate that the acute neuronal response was mediated by axon collaterals that extended from the dura into the pia (Kosaras et al., 2009) at the site where the cortex was stimulated.
Bolay et al. (2002) showed that blood flow in middle meningeal artery increases after CSD, and that this CSD effect is blocked by ablation of the ipsilateral SPG. We found that ablation of the ipsilateral SPG had no effect on the induction or duration of sustained activation of meningeal nociceptors by CSD. We therefore suggest that neither the ipsilateral SPG, nor the sustained vasodilatation of dural blood vessels, are involved in the sustained activation of meningeal nociceptors by CSD. If anything, dilatation of dural blood vessels and increased blood flow in the medial meningeal artery could conceivably be driven by the sustained firing of meningeal nociceptors.
The observation that some meningeal nociceptors become active some 14 min after a wave of CSD impinged on their receptive field may be the most clinically promising finding. Intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.
Footnotes
This research was supported by National Institutes of Health Grants NS-051484 and NS-035611 (R.B.) and NS-46502 (D.L.).
References
- Belmonte C, Giraldez F. Responses of cat corneal sensory receptors to mechanical and thermal stimulation. J Physiol. 1981;321:355–368. doi: 10.1113/jphysiol.1981.sp013989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessou P, Perl ER. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. J Neurophysiol. 1969;32:1025–1043. doi: 10.1152/jn.1969.32.6.1025. [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltrán-Parrazal L, López-Valdés HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–4151. doi: 10.1152/jn.00028.2007. [DOI] [PubMed] [Google Scholar]
- Brinley FJ, Jr, Kandel ER, Marshall WH. Potassium outflux from rabbit cortex during spreading depression. J Neurophysiol. 1960;23:246–256. doi: 10.1152/jn.1960.23.3.246. [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: A race against the development of central sensitization. Ann Neurol. 2004;55:27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol. 2004;55:19–26. doi: 10.1002/ana.10786. [DOI] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K. Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol. 2008;86:379–395. doi: 10.1016/j.pneurobio.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Welch KM, Aurora S, Vikingstad EM. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548–554. doi: 10.1001/archneur.56.5.548. [DOI] [PubMed] [Google Scholar]
- Cervero F. Visceral nociceptors. In: Belmonte C, Cervero F, editors. Neurobiology of nociceptors. Oxford: Oxford UP; 1996. pp. 220–240. [Google Scholar]
- Charles A, Brennan K. Cortical spreading depression-new insights and persistent questions. Cephalalgia. 2009;29:1115–1124. doi: 10.1111/j.1468-2982.2009.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiba L, Paschen W, Mies G. Regional changes in tissue pH and glucose content during cortical spreading depression in rat brain. Brain Res. 1985;336:167–170. doi: 10.1016/0006-8993(85)90430-5. [DOI] [PubMed] [Google Scholar]
- D'Andrea G, Cananzi AR, Joseph R, Morra M, Zamberlan F, Ferro Milone F, Grunfeld S, Welch KM. Platelet glycine, glutamate and aspartate in primary headache. Cephalalgia. 1991;11:197–200. doi: 10.1046/j.1468-2982.1991.1104197.x. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Schaible HG, Averbeck B, Richter F. Is there a correlation between depression, neurogenic inflammation, and nociception that might cause migraine headache? Ann Neurol. 2001;49:7–13. [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M. Transient hyperemia succeeds oligemia in the wake of cortical spreading depression. Brain Res. 1993;602:350–353. doi: 10.1016/0006-8993(93)90701-n. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993;612:61–69. doi: 10.1016/0006-8993(93)91644-8. [DOI] [PubMed] [Google Scholar]
- Gallar J, Pozo MA, Tuckett RP, Belmonte C. Response of sensory units with unmyelinated fibres to mechanical, thermal and chemical stimulation of the cat's cornea. J Physiol. 1993;468:609–622. doi: 10.1113/jphysiol.1993.sp019791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders, second edition. Cephalalgia. 2004;24(Suppl 1):S1–S160. doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- James MF, Smith JM, Boniface SJ, Huang CL, Leslie RA. Cortical spreading depression and migraine: new insights from imaging? Trends Neurosci. 2001;24:266–271. doi: 10.1016/s0166-2236(00)01793-8. [DOI] [PubMed] [Google Scholar]
- Kosaras B, Jakubowski M, Kainz V, Burstein R. Sensory innervation of the calvarial bones of the mouse. J Comp Neurol. 2009;515:331–348. doi: 10.1002/cne.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GA, Michalicek J, Storer RJ, Zagami AS. Effect of cortical spreading depression on activity of trigeminovascular sensory neurons. Cephalalgia. 1999;19:631–638. doi: 10.1046/j.1468-2982.1999.019007631.x. [DOI] [PubMed] [Google Scholar]
- LaMotte RH, Campbell JN. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. J Neurophysiol. 1978;41:509–528. doi: 10.1152/jn.1978.41.2.509. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab. 1992;12:223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Olesen J. Regional cerebral blood flow during migraine attacks by xenon-133 inhalation and emission tomography. Brain. 1984;107:447–461. doi: 10.1093/brain/107.2.447. [DOI] [PubMed] [Google Scholar]
- Leão A. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Leão AA. Spreading depression. Funct Neurol. 1986;1:363–366. [PubMed] [Google Scholar]
- Levy D, Strassman AM. Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol. 2002;88:3021–3031. doi: 10.1152/jn.00029.2002. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58:698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988;457:226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE. Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res. 1996;740:268–274. doi: 10.1016/s0006-8993(96)00874-8. [DOI] [PubMed] [Google Scholar]
- Mense S, Stahnke M. Responses in muscle afferent fibres of slow conduction velocity to contractions and ischaemia in the cat. J Physiol. 1983;342:383–397. doi: 10.1113/jphysiol.1983.sp014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA. Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology. 1993;43:S16–20. [PubMed] [Google Scholar]
- Moskowitz MA, Nozaki K, Kraig RP. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci. 1993;13:1167–1177. doi: 10.1523/JNEUROSCI.13-03-01167.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Friberg L, Olsen TS, Iversen HK, Lassen NA, Andersen AR, Karle A. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol. 1990;28:791–798. doi: 10.1002/ana.410280610. [DOI] [PubMed] [Google Scholar]
- Olesen J, Goadsby P, Ramadan NM, Tfelt-Hansen P, Welch KMA. The headaches, third edition. Philadelphia: Lippincott Williams and Wilkins; 2005. [Google Scholar]
- Rapoport SI, Marshall WH. Measurement of cortical pH in spreading cortical depression. Am J Physiol. 1964;206:1177–1180. doi: 10.1152/ajplegacy.1964.206.5.1177. [DOI] [PubMed] [Google Scholar]
- Schock SC, Munyao N, Yakubchyk Y, Sabourin LA, Hakim AM, Ventureyra EC, Thompson CS. Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res. 2007;1168:129–138. doi: 10.1016/j.brainres.2007.06.070. [DOI] [PubMed] [Google Scholar]
- Spencer SE, Sawyer WB, Wada H, Platt KB, Loewy AD. CNS projections to the pterygopalatine parasympathetic preganglionic neurons in the rat: a retrograde transneuronal viral cell body labeling study. Brain Res. 1990;534:149–169. doi: 10.1016/0006-8993(90)90125-u. [DOI] [PubMed] [Google Scholar]
- Stovner L, Hagen K, Jensen R, Katsarava Z, Lipton R, Scher A, Steiner T, Zwart JA. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27:193–210. doi: 10.1111/j.1468-2982.2007.01288.x. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Levy D. Response properties of dural nociceptors in relation to headache. J Neurophysiol. 2006;95:1298–1306. doi: 10.1152/jn.01293.2005. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]