

Ureas are normally rather inert towards alcohols, amines, and thiols: they require high temperatures, acidic or basic conditions, or metal catalysis, to undergo nucleophilic substitution reactions.[1] Whilst this feature makes them robust protecting groups for aromatic and aliphatic amines, it somewhat limits their subsequent utility. Herein we demonstrate that, in stark contrast to this general behavior, hindered trisubstituted ureas undergo efficient substitution reactions with a range of O, N, and S nucleophiles under neutral conditions, and that in some cases reactions proceed to completion in less than an hour at 20°C.
We have recently reported on the development of a PdII-catalyzed ortho-carbonylation of alkyl aryl ureas, during which we noted that N,N-diisopropyl urea 1 underwent slow hydrolysis to aniline 2 at 100°C (Scheme 1), whereas the corresponding dimethyl and diethyl urea analogues failed to react.[2] The key structural features responsible for this remarkable difference in reactivity have now been elucidated by carrying out methanolysis on a range of simple urea derivatives (Table 1).
Scheme 1.
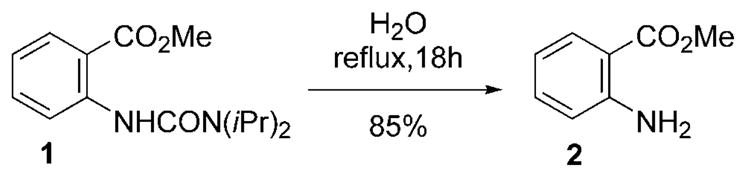
Neutral hydrolysis of aryl diisopropyl urea 1.
Table 1.
Solvolysis of ureas in neutral methanol. Bn =benzyl.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | 3 | R | R′ | R″ | T [°C] | t [h] | Yield of 4 [%][a] |
| 1 | 3 a | Ph | Me | Me | 70 | 18 | <1 |
| 2 | 3 b | Ph | Et | Et | 70 | 18 | <2 |
| 3 | 3 c | Ph | iPr | iPr | 70 | 18 | 81 |
| 4 | 3 d | Ph | H | tBu | 70 | 18 | 0 |
| 5 | 3 e | Ph | Me | tBu | 70 | <5 min | >99 |
| 6 | 3 e | Ph | Me | tBu | 50 | 1 | >99 |
| 7 | 3 f | Ph | Et | tBu | 50 | 1 | >99 |
| 8 | 3 f | Ph | Et | tBu | 20 | 18 | >99 |
| 9 | 3 g | Ph | nPr | tBu | 50 | 1 | >99 |
| 10 | 3 g | Ph | nPr | tBu | 20 | 18 | >99 |
| 11 | 3 h | Ph | iPr | tBu | 20 | 1 | >99 |
| 12 | 3 i | Bn | iPr | iPr | 70 | 18 | 33 |
| 13 | 3 i | Bn | iPr | iPr | 70 | 72 | 85 |
| 14 | 3 j | tBu | iPr | iPr | 70 | 18 | 26 |
| 15 | 3 j | tBu | iPr | iPr | 70 | 72 | 85 |
| 16 | 3 k | tBu | iPr | tBu | 20 | 1 | >99 |
Yield of isolated product.
As with hydrolysis, the aniline-based N,N-dimethyl (3a) and N,N-diethyl ureas (3b) were unreactive, whereas after 18 hours at reflux the more hindered N,N-diisopropyl urea (3c) gave the corresponding carbamate in 81% yield (Table 1, entries 1–3). The requirement for N,N disubstitution at the leaving group is evident from the extreme contrast in reactivity of the tBu-N-H substrate 3d with the tBu-N-Me substrate 3e (Table 1, entries 4 and 5), the latter underwent methanolysis in minutes at 70°C. As the Me group in 3e was changed for the increasingly bulky Et (3 f), nPr (3g), and iPr (3h) substituents (Table 1, entries 7–11), the reactivity increased further; indeed the iPr derivative 3h underwent quantitative methanolysis in less than an hour at 20°C. This phenomenon was not just limited to aryl ureas as the benzylamine (3i) and tert-butylamine (3k) examples attest (Table 1, entries 12–16). The latter derivative is of similar reactivity to 3h, and underwent quantitative methanolysis in under an hour at 20°C.
An explanation for this reactivity was initially sought by comparison of the physical data of 3a and 3c. The IR stretching frequencies and 13C NMR signals of the C=O groups showed no significant differences (νmax = 1641 vs. 1635 cm−1 and δ =155.8 vs. 154.5 ppm, respectively). Their single-crystal X-ray structures[13] showed no significant differences in C–N bond lengths, and DFT calculations on the urea compounds as well as their tetrahedral methanol adducts showed few differentiating structural features.[3]
A more extensive mechanistic investigation was then undertaken by choosing a pair of reasonably reactive substrates (3c and 3 f) and exploring the kinetics of their methanolysis. A Hammett analysis (see the Supporting Information) of the pseudo-first-order rates of solvolysis (MeOH, 70°C) of 3c and its Ph-substituted analogues (p/m MeO, Br, and NO2) revealed a weak activating effect of the electron-withdrawing substitutents (ρ =0.7 ± 0.1, R2 =0.92; Figure 1). Irreversible methanolysis of 3 f in toluene at 35°C, under pseudo-first-order conditions, indicated a fractional dependence on MeOH concentration (−d[3 f]/dt =3.2 0 10−5 [3 f][MeOH]0.32), and no dependence on the concentration of tBuN(H)Et, with a predicted half-life of 2 hours for 3 f in pure MeOH.
Figure 1.
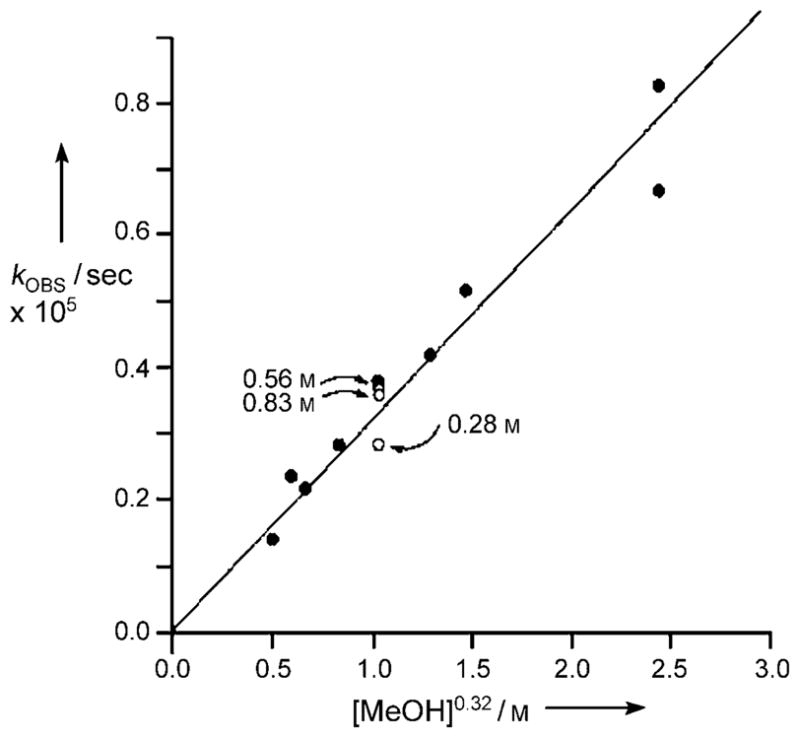
Methanolysis of 3 f (0.11M) in toluene at 35 °C. Open circles: reaction in the presence of added tBuN(H)Et (0.28 to 0.83M).
The mechanism of hydrolysis of N-aryl ureas has been the subject of a number of detailed studies,[4,5,6] with a general consensus that N-aryl isocyanates are generated as transient intermediates through the expulsion of R2NH from a zwitterion of the form Ar–N=C(O−)–NR2H+. Pioneering work by O’Connor and co-workers,[4] led to the suggestion that water mediates a “proton switch”[7,8] (see A; Scheme 2) to generate the zwitterion from the urea at neutral pH, or to generate the R2N-protonated urea under acidic conditions (pH ≤ 6.5). In the most recent study, Capasso and co-workers[6] presented a unified mechanism to account for reactions at low, neutral, and high pH: all of which proceed through the zwitterion, with buffer species (carboxylic acids, hydrogen phosphates etc.) mediating the proton switch. Nonetheless, there are alternative interpretations of the data, for example Laudien and Mitzner[5] have suggested that a simple addition/elimination mechanism occurs under both acidic and basic hydrolysis conditions (A-2, B-2 mechanisms), without any involvement of an isocyanate. Recently, Clayden and Hennecke[9] reported on the butanolysis of N,N′-dimethyl-N′-alkyl ureas at 118°C and suggested the intermediacy of alkyl isocyanates.
Scheme 2.

N-phenyl isocyanate 5 liberation from 3 f through a proton switch by MeOH (A), and by amine transfer (B) to p-bromophenyl isocyanate 7.
The finding of increased rates of methanolysis of N-phenyl ureas 3a–3h as the steric hindrance of the N′,N′ substituents increases weighs against an analogous addition/elimination mechanism. Certainly, nucleophilic attack would be more hindered and the greater nucleofugacity of an anilinium over N,N-dialkylammonium moiety would give a carbamate of the form R2NCO2Me, rather than 4.[10] The data is, however, consistent with the increased basicity of the dialkylamino group for participation in the proton switch, a substantial steric decompression[14] upon liberation of R2NH and N-phenyl isocyanate 5, and a positive Hammett ρ-value (+0.7) arising from a proton transfer from PhNH. The isocyanate 5 may be liberated directly, or through the much postulated zwitterionic precursor 6, the latter being driven by relief of allylic strain in 6. Monomeric methanol can facilitate the proton switch (A; Scheme 2) in an identical manner to that proposed for water;[4] the fractional order in MeOH/toluene reflects the tendency for alcohols to aggregate by hydrogen bonding in hydrocarbon media, nominally as a cyclic trimer.[11] Importantly, the lack of any rate suppression upon methanolysis of 3 f by added tBuN(H)Et suggests that the generation of the isocyanate or zwitterion is rate limiting.
The facile liberation of isocyanate from urea 3 f under neutral conditions in toluene (0.2M) was confirmed by the addition of 0.2M of N-p-bromophenyl isocyanate (7, Scheme 2), which generated an equilibrium mixture with isocyanate 5 and urea 8 (K =1.0). However, the rate of this equilibration (≤1 min at 20°C; 3:2 ratio of 8/3 f) is faster than would be predicted based on the kinetics of methanolysis at 35°C and suggests a direct reaction (B; Scheme 2) between isocyanate (5/7) and urea (3 f/8).[12]
A more specific result supporting the intermediacy of isocyanate 5 during methanolysis came from the reaction of 3 f (0.1M) with a mixture of MeOH (1M), EtOH (1M), and PrOH (1M) in toluene at 35°C, to give the corresponding carbamates 4Me/4Et/4Pr (Scheme 3). Control experiments confirmed that there is no equilibration under these conditions and the partitioning between the three carbamates is identical, within experimental error, to that generated by the reaction of a reference sample of N-phenyl isocyanate 5 under the same conditions.
Scheme 3.
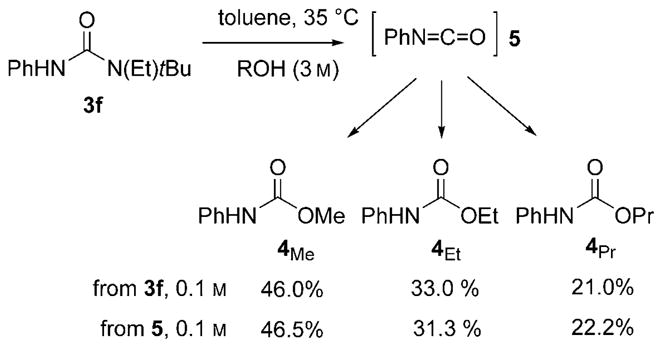
Partitioning of N-phenyl isocyanate 5, which was generated in situ, with a 1:1:1 mixture of MeOH/EtOH/PrOH (3M).
Alcohols, and indeed any species containing an X–H unit in which the X group bears an available lone pair of electrons, can in principle act to effect a proton switch in trisubstituted urea 3 f and a range of such species were found to react readily to give the corresponding carbamates in excellent yields (Table 2). The substantial reactivity of the N-phenyl isocyanate intermediate means that even weak nucleophiles such as phenol (pKa = 9.9) and thiophenol (pKa = 6.6) efficiently give the corresponding carbamates. Entries 3–5 of Table 2 show the synthetic potential of these bulky urea compounds as precursors with reactivity reminiscent of acyl halides, for the conversion of a single amine into derivatives of three of the most common nitrogen protecting groups under neutral conditions.
Table 2.
Synthetic potential of trisubstituted ureas as masked isocyanates.
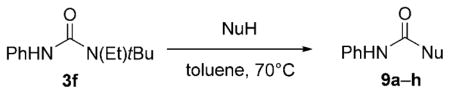 | ||||
|---|---|---|---|---|
| Entry | NuH (equiv) | t [h] | Product | Yield [%][a] |
| 1 | H2O[b] | 1 | 9 a[c] | >99 |
| 2 | MeOH (1.1) | 6 | 9 b | >99 |
| 3 | tBuOH[b,d] | 8 | 9 c | 70 |
| 4 | PhCH2OH (1.1) | 8 | 9 d | 94 |
| 5 | 9-fluorenylmethanol (2) | 5 | 9 e | 78 |
| 6 | PhOH (2) | 18 | 9 f | 72 |
| 7 | PhSH (2) | 18 | 9 g | 62 |
| 8 | tBuNH2 (1.1) | 5 | 9 h | >99 |
Yield of isolated product.
Used as a solvent.
The aniline generated through decarboxylation is trapped in situ to yield 1,3-diphenylurea.
Reaction was performed at 90 °C. Nu =nucleophile.
In conclusion, we have demonstrated the unique ability of hindered trisubstituted ureas to undergo rapid and high-yielding acyl substitution with simple nucleophiles (amines, alcohols, thiols) under neutral conditions. The mechanism of the reaction appears to involve a nucleophile-mediated proton-switch to generate either a zwitterionic precursor (6, Scheme 2), or directly, an isocyanate (5, Scheme 2), which is then captured in a subsequent reaction with the nucleophile. The remarkable reactivity of hindered trisubstituted ureas will be of interest to synthetic chemists in the preparation of amine derivatives. For example, a single urea is efficiently converted under neutral conditions into N-protected aniline derivatives incorporating three commonly employed amine protecting groups: Cbz, Boc, and Fmoc (Table 2, entries 3–5; Boc =tert-butoxycarbonyl, Cbz =benzyloxycarbonyl, Fmoc =9-fluorenylmethyloxycarbonyl). Finally, the crystalline nature of these hindered ureas make them highly convenient as reagents for in situ liberation of isocyanates, simply requiring release through a proton switch.
Footnotes
We thank Dr. M. Haddow for X-ray crystallography, the EPSRC (GR/R02382 and E061575) and AstraZeneca for support, the joint EPSRC/AstraZeneca/GlaxoSmithKline/Pfizer Organic Studentship Initiative, and the NIH (GM-60578). G.C.L.-J. is a Royal Society Wolfson Research Merit awardee.
Contributor Information
Marc Hutchby, School of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS (UK).
Dr. Chris E. Houlden, School of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS (UK)
Dr. J. Gair Ford, AstraZeneca PR&D, Silk Road, Macclesfield, Cheshire, SK10 2NA (UK)
Dr. Simon N. G. Tyler, AstraZeneca, Global Process R&D, Severn Road, Hallen, Bristol, BS10 7ZE (UK)
Prof. Dr. Michel R. Gagné, Department of Chemistry, Caudill and Kenan Laboratories, UNC-Chapel Hill, NC 27599-3290 (USA)
Prof. Dr. Guy C. Lloyd-Jones, Email: guy.lloyd-jones@bris.ac.uk, School of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS (UK), Fax: (+44)117-929-8611
Prof. Dr. Kevin I. Booker-Milburn, Email: k.booker-milburn@bristol.ac.uk, School of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS (UK), Fax: (+44)117-929-8611
References
- 1.For selected examples of the generally forcing conditions needed for the cleavage of urea, see: Akester J, Cui J, Fraenkel G. J Org Chem. 1997;62:431. doi: 10.1021/jo961972l.Kaminskaia NV, Kostić NM. Inorg Chem. 1997;36:5917. doi: 10.1021/ic961500p.Blakeley RL, Treston A, Andrews RK, Zerner B. J Am Chem Soc. 1982;104:612.Kaminskaia NV, Kostić NM. Inorg Chem. 1998;37:4302. doi: 10.1021/ic980065r.Clayden J, Dufour J, Grainger DM, Helliwell M. J Am Chem Soc. 2007;129:7488. doi: 10.1021/ja071523a.Osborn HMI, Williams NAO. Org Lett. 2004;6:3111. doi: 10.1021/ol040042i.Hassner A, Yagudayev D, Pradhan TK, Nudelman A, Amit B. Synlett. 2007:2405.Wang J, Li Q, Dong W, Kang M, Peng S. Appl Catal A. 2004;261:191.Shivarkar AB, Gupte SP, Chaudhari RV. J Mol Catal A. 2004;223:85.
- 2.Houlden CE, Hutchby M, Bailey CD, Ford JG, Tyler SNG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. Angew Chem. 2009;121:1862. doi: 10.1002/anie.200805842. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:1830. [Google Scholar]
- 3.The 10 lowest energy conformers (from AM1 calculations) of 3a and 3c were fully optimized using DFT methods (B3LYP-6-31G*) on MacSpartan 2008 to generate the lowest energy structure; uncorrected energies were utilized. While the methanolysis of 3c was more favorable than that of 3a by 3 kcalmol−1, its tetrahedral intermediate was also 2.5 kcalmol−1 higher in energy, thereby predicting a slower methanolysis of 3c than 3a by the addition/elimination pathway, which is in contrast to our observation.
- 4.a) O’Connor CJ, Barnett JW. J Chem Soc Perkin Trans. 1973;2:1457. [Google Scholar]; b) Giffney CJ, O’Connor CJ. J Chem Soc Perkin Trans. 1976;2:362. [Google Scholar]; c) Mollett KJ, O’Connor CJ. J Chem Soc Perkin Trans. 1976;2:369. [Google Scholar]
- 5.a) Laudien R, Mitzner R. J Chem Soc Perkin Trans. 2001;2:2226. [Google Scholar]; b) Laudien R, Mitzner R. J Chem Soc Perkin Trans. 2001;2:2230. [Google Scholar]
- 6.Salvestrini S, Di Cerbo P, Capasso S. J Chem Soc Perkin Trans. 2002;2:1889. [Google Scholar]
- 7.Williams A, Jencks WP. J Chem Soc Perkin Trans. 1974;2:1753. [Google Scholar]
- 8.a) Alexandrova AN, Jorgensen WL. J Phys Chem B. 2007;111:720. doi: 10.1021/jp066478s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Callahan BP, Yuan Y, Wolfenden R. J Am Chem Soc. 2005;127:10828. doi: 10.1021/ja0525399. [DOI] [PubMed] [Google Scholar]
- 9.Clayden J, Hennecke U. Org Lett. 2008;10:3567. doi: 10.1021/ol801332n. [DOI] [PubMed] [Google Scholar]
- 10.Amides, carbamates, and tetrasubstituted ureas fail to react under methanolysis conditions (see the Supporting Informaition).
- 11.a) Mandado M, Graña AM, Mosquera RA. Chem Phys Lett. 2003;381:22. and references therein. [Google Scholar]; b) Satchell DPN, Satchell RS. Chem Soc Rev. 1975;4:231. [Google Scholar]
- 12.Ozaki S. Chem Rev. 1972;72:457. [Google Scholar]
- 13.CCDC 749529 (for compound 3c) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Compound 3a is a known compound: Lewis RJ, Camilleri P, Kirby AJ, Marby CA, Slawin AA, Williams DJ. J Chem Soc Perkin Trans. 1991;2:1625.
- 14.N′,N′-dialkyl ureas of 2,6-xylidine undergo slow dissociation in anisole (40–140°C) to generate equilibrium mixtures of N-(2,6-dimethylphenyl)isocyanate and the corresponding dialkylamine: Stowell JC, Padegimas SJ. J Org Chem. 1974;39:2448.