

Abstract
Removal of Bβl-42 from fibrinogen by Crotalus atrox venom results in a molecule lacking fibrinopeptide B and part of a thrombin binding site. We investigated the mechanism of polymerization of desBβ1-42 fibrin. Fibrinogen trinodular structure was clearly observed using high resolution noncontact atomic force microscopy. E-regions were smaller in desBβ1-42 than normal fibrinogen (1.2 nm ± 0.3 vs. 1.5 nm ± 0.2), whereas there were no differences between the D-regions (1.7 nm ± 0.4 vs. 1.7 nm ± 0.3). Polymerization rate for desBβ1-42 was slower than normal, resulting in clots with thinner fibers. Differences in oligomers were found, with predominantly lateral associations for desBβ1-42 and longitudinal associations for normal fibrin. Clot elasticity as measured by magnetic tweezers showed a G′ of ∼1 Pa for desBβ1-42 compared with ∼8 Pa for normal fibrin. Spring constants of early stage desBβ1-42 single fibers determined by atomic force microscopy were ∼3 times less than normal fibers of comparable dimensions and development. We conclude that Bβ1-42 plays an important role in fibrin oligomer formation. Absence of Bβ1-42 influences oligomer structure, affects the structure and properties of the final clot, and markedly reduces stiffness of the whole clot as well as individual fibrin fibers.
Introduction
Fibrinogen is composed of two subunits, each comprising three polypeptide chains (Aα,Bβ,γ), held together by a network of disulfide bonds. X-ray crystallography data and electron microscopy studies show that fibrinogen has a trinodular structure, with a central (E) region that consists of amino termini of all 6 polypeptide chains and 2 distal (D) regions containing the carboxyl termini of the Bβ-, and γ-chains (1–5). The Aα-chains extend from the D-regions to form relatively flexible αC-extensions. The whole molecule is 45 nm in length (1,6). During conversion to fibrin, thrombin cleaves the Aα-chain at position 16 to produce fibrinopeptide A (FpA). This exposes a binding site in the E-region that will interact with a binding pocket in the D-region. Similarly, at a slower rate, the Bβ-chain is cleaved by thrombin at position 14 to produce fibrinopeptide B (FpB), which exposes a second polymerization site in the E-region (7–9).
Hydrolysis of fibrinogen by protease III from the venom of Crotalus atrox results in the formation of desBβl-42 fibrinogen lacking a cleavable B peptide (10,11). The polymerization rate of desBβl-42 fibrinogen as assessed by clot turbidity on incubation with thrombin (cleaves both FpA and FpB) and reptilase (cleaves only FpA) has been shown to be slower than that of native fibrinogen (11,12). Removal of Bβ1-42 also reduced thrombin binding to fibrin (12). Based on this, it has been surmised that the sequence comprised by 1-42 in the Bβ-chain contributes to the A polymerization site and provides a secondary thrombin-binding site on fibrin (12). However, the underlying mechanisms for the altered polymerization characteristics of desBβl-42 fibrin remain largely unknown.
In this study we investigated the role of Bβ1-42 in fibrin formation using high resolution imaging by atomic force microscopy (AFM) and new measures of elasticity with magnetic tweezers and AFM. We investigated fibrin formation in its native environment, and visualized fibrin at the nanometre scale using noncontact AFM. We focused on the initial stages of fibrin polymerization and show the formation of early oligomers. The oligomers follow abnormal aggregation patterns in desBβl-42 fibrin, leading to major differences in polymerization and elasticity of fibrin, at a single fiber level as well as in the whole clot. These data provide what we believe are novel insights into the early phase of fibrin polymerization and highlight the importance of Bβ1-42 in these processes.
Materials and Methods
Materials
DesBβ1-42 and normal fibrinogen were prepared as described (10,11) in Buffer 1 (150 mM NaCl, 2.5 mM CaCl2, and 10 mM Hepes, at pH 7.4), divided in several aliquots, and stored frozen at −80°C until analysis, with a final concentration of 0.9 mg/mL in the reaction mixture, unless stated otherwise. Human α-thrombin was obtained from American Diagnostica (Stamford, CT) and was diluted in 0.05 M Tris-HCl, 0.1 M NaCl, pH 7.5, to a concentration of 291 U/mL, divided in aliquots, and stored at −80°C. Before its use, thrombin was diluted to reach the required concentration using Buffer 2 (0.13 M NaCl and 10 mM Hepes, pH 7.4). All other reagents were of analytical grade.
SDS-polyacrylamide gel electrophoresis
Protein samples were prepared in sample loading buffer containing sodium-dodecylsulfate (SDS) by heating to 95°C for 5 min. Gel electrophoresis was carried out using a NuPAGE unit (Invitrogen, Paisley, UK). Bis-Tris gels (1.5 × 10 mm well; Invitrogen, Carlsbad, CA) with a 4%–12% polyacrylamide gradient were run at 200 V for 60 min. Gels were stained with Gelcode blue protein stain (Pierce, Rockford, IL) and photographed digitally using an Alpha Innotech (San Leandro, CA) gel documentation system.
Fibrin polymerization by turbidity
In a 96-well plate, 90 μL fibrinogen in Buffer 1 was mixed with 10 μL thrombin in Buffer 2 to achieve final concentrations of 0.9 mg/mL fibrinogen and 1 U/mL thrombin. Calcium chloride had a final concentration of 2.25 mM in the reaction mixture. Fibrin polymerization was monitored using a Biotek ELX808 microplate reader supplied by Labtech International (Ringmer, UK), at a wavelength of 340 nm every 10 s for 1 h at 37°C using a temperature controlled program.
AFM imaging of clot formation
The AFM, a mechanical scanning probe technique, can be used to image the surface topography of biological samples with molecular resolution in air or liquid. In addition, AFM is suitable for measurement of intermolecular forces with picoNewton (pN) resolution in aqueous media (13–15). It can also be used for nanoscale manipulation. Imaging resolution is governed by the sharpness of the AFM probe tip and tip-sample interaction forces, whereas high force resolution results from the small cantilever spring constants to which the probe tip is attached. The AFM has been used extensively to study soft molecules in biological systems (15,16). In this study, we used the AFM in amplitude-detection tapping mode (TM-AFM) (17) to image fibrin polymerization at different time points. In tapping mode the AFM tip is oscillating at a high frequency in intermittent contact with the surface, which decouples the lateral shear forces generated as the tip moves over the surface, enabling stable imaging of weakly bound biomolecules.
A Nanoscope IIIa MultiMode AFM or Nanoscope IV Dimension 3100 (Veeco Instruments, Santa Barbara, CA) were used for all imaging. Tapping mode was used with the highest possible set point to avoid applying forces that might disturb or damage the soft biological structures, while providing clear high resolution images. As a guide to the time points for AFM we carried out turbidity assays (see above) under identical conditions and chose time points from the lag-phase, rapid growth region, and plateau phase of the turbidity curve (and other points in between, data not shown). To prepare samples for imaging, we placed 9 μL fibrinogen (at the same concentrations described above for turbidity analysis) on a freshly cleaved mica disc into which 1 μL of thrombin (also prepared as above for the turbidity experiments) was injected. Samples were incubated for the required time in a humid atmosphere to prevent drying. The reaction was stopped by rinsing away the drop with Milli-Q water and the sample was dried with nitrogen gas. Samples were imaged by TM-AFM in air.
Noncontact AFM imaging
This mode, otherwise known as attractive regime TM-AFM, has been used previously to obtain molecular resolution images of soft biomolecules such as antibodies (18,19) and streptavidin (20) but is not widely used due to some experimental difficulties and limitations, described below. To our knowledge this mode has not been used previously in the study of fibrinogen.
The theory behind tapping mode is an active area of research, and several good studies describing the complex behavior (17,21) have been published. In tapping mode AFM a combination of long range attractive and short range repulsive forces act on the oscillating probe, where the forces are distance dependent according to a power-law. This leads to nonlinear dynamic tip behavior, manifested in the co-existence of two stable oscillation states, termed “High and Low” amplitude (or the H- and L-state). The L-state is dominated by long range attractive forces where the probe is essentially noncontact, whereas the H-state is dominated by short range repulsive forces. The size of the low amplitude branch was maximized by operating at low amplitude, Ao < 10 nm (typically an RMS voltage of <0.3 V), and so the average position of the tip is therefore closer to the sample. At higher amplitudes the low branch shrank, virtually disappearing when Ao > 20 nm, so small amplitudes were required to achieve noncontact imaging. Another crucial parameter in determining tip-sample interaction forces was the drive frequency. Although both the L-state and H-state were stable, the state that the tip initially took depended on the drive frequency chosen. In frequency sweep experiments with the probe close to a surface, approaching resonance from lower frequencies and then well beyond resonance, the probe oscillated in the H-state. In the opposite direction, when sweeping down to resonance from higher frequencies, the probe oscillated in the L-state, before returning to the H-state at resonance (22). Attractive forces had the effect of lowering the resonant frequency, so if feedback in the L-state was required, then it was beneficial to operate at a drive frequency above resonance, as opposed to at or just below resonance for repulsive tapping mode. The decrease in resonant frequency as the attractive force was experienced was therefore translated into a reduction in amplitude, detected by the AFM and maintained by the feedback loop.
Although both low and high states were stable, the transition between the two could occur with a very small perturbation, for example a momentary contact with the sample due to scanning too fast for the feedback loop to compensate. These events would normally go unnoticed in the H-state, and manifested as a small spot of noise. The ease of moving from one state to another was determined by the relative contribution of attractive and repulsive interactions, a detailed description of which can be found in the study by Garcia and San Paolo (23). Once a feedback perturbation had flipped the oscillation into the H-state, to regain the L-state it was usually necessary to increase the set point so that the probe lifted away from the surface momentarily, before reducing the set point very gradually until the L-state was re-engaged.
Based on above considerations, the conditions for operating a stable noncontact mode were a small amplitude <10 nm, drive frequency above resonance to guide the probe into L-state oscillation, a very light set point, low scan speeds (≤1 Hz), small scan sizes (≤2 μm), and very low topography samples to reduce feedback perturbations and maintain the L-state. These criteria were perfectly met by fibrin(ogen) monomers and early oligomers adsorbed to a flat substrate. For the same reasons, noncontact mode was unsuitable for imaging the later stage clot formation that had a much rougher topography and large features requiring larger scan sizes and hence higher tip velocity.
AFM data analysis
The topographic images were analyzed with standard Veeco Nanoscope AFM software; they were line flattened and all presented with the same z-scale. After leveling, AFM images were normally presented with average height normalized, which tended to obscure a growth process from a fixed baseline. For this reason, the linear z-scale brightness level was shifted so that the lowest points in the images (i.e., the mica substrate) were aligned. In the high resolution images, the height of each domain in single molecule images was measured (a representative selection taken from 10 images) using line profile cross sections in the AFM software. These measurements were plotted as histograms, with Gaussian fitting of the height distributions applied to quantify differences in the molecular topography that were also obvious by visual comparison of the images.
Magnetic tweezers
Magnetic tweezers use magnetic fields to control the position of nano- and microscale magnetic particles. The concept began in 1949 with Crick and Hughes (24), who used a permanent magnet to move magnetic beads held inside living cells. Since then, numerous 1D, 2D, and 3D designs have been reported (25–27), and their applications are widespread. The general principal behind the operation of magnetic tweezers is that an external magnetic field can exert a force on a magnetic particle according to Eq. 1:
| (1) |
where m represents magnetic moment, and B represents magnetic field.
A 3D magnetic tweezers device was constructed with an 8-pole piece design similar to that of Huang et al. (28). With a single electromagnet, a magnetic particle can only be moved in a single direction (the electromagnet always attracts, regardless of polarity). Two electromagnets enable full 1D control, and four electromagnets give 2D control. Adding a third set to give 3D control raises the problem of retaining the optical axis of the microscope, and so more sophisticated designs are required. In this study, only the 1D aspect was required. Each of the eight soft-iron cored electromagnets was wrapped with 480 turns of copper wire. The electromagnets were held in a soft iron frame, and positioned over an Olympus IX71 inverted microscope. An ultralong working distance ×40 objective was used to observe the sample. A standard CCD camera captured images from the microscope. The electromagnets were connected to a bank of voltage amplifiers, which were in turn controlled by a National Instruments (Austin, TX) SCB-68 signal box. The signals used to control the electromagnets were generated from custom written LabView 7.1 (National Instruments) programs. Each pair of electromagnets was also wrapped in copper tubing, in which water at 10°C could be circulated. A thermocouple and Hall Probe could also be used to measure the sample region's temperature and magnetic field levels (both of which interface with the software via an RS232 port). LabView was used to control the voltage levels sent to the electromagnets, via the amplifiers. The software also analyzed the images taken from the CCD camera, and allowed a particle to be tracked in real-time. The x, y, and z positions of the particle were saved as ASCII data, avoiding the need to store large quantities of video footage. Force calibration was achieved by observing the velocity of a magnetic particle inside a fluid of known viscosity. Samples of 100% glycerol were used for this purpose. Custom written software sent voltages to the electromagnets, which moved the bead up, down, left, and right (in x and y) at different voltage levels. This, along with Stokes law (Eq. 2), enabled plots of voltage Vs force to be produced. These data were then programmed back into the software, enabling control of the force, rather than voltage
| (2) |
Superparamagnetic particles (Dynal, Oslo, Norway), measuring 4.5 μm in diameter were used. These possess a core of Fe2O3 single domain crystals, and have no remnant magnetic moment in the absence of an external magnetic field.
Knowing the particle radius, having calibrated the force, and measuring the particle's displacement, we obtained the elastic modulus of the sample through Eqs. 3 and 4:
| (3) |
where J represents compliance (Pa−1), and
| (4) |
where G′ represents elastic modulus (Pa).
In this study we investigated differences in elasticity between normal and desBβ1-42 fibrinogens. Fibrinogen (0.55 mg/mL) and thrombin (1 U/mL) were mixed with magnetic particles (suspended in water), and quickly placed inside a glass capillary tube. As the fibrin network formed, particles were trapped and a suitable one was selected to be tested (away from capillary walls, away from other particles). Initial experiments applied forces in 10 pN steps up to 100 pN to probe the elastic response of the network. For each fibrinogen variant, several samples were tested and the results averaged. The force steps were applied after 1 h of polymerization when structure and elasticity is thought to have stabilized. All displacements up to 100 pN were fully recoverable. A second set of experiments applied a single force in 30 s time intervals over a period of 1 h to investigate the increase in stiffness during polymerization.
AFM force spectroscopy
AFM has been used previously by Guthold et al. (4) to investigate the lateral force required to rupture individual fibrin fibers (29). The approach described in this study used AFM to directly measure the elasticity of individual fibrin fibers, and groups of fibers, suspended across an 800 nm gap in a semiquantitative fashion. The force curve produced is a plot of deflection (and hence force) as the tip was approached and came into contact with the sample, then retracted away (30,31). On a hard surface, the probe stopped moving on contact, and the cantilever deflected with a slope of unity. On a compliant surface, on contact the tip continued moving forward to some extent as the force increases, and the surface was indented. A measure of indentation, or in this case the stretching of the fiber, against force, provides a measure of fiber elasticity. Extraction of a quantitative elastic modulus is currently difficult to calculate due to various experimental uncertainties that need refining. However, a measure of the spring constant of the single fibrin fibers was accessible, and directly comparable between samples. Fibrin samples were prepared as for the AFM time point imaging, with the reaction stopped after 2 min. At this stage well defined fibers had formed, but the density was low, such that single fibers laid across the 800 nm voids in the substrate. Fibrin spring constants were presented as an average of 20 measurements, taken from three replicate experiments for each sample. Fiber diameters were measured from AFM images. The spring constant of the cantilevers were determined using the thermal noise method (32).
Results
Fibrinogen preparations
The fibrinogen polypeptide chains were separated according to molecular mass on reducing SDS polyacrylamide electrophoresis. Three main bands according to the Aα-, Bβ- and γ-chains were observed (Fig. 1, inset). The deletion of the N-terminal 42 residues from the Bβ-chain by the venom enzyme can be observed as a shortening of the polypeptide and hence a faster migrating band for Bβ, almost as fast as that of the γ-chain (Fig. 1, inset, lane 3). Although the band for the Aα-chain was somewhat heterogeneous in desBβ1-42 compared with normal, this was negligible compared to the effect of the venom protease treatment on the Bβ-chain of fibrinogen. The Bβ- and γ-polypeptide bands were identical in intensity comparing desBβ1-42 with normal fibrinogen.
Figure 1.

SDS-polyacrylamide gel electrophoresis and polymerization by turbidity for normal and desBβ1-42 fibrin(ogen). The curves are representative of four repeats each and show the differences in kinetics and the rate of clot formation for desBβ1-42 compared with normal fibrin. Closed bullet points on the x axis indicate time points used for AFM. (Inset) SDS-polyacrylamide gel electrophoresis of normal and desBβ1-42 fibrinogen. Lane 1 shows a molecular mass marker (from top 106.5, 97.6, 50.2, 36.9, 28.9, and 19.9 kDa), Lane 2 shows the three polypeptide chains of normal fibrinogen, from top Aα, Bβ, and γ, whereas in Lane 3 the desBβ1-42 fibrinogen showed a lighter Bβ-chain that migrated faster, approaching the band for the γ-chain. Lane 4 is a prestained molecular mass marker (Novagen Perfect Protein, from top 150, 100, 75, 50, 35, 25 kDa) with a contrast enhanced copy to the right to clarify the marker bands. The Lane 1 marker appeared to be running slightly slow, probably due to its position near the gel edge.
Fibrin polymerization rates
Using turbidity analysis we observed a decreased polymerization rate for clots made from desBβ1-42 fibrinogen. The lag phase was longer (3–4 min), and the process did not reach a plateau even after >50 min of incubation (Fig. 1). For clots made from normal fibrinogen, the lag phase was shorter (1–2 min), polymerization occurred faster and a plateau in turbidity generation was reached within 10 min.
AFM imaging of polymerizing fibrin
We investigated polymerization of the fibrin variants using AFM imaging to monitor progress in clot formation at time points derived from the turbidity experiments (Fig. 1). TM-AFM images showed that major differences already occurred in the early stages of polymerization for desBβ1-42 when compared with normal fibrin (Fig. 2). Polymerization of desBβ1-42 fibrin was impaired at all time points compared to normal fibrin. After 1 min incubation with thrombin, desBβ1-42 fibrin showed short, isolated fibers, when a recognizable fiber network already existed for normal fibrin. By 3 min, desBβ1-42 developed a fibrous structure, and normal fibrin fibers continued to increase in diameter. At 10 and 20 min the contrast between the two samples was stark; fibers continued to grow and thicken in normal fibrin, whereas in desBβ1-42 fibrin the maximum fiber diameter did not increase much above the range 150–200 nm. The 20 min desBβ1-42 sample was characterized by an increasing density of finer fibers, and normal fibrin had a lower number of more substantial fibers (Fig. 2). Clot development at this time point appeared to involve rearrangement into thicker fiber bundles for normal fibrin whereas this effect was not apparent for desBβ1-42.
Figure 2.

Fibrin clot structure of desBβ1-42 and normal fibrin by atomic force microscopy. AFM images at different time points during the polymerization process. (A, C, E, and G) Normal fibrin; (B, D, F, and H) desBβ1-42, at time points 1, 3, 10, and 20 min, respectively. Each image is representative of at least 10 images per fibrinogen variant, and all are 10 μm images with the same z-scaling. Note: Image in g is a digital zoom from a 50 μm image, and hence has lower pixel resolution. Scale bar, 2 μm.
Molecular resolution AFM Imaging
When traditional tapping mode AFM was used to image individual fibrinogen molecules, the trinodular molecular structure was sometimes visible in the best images, but often this detail was obscured and image quality was highly inconsistent. Fig. 3 A is a 1 μm tapping mode image of fibrinogen 30 s after thrombin injection. The molecules mostly appeared as ∼45 nm strands, occasionally with the trinodular structure visible, but often with little evident substructure. This image quality is comparable to previous reports in the literature (33). This length is the same as the natural length of fibrinogen, and is found when fibrinogen is adsorbed to hydrophilic substrates such as mica (33,34). Resolving the globular domains within fibrinogen is important if we wish to investigate the detail of oligomer and protofibril formation. We therefore used noncontact mode (NC-AFM) to determine the structure of fibrinogen molecules (Figs. 3 and 4), fibrin monomers (Figs. 3 and 5) and early stage fibrin oligomers at high resolution (Fig. 5).
Figure 3.

Noncontact AFM imaging of fibrinogen. (A) Tapping mode topographic image of fibrin monomers on mica 30 s after thrombin injection. (B) Noncontact topographic (left) and phase image (right) of an area offset 500 nm to the left of A, hence half the image has been imaged previously in tapping mode (within the dashed line), and half is imaged for the first time. An arrow pointing to the same group of molecules is included for guidance. This shows the irreversible damage to the soft molecules caused by imaging in tapping mode. The noncontact mode was unstable and switched repeatedly to H-state tapping (labeled 1–4). The phase image has been line leveled, and has a z-scale of 5°. Sections of the unleveled raw phase image are overlaid on the left and right edge (z-scale = 150°), in which dark contrast indicates noncontact mode, switching to tapping mode in the lighter contrast areas with a 70° shift in phase angle. Scale bar, 100 nm (1 μm images). (C) Zoomed 100 nm images of individual molecules together with overlaid line scans of three molecules, highlighting the flattened molecule in tapping mode (first column), the same molecules subsequently imaged in noncontact mode (second column), contrasted with molecules imaged in noncontact mode for the first time. (D–F) Presentation of a higher contrast inverted color scale to highlight αC regions. (D) Fibrinogen molecules, with no αC regions visible, contrasting with E and F that were taken 1 min after thrombin. αC regions were visible extending from most of the isolated molecules, tethering two molecules together, or extending from various points on the oligomers in E. αC regions were also visible in the noncontact molecules of C. Scale bar in c–e, 50 nm. The z-scale of 5 nm is the same in all topographic images.
Figure 4.

High resolution NC-AFM imaging of normal and desBβ1-42 fibrinogen single molecules. (A) normal fibrinogen. (B) desBβ1-42 fibrinogen. Trinodular structure was observed for each fibrinogen molecule, with a central E-region (open arrows) and two distal D-regions (closed arrows). Scale bars, 50 nm. (C–E) Molecular heights of fibrinogen E- and D-regions by NC-AFM. (C) Bar chart of E- and D-region height distribution in desBβ1-42 fibrinogen. (D) Bar chart of E- and D-region height distribution in normal fibrinogen. (E) Average height plots for E- and D-region in both fibrinogen variants (broken light gray line represents the D-region molecular height distribution for desBβ1-42 fibrin, broken dark gray line that of normal fibrin, unbroken dark gray line the E-region molecular height distribution for normal fibrin, and the unbroken light gray line that of desBβ1-42 fibrin).
Figure 5.

Molecular interactions and oligomer formation during early polymerization using high resolution NC-AFM. Fibrin formation was triggered with thrombin in the presence of 2.25 mM calcium, the reaction stopped after 30 s for normal fibrin and 2 min for desBβ1-42 fibrin by dilution in buffer, and the formation of oligomers was visualized by NC-AFM. (A) Mainly longitudinal oligomers were observed in normal fibrin (zoomed images in panels on right). (B) DesBβ1-42 fibrin showed formation of laterally associated as well as longitudinal oligomers (zoomed images in panels on right). Scale bar on enlargements, 50 nm.
Damage to the fibrinogen molecules inflicted during H-state tapping is illustrated in Fig. 3 B, where the field of view has been offset 500 nm to the left (and slightly up) and an L-state (attractive regime) noncontact image acquired with the same tip. Within the previous scan area (indicated by the dashed white line, on the right of the image) the fibrinogen molecules had a definition similar to the tapping mode image, flattened and indistinct. In contrast, the molecules that had not been imaged previously in tapping mode appeared more pronounced, with much higher definition, and with a perfectly clear trinodular structure. Zooms of 100 nm highlighting individual molecules are shown in Fig. 3 C, together with line-scan cross sections of the molecules for direct height comparison, with examples of tapping mode (TM), the same molecules then imaged in noncontact mode (nc) (TM → nc), and molecules imaged for the first time in noncontact mode. This showed that the noncontact mode did not have an inherently higher resolution, but improved image quality on this particular sample by not damaging the soft molecular structure. H-state tapping mode irreversibly deformed and flattened the molecules. The energy dissipated into the molecule in the L-state has been shown to be ∼10× smaller than the H-state, and is also in the negative direction (35).
Fig. 3 B also shows the initial instability in noncontact imaging before operating parameters have been fully optimized, with the desired L-state flipping to the H-state on four occasions (labeled 1–4), before returning to the L-state on set point adjustment (several blank scan lines could be detected on each H-L state transition, as the set point is momentarily increased to lift the tip off the surface). NC-AFM operation was verified and controlled by monitoring the phase-lag image (a measure of energy dissipation) as follows. In line leveled phase images, NC-AFM was characterized by phase shifts on the molecules +0.5 to +1.2° of the background mica. If the mode switched to the H-state, the phase contrast difference increased substantially as more energy was dissipated into the molecules relative to the background, with phase shifts of >+5°. This was observed clearly in the phase image of Fig. 3 B, where the four H-state tapping regions showed bright phase contrast fibrinogen molecules, which were obvious in comparison to the low contrast L-state molecules. Molecules in the corresponding H-state regions of the topographic image appeared flatter and blurred, with little structure visible. Fig. 3 B is bounded on either side by a vertical slice of the raw unleveled phase data. Here the real sharp swings in phase were more clearly seen. The brighter contrast region showed an absolute real phase shift of ∼−55° (tapping) and the dark contrast was −135° (noncontact). A general rule is that phase shifts >90° are noncontact and <90° are tapping (35). It should be noted that in the Veeco Instruments software, a phase angle of +90° (light contrast) → 0° → −90° (dark contrast) is equivalent to a real phase shift of 0°→ −90°→ −180°. In the line leveled phase image acquired in the H-state a brighter contrast indicated more energy dissipation (hence more damage to the molecular structure), but in noncontact mode bright contrast indicated less energy dissipation. A relationship has been shown between the phase shift and energy dissipation, but a full understanding is still lacking (35).
Noncontact AFM reveals αC-regions
As a demonstration of the image quality, it is possible to achieve noncontact images of fibrin at 1 min after thrombin injection as shown in Fig. 3 E (a collection of molecules, each image size = 100 nm) and Fig. 3 F (a 500-nm field showing a variety of monomers and oligomers). For comparison, images of fibrinogen molecules before thrombin injection are shown in Fig. 3 D. In the images after thrombin, either a clear purple strand, or purple patch, could be seen extending from most of the visible outer D-regions, which we interpret as the αC region. In some instances (Fig. 3 E) the αC region can be seen extending between two separate monomers. These figures have been presented in a nonstandard color table (yellow-purple-blue) to emphasize the very small strand in purple, as distinct from the yellow background and blue fibrin. This is the first time, to our knowledge, that the αC region has been imaged by AFM. αC regions are thought to be involved mainly in later stage lateral aggregation and cross-linking of fibrils (36). In fibrinogen the ends of the αC regions are docked to the central E-region, which are then released by the action of thrombin (discussed in more detail below). The images indicated that αC regions are involved with the initial stages of clot formation (to some extent at least) from the earliest time points. Higher resolution AFM images may be possible using sharper nonstandard probes together with noncontact imaging.
Noncontact imaging of normal and desBβ1-42 fibrinogen
We used NC-AFM to obtain high resolution images of the fibrinogen molecules before polymerization, visualizing, and measuring the domain structures of the molecule. Single fibrinogen molecules were imaged without polymerization at high resolution by diluting to ≈1.35 μg/mL with Buffer 1. The images were in good agreement with those observed by previous investigators using AFM (37), but of higher resolution and clarity. Both fibrinogens shared the same D-E-D structure of ∼45 nm in length (Fig. 4, A and B). In desBβ1-42 fibrinogen, the E-region was significantly smaller than the D-region with an average height of 1.2 nm ± 0.3 (n = 44) compared with 1.7 nm ± 0.2 (n = 88, p < 0.0001) respectively (Fig. 4 C). In normal fibrinogen, this difference in height between the E- and D-regions was less marked although still statistically significant (1.5 nm ± 0.2, n = 30 vs. 1.7 nm ± 0.3 respectively, n = 60, p = 0.002) (Fig. 4 D). The E-region of desBβ1-42 was smaller than observed with normal fibrinogen, (p < 0.0001) whereas the D-regions did not differ in size between the desBβ1-42 and normal fibrinogen (Fig. 4 E). Our height measurements are comparable with heights of fibrinogen D- and E-regions reported previously. Tapping mode images presented by Marchin and Berrie (38) showed a 1.7 ± 0.6 nm height for fibrinogen on mica (though with a globular rather than trinodular structure) and 1.1 nm ± 0.1 nm on graphite, whereas Agnihotri and Siedlecki (39) found trinodular structures on mica with heights of D-regions ranging between 1.3 and 2.1 nm and those of E-regions between 1.2 and 2.2 nm. Ohta et al. (40) reported heights of ∼1.5 nm for the D-region and ∼1.2 nm for E-region, although the images presented did not appear to show a trinodular structure. We have found that the measured molecular height was variable dependent on imaging conditions. For example, our fibrin molecules imaged by noncontact mode in Fig. 3 are ∼3.8 nm in height. When imaging in Buffer 1 on mica we have found a height of 3.5 nm (data not shown), whereas on graphite in air it was 0.7 nm (data not shown). It is clear that the measured height could be dependent on a large number of variables, including humidity (in air), buffer ionic strength, imaging set point, substrate, presence of background contamination, and preparation history. A full systematic study is required to fully understand these effects. Despite the variability in reported heights with changing conditions, we are confident in the difference in height between normal and desBβ1-42 fibrinogen E-regions, as the conditions used and the heights of the D-regions were identical.
Formation of fibrin oligomers
We next used high resolution NC-AFM to investigate the start of oligomer formation after incubation of fibrinogen with thrombin (Fig. 5). Among individual fibrin monomers, we observed the first oligomer structures. The polymerization reaction was stopped after 30 s for normal fibrin and 2 min for desBβ1-42 fibrin, by dilution with the same buffer where the final concentration of fibrinogen in the solution was 0.7 μg/mL. The different time points were necessary due to the relatively long turbidity lag phase of the desβ1-42 fibrin. The NC-AFM molecular resolution imaging requires a low density of molecules, and high dilutions were also required to clearly separate individual monomers and oligomers for identification and analysis purposes. At 30 s a substantial proportion of normal fibrin oligomers had formed, but in desBβ1-42 these oligomers were rarely observed, only monomers of fibrin. Conversely, at 2 min, a reasonable number of oligomers could be observed in the desBβ1-42 sample, but the normal fibrin had clotted to such an extent that NC-AFM was no longer possible. As well as the difference in kinetics of oligomer formation, there was also a difference in oligomer structure. Normal fibrin molecules interacted mostly in a longitudinal manner through D-D interactions (Fig. 5 A, zoomed panels). They also interacted in half-staggered overlapping (D-E-D) arrangements. In contrast to this, polymerizing desBβ1-42 fibrin showed different types of oligomer arrangements that often involved molecules laterally stacking (Fig. 5 B, zoomed panels) and aggregating together. For the purpose of quantitative analysis we defined longitudinal oligomers as structures composed of three or more monomers that associate in linear chains, either by D-D end-to-end interactions or by D-E-D staggered association. Laterally associating oligomers were defined as structures arising from side-ways association of three or more monomers. Analysis of all oligomer assemblies across 50 images yielded the following counts: 85 (73%) longitudinal associations or linear chains and 31 (27%) lateral associations for desBβ1-42 fibrin; 221 (99%) longitudinal and 3 (1%) lateral associations for normal fibrin. All larger aggregate structures were disregarded if it was not clear how the individual molecules were aligned.
Fibrin clot elasticity by magnetic tweezers
Several repeat measurements for desBβ1-42 and normal fibrin samples were taken, and the G′ plots averaged. For each force pulse applied, the particle moved elastically, and remained in position until the force was removed. It then returned to its original position (Fig. 6 A). It was observed that the fibrin network obeyed Hooke's law up to forces of ∼60 pN (Fig. 6 B). After this, the system appeared to strain harden and the displacement for a given force reduced (electromagnet saturation at >100 pN only). For all forces and time durations used, no viscous component was observed other than that due to water, except for the very early stages of polymerization (<2 min) before the clot volume had fully gelled. Using force pulses at 30 s intervals over a period of 1 h, the elastic modulus G′ of both sample types increased and reached a plateau. The maximum G′ of desBβ1-42 fibrin was lower at ∼1 Pa compared with ∼8 Pa for normal fibrin (Fig. 6 C), showing that desBβ1-42 fibrin is less rigid than normal fibrin.
Figure 6.

Fibrin elasticity by magnetic tweezer experiments. Panel A: A force was applied to a magnetic particle incorporated in the fibrin clot. The particle moved and distorted the fibrin network, which then restored the particle to its original location on removal of force. Panel B: Elasticity measurements of normal and desBβ1-42 fibrin. Force pulses in 10pN increments were applied and the displacement of the magnetic particle measured. Hooke's law was obeyed up to 60pN. Panel C: Measurements of G’ over 1 h for normal and desBβ1-42 fibrin. Force pulses of 80 pN were used in 5 s durations, every 30 s.
Single fiber elasticity by AFM force spectroscopy
Force-distance curves collected on suspended single fibers and multifibers for the two samples were used to determine the force versus indentation curves (Fig. 7). Also shown for comparison is the deformation response of a nonsuspended fiber, and that of the clean substrate (hard response). The curves were aligned so that 0 on the z axis denotes the first point of contact. On very soft surfaces (the suspended fibers) there was a small uncertainty about the location of this contact point. We have chosen the first point where the cantilever deflection measurably moved positive of the zero deflection/force line. Calculating the initial slope of these curves showed that the spring constant of the normal fibers was 0.03 ± 0.01 N/m compared to 0.01 ± 0.004 N/m for desBβ1-42 fibers for 20 measurements on each sample. The individual curves shown in Fig. 7 are typical responses. The diameters of the fibers measured at 2 min in Fig. 7 were 110 nm for desBβ1-42 and 85 nm for normal fibrin. At this early time point, the largest desBβ1-42 fibers chosen were slightly larger than the more numerous normal fibrin fibers. However, at subsequent time points the normal fibrin grew in diameter more rapidly than the desBβ1-42. Despite this desBβ1-42 fiber being slightly thicker, it was ∼3 times more elastic. Fiber diameter is an important parameter if a quantitative elastic modulus is required, but in this study the larger desBβ1-42 fibrin fiber was substantially more elastic, so a relative comparison was sufficient. The true difference in elastic modulus will therefore be greater than three times. These data show that the reduced stiffness of desBβ1-42 fibrin is inherent to the individual desBβ1-42 fiber, and suggests that structural differences in the early stage fiber formation process are the cause for this.
Figure 7.
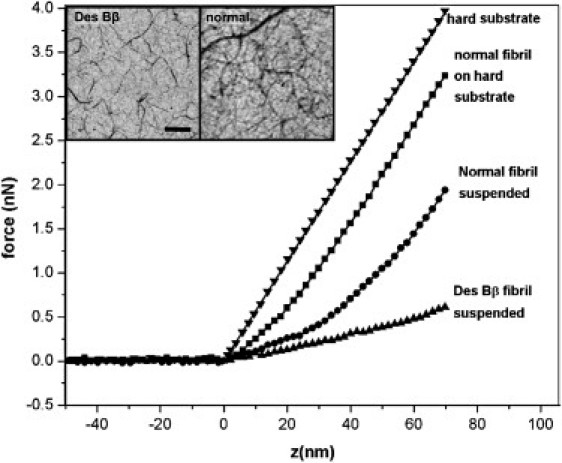
AFM force-distance approach curves comparing the elasticity of individual suspended fibers of desBβ1-42 with normal fibrin. A fiber suspended across an 800 nm gap was stretched at the center of the suspended length. A simple spring constant for the fiber was calculated from the curve, and averaged across 20 repeat measurements for each sample. Normal fibrin is 0.03 ± 0.01 N/m compared to 0.01 ± 0.004 N/m for desBβ1-42. The diameters of the measured fibers chosen were comparable at the 2-min time point used for both samples (in the case of the above curves the desBβ1-42 fiber was slightly thicker). Force curves on the clean substrate, and on a supported fiber, are shown for comparison. Inset shows images of normal and desBβ fibrin at 2 min. Scale bar, 1 μm.
Discussion
DesBβ1-42 fibrinogen is produced by cleavage of fibrinogen by protease III from the venom of the western diamondback rattlesnake (Crotalus atrox). A bite from Crotalus atrox is associated with a severe bleeding diathesis and the effect of the venom on fibrinogen is one of the mechanisms by which this occurs. In this study, using biophysical techniques to investigate the early stages of clot formation, we provide evidence for a mechanism by which the lack of the 42 N-terminal residues of the Bβ-chain changes the structure and stiffness of the fibrin clot. We used high resolution atomic force microscopy to compare fibrin formation of desBβ1-42 with that of normal fibrinogen. The high resolution of NC-AFM showed differences between the two species of fibrinogen at the molecular level. The deletion of 42 amino acids from the N-terminus of the β-chain was detectable as a significant difference in molecular height of the E-region of fibrinogen. We also observed clear differences in behavior of initial oligomer formation for clotting desBβ1-42 fibrin, with increased lateral rather than longitudinal oligomer associations as observed for normal fibrin. Altered oligomer formation was associated with the formation of thinner and more branched fibers in desBβ1-42 as shown on lower magnification TM-AFM. In addition, we demonstrated an eightfold difference in stiffness for desBβ1-42 fibrin using magnetic tweezers, and shown by AFM that the difference in stiffness is inherent to the mechanical characteristics of the individual desBβ1-42 fibrin fibers.
We investigated the initial interactions involved in fibrin oligomer formation using NC-AFM experiments at high resolution. Previously, oligomer formation has been investigated by transmission electron microscopy, gel filtration, and light scattering techniques (41–44). Marchant et al. (33) used AFM to investigate early protofibril formation, however, these images did not reach the same resolution or clarity as those presented in this study. Our NC-AFM images are the first, to our knowledge, of fibrin oligomers showing the individual fibrin monomer domain structures. There was a clear difference between the type of oligomer structures in desBβ1-42 compared with normal fibrin, whereby desBβ1-42 fibrin oligomers were more frequently composed of laterally associating oligomers and normal fibrin oligomers showed more longitudinal associations. Longitudinal, single stranded (as opposed to double-stranded) oligomers have been described previously for normal fibrin by light scattering (45). Dimers and trimers of longitudinally associating molecules have also been observed for fibrinogen and FXIIIa cross-linked fibrinogen by transmission electron microscopy (46). D-D interactions that could provide the basis for these structures have been observed by electron microscopy (47). In many of these studies, as in our study, the single stranded oligomer structures appeared to occur alongside double-stranded oligomers in thrombin-treated fibrinogens. These findings challenge the concept that fibrin polymerization entirely commences with protofibril formation of half-staggered overlapping fibrin molecules, and indicate that other interactions such as longitudinal oligomers of molecules associating end to end and laterally associating oligomers may play a role in the early polymerization processes. Using AFM, Marchant et al. (33) measured a period of 22.5 nm along a narrow fibril, although the images were not clear enough to distinguish individual domains within the fibril. Our images indicate that this periodicity is due to the D-E, E-D spacing in end to end polymerized monomers. It has been suggested that the single stranded oligomer species precede those of a double-stranded nature (45). Protofibril formation through D-E-D interactions then follow the formation of oligomers. The nonlinear structure of desBβ1-42 oligomers can be expected to impair longitudinal growth of initial protofibrils (Fig. 8), resulting in an extended lag phase and delay in first observable fibers in lower magnification images. Additionally, the early involvement of αC interactions during the formation of desBβ1-42 oligomers could reduce their availability during lateral aggregation of protofibrils, resulting in the formation of thinner fibers in the final clot. Hence, the disordered oligomer and protofibril formations of desBβ1-42 fibrin likely impede overall fibrin gelation, leading to a longer lag phase in turbidity experiments and a fibrin network of thinner, more branched fibers.
Figure 8.

Diagram of oligomer formation in normal and desBβ1-42 fibrin. αC regions are tethered alongside coiled coil and E-region in normal fibrinogen due to binding to fibrinopeptides (Fp)A and in particular (Fp)B (56). In desBβ1-42, which lacks FpB, αC regions are prematurely disengaged immediately after cleavage of FpA, leading to lateral associations during early oligomer formation. Disordered oligomer formation in desBβ1-42 fibrin impedes normal fiber formation, producing thinner fibers with reduced stiffness. FpA is indicated by thin lines and FpP by thick lines.
The effects of deletion of the Bβ1-42 sequence on fibrin oligomer formation and polymerization could involve several mechanisms. In normal fibrin, the new N-terminus of the β-chain exposed by cleavage of FpB contributes a binding site (knob “B”) on the E-region for a complementary binding hole (“b”) in the C terminus of the β-chain located in the D-region on another fibrin molecule (6,48,49). The precise role of ”Bb” interaction in fibrin polymerization is currently a debated issue (50–52), but the complete absence of knob B in desBβ1-42 due to deletion of this part of the molecule could have significant effects on fibrin polymerization. The finding of shorter, more branched protofibrils and fibers in desBβ1-42 is in agreement with the frequently hypothesized role of FpB release in lateral aggregation of fibrin during the production of thicker fibers (53,54). However, our observation of differences in fibrin oligomer formation is probably more complex and could involve additional mechanisms that may be explained by involvement of the αC-region. The C-terminal region of the α-chain (or αC) forms a relatively long extension from the D-region of fibrinogen and is composed of a flexible chain terminating in a globular domain (36,55). Normally, this region is tethered alongside the coiled coil with the globular domain aligned close to the E-region. During the conversion to fibrin by thrombin, the αC-regions are disengaged from this aligned position and are thought to take part in αC-αC interactions that contribute to lateral aggregation of the fibrin fibers. The αC disengagement is thought to occur at the same time as FpB release by thrombin. A recent study by Litvinov et al. (56) provides evidence using laser tweezers that the C-terminal domain of the α-chain interacts directly with the fibrinopeptides, FpB in particular, suggesting that FpB release itself may contribute to disengagement of αC. In desBβ1-42 fibrinogen, absence of FpB could reduce affinity of the αC region to the E-region and lead to early disengaged conformation immediately after cleavage of FpA. If confirmed, it is therefore possible that disengaged αC-regions of desBβ1-42 fibrinogen favor lateral oligomer formation through αC-αC interactions, whereas longitudinal association is favored in normal fibrin (Fig. 8).
The Bβ15-42 sequence contributes a nonsubstrate binding site on the E-region (12). It is likely that the corresponding sequence on the α-chain also contributes nonsubstrate binding for thrombin, as binding has been reported to be affected by a synthetic peptide analog of α27-50 (57). Nonsubstrate thrombin binding sites are distinct in character from the substrate interaction sites on the E-region of fibrinogen that lead to fibrinopeptide cleavage. There are two possible implications of this for desBβ1-42 fibrin formation. First, absence of the nonsubstrate thrombin binding site in desBβ1-42 fibrin could lead to quick release of thrombin from the fibrin molecule after FpA cleavage (12). Second, despite the molar paucity of thrombin over fibrin, nonsubstrate thrombin binding to the E-region of normal fibrin may sterically impede lateral oligomer associations to favor longitudinal orientation of the oligomers. This may particularly occur in the initial phases of polymerization when a relatively small fraction of the fibrin(ogen) molecules is taking part. In desBβ1-42 fibrin, where this nonsubstrate binding site is absent, there is no such sterical constraint and lateral associations of the oligomers are possible, likely through disengaged αC-αC interactions as discussed above.
It is of note that the AFM images of the forming clots corresponded well with the turbidity curves in both fibrinogen samples, apart from normal fibrin at 20 min after the initiation of clot formation. The AFM images of this sample showed fibrin fibers with much larger diameter than those obtained after 10 min of clot formation, when the turbidity curve had already reached its maximum. Whereas every other time point in normal fibrin and all time points in desBβ1-42 fibrin concurred with the commonly held conception that the maximum turbidity of a fibrin clot relates directly to the mass/length ratio and hence diameter of the individual fibers (42,58), the normal fibrin after 20 min did not. This data suggests that the reported association between turbidity and fiber thickness may not necessarily hold for fibers with very large diameter, larger than the wavelength of the light used for turbidity measurements. Such limitations should be kept in mind when using light scattering techniques to study fibrin structure.
Magnetic tweezers were used to characterize elastic properties of the fibrin clot by detecting movement of a magnetic particle trapped in the fibrin clot. The elastic properties measured by this method is dependent on the number of fibers in which the magnetic particle is trapped and is also related to adjacent branching fibers that can affect the behavior of the particle. In this respect, characterization of elastic properties of fibrin by magnetic tweezers is more akin to analysis of the elastic behavior of a whole fibrin clot as shown using a torsion pendulum (55,59), rather than that of single fibers by optical tweezers (60) or AFM (4). The estimates for fibrin rigidity from magnetic tweezers presented in this study, range between 10 and 80 dyne/cm2 (1–8 Pascal/cm2). Deletion of the 1-42 sequence in the β-chain reduced fibrin stiffness by ∼8-fold. Measurement of the rigidity of single fibers within the clot by AFM force spectroscopy also showed a significant decrease for desBβ1-42 fibers compared with normal fibrin. These data show that elastic properties of desBβ1-42 fibrin are not only determined by differences in overall fibrin clot structure, but that inherent mechanical properties of the individual fibrin fibers play a role. This data is in support of effects of Bβ1-42 on fiber structure and mechanisms of fibrin polymerization. It provides evidence in support of differences in the internal structure of the desBβ1-42 fibers, and hence to a change in the mechanism of fibrin polymerization at the earliest stage.
Using nanoscale biophysical analysis we have shown differences in fibrin polymerization and elastic properties of clots made from desBβ1-42 and normal fibrinogen. Mechanisms underpinning these effects on structure and function involve differences in oligomer formation. These findings show what we consider to be novel aspects of desBβ1-42 fibrin formation and highlight the important role of the amino terminal region of the fibrinogen β-chain in normal fibrin formation.
Acknowledgments
Author contributions: R.H.A. carried out main experiments and data analysis, and wrote the manuscript. S.D.C. designed the research, developed AFM experimental protocols, analyzed and interpreted data, and wrote the manuscript. R.H. designed and built the magnetic tweezers and carried out the tweezers elasticity experiments. R.A. provided critical discussion. M.W.M. contributed essential reagents and critical discussion. A.S. initiated collaboration and interpreted data. P.G. initiated collaboration and interpreted data. R.A.S.A. designed the research, interpreted data, and wrote the manuscript.
This study was supported by the Egyptian Higher Ministry of Education (R. Abou-Saleh), the Department of Health (grant G121/71 to R. Ajjan), and the British Heart Foundation (FS/2000023).
References
- 1.Doolittle R.F. X-ray crystallographic studies on fibrinogen and fibrin. J. Thromb. Haemost. 2003;1:1559–1565. doi: 10.1046/j.1538-7836.2003.00278.x. [DOI] [PubMed] [Google Scholar]
- 2.Doolittle R.F. Structural basis of the fibrinogen-fibrin transformation: contributions from x-ray crystallography. Blood Rev. 2003;17:33–41. doi: 10.1016/s0268-960x(02)00060-7. [DOI] [PubMed] [Google Scholar]
- 3.Doolittle R.F. Determining the crystal structure of fibrinogen. J. Thromb. Haemost. 2004;2:683–689. doi: 10.1111/j.1538-7933.2004.00664.x. [DOI] [PubMed] [Google Scholar]
- 4.Guthold M., Liu W., Stephens B., Lord S.T., Hantgan R.R. Visualization and mechanical manipulations of individual fibrin fibers suggest that fiber cross section has fractal dimension 1.3. Biophys. J. 2004;87:4226–4236. doi: 10.1529/biophysj.104.042333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosesson M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z., Kollman J.M., Pandi L., Doolittle R.F. Crystal structure of native chicken fibrinogen at 2.7 Å resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
- 7.Blombäck B. Fibrinogen and fibrin–proteins with complex roles in hemostasis and thrombosis. Thromb. Res. 1996;83:1–75. doi: 10.1016/0049-3848(96)00111-9. [DOI] [PubMed] [Google Scholar]
- 8.Brummel K.E., Butenas S., Mann K.G. An integrated study of fibrinogen during blood coagulation. J. Biol. Chem. 1999;274:22862–22870. doi: 10.1074/jbc.274.32.22862. [DOI] [PubMed] [Google Scholar]
- 9.Profumo A., Turci M., Damonte G., Ferri F., Magatti D. Kinetics of fibrinopeptide release by thrombin as a function of CaCl2 concentration: different susceptibility of FPA and FPB and evidence for a fibrinogen isoform-specific effect at physiological Ca2+ concentration. Biochemistry. 2003;42:12335–12348. doi: 10.1021/bi034411e. [DOI] [PubMed] [Google Scholar]
- 10.Pandya B.V., Budzynski A.Z. Anticoagulant proteases from Western diamondback rattlesnake (Crotalus atrox) venom. Biochemistry. 1984;23:460–470. doi: 10.1021/bi00298a010. [DOI] [PubMed] [Google Scholar]
- 11.Pandya B.V., Cierniewski C.S., Budzynski A.Z. Conservation of human fibrinogen conformation after cleavage of the Bβ-chain NH2 terminus. J. Biol. Chem. 1985;260:2994–3000. [PubMed] [Google Scholar]
- 12.Siebenlist K.R., DiOrio J.P., Budzynski A.Z., Mosesson M.W. The polymerization and thrombin-binding properties of des-(Bβ1–42)-fibrin. J. Biol. Chem. 1990;265:18650–18655. [PubMed] [Google Scholar]
- 13.Bustamante C., Erie D.A., Keller D. Biochemical and structural applications of scanning force microscopy. Curr. Opin. Struct. Biol. 1994;4:750–760. [Google Scholar]
- 14.Bustamante C., Rivetti C., Keller D.J. Scanning force microscopy under aqueous solutions. Curr. Opin. Struct. Biol. 1997;7:709–716. doi: 10.1016/s0959-440x(97)80082-6. [DOI] [PubMed] [Google Scholar]
- 15.Moreno-Herrero F., Perez M., Baro A.M., Avila J. Characterization by atomic force microscopy of Alzheimer paired helical filaments under physiological conditions. Biophys. J. 2004;86:517–525. doi: 10.1016/S0006-3495(04)74130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radmacher M., Fritz M., Kacher C.M., Cleveland J.P., Hansma P.K. Measuring the viscoelastic properties of human platelets with the atomic force microscope. Biophys. J. 1996;70:556–567. doi: 10.1016/S0006-3495(96)79602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia R., Perez R. Dynamic atomic force microscopy methods. Surf. Sci. Rep. 2002;47:197–301. [Google Scholar]
- 18.San Paulo A., Garcia R. High-resolution imaging of antibodies by tapping-mode atomic force microscopy: attractive and repulsive tip-sample interaction regimes. Biophys. J. 2000;78:1599–1605. doi: 10.1016/S0006-3495(00)76712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomson N.H. The substructure of immunoglobulin G resolved to 25 kDa using amplitude modulation AFM in air. Ultramicroscopy. 2005;105:103–110. doi: 10.1016/j.ultramic.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 20.Pignataro B., Chi L., Gao S., Anczykowski B., Niemeyer C. Dynamic scanning force microscopy study of self-assembled DNA-protein nanostructures. Appl. Phys. A: Mater. Sci. Process. 2002;74:447–452. [Google Scholar]
- 21.Paulo A.S., Garcia R. Unifying theory of tapping-mode atomic-force microscopy. Phys. Rev. B. 2002;66:041406. [Google Scholar]
- 22.Stark R.W., Schitter G., Stemmer A. Tuning the interaction forces in tapping mode atomic force microscopy. Phys. Rev. B. 2003;68:085401. [Google Scholar]
- 23.Garcia R., San Paulo A. Dynamics of a vibrating tip near or in intermittent contact with a surface. Phys. Rev. B. 2000;61:13381–13384. [Google Scholar]
- 24.Crick F.H.C., Hughes A.F.W. Physical properties of cytoplasm, a study by means of the magnetic particle method. Exp. Cell Res. 1949;1:36–80. [Google Scholar]
- 25.Amblard F., Yurke B., Pargellis A., Leibler S. A magnetic manipulator for studying local rheology and micromechanical properties of biological systems. Rev. Sci. Instrum. 1996;67:818–827. [Google Scholar]
- 26.Gosse C., Croquette V. Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys. J. 2002;82:3314–3329. doi: 10.1016/S0006-3495(02)75672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosu B.G., Jakab K., Banki P., Toth F.I., Forgacs G. Magnetic tweezers for intracellular applications. Rev. Sci. Instrum. 2003;74:4158–4163. [Google Scholar]
- 28.Huang H., Dong C.Y., Kwon H.S., Sutin J.D., Kamm R.D., So P.T. Three-dimensional cellular deformation analysis with a two-photon magnetic manipulator workstation. Biophys. J. 2002;82:2211–2223. doi: 10.1016/S0006-3495(02)75567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu W., Jawerth L.M., Sparks E.A., Falvo M.R., Hantgan R.R. Fibrin fibers have extraordinary extensibility and elasticity. Science. 2006;313:634. doi: 10.1126/science.1127317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bottomley L.A., Coury J.E., First P.N. Scanning probe microscopy. Anal. Chem. 1996;68:R185–R230. [Google Scholar]
- 31.Hassan E.A., Heinz W.F., Antonik M.D., D'Costa N.P., Nageswaran S. Relative microelastic mapping of living cells by atomic force microscopy. Biophys. J. 1998;74:1564–1578. doi: 10.1016/S0006-3495(98)77868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohler B. Cantilever spring constant calibration using laser Doppler vibrometry. Rev. Sci. Instrum. 2007;78:063701. doi: 10.1063/1.2743272. [DOI] [PubMed] [Google Scholar]
- 33.Marchant R.E., Kang I., Sit P.S., Zhou Y., Todd B.A. Molecular views and measurements of hemostatic processes using atomic force microscopy. Curr. Protein Pept Sci. 2002;3:249–274. doi: 10.2174/1389203023380611. [DOI] [PubMed] [Google Scholar]
- 34.Tunc S., Maitz M.F., Steiner G., Vazquez L., Pham M.T. In situ conformational analysis of fibrinogen adsorbed on Si surfaces. Colloids Surf. B. 2005;42:219–225. doi: 10.1016/j.colsurfb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Martinez N.F., Garcia R. Measuring phase shifts and energy dissipation with amplitude modulation atomic force microscopy. Nanotechnology. 2006;17:S167–S172. doi: 10.1088/0957-4484/17/7/S11. [DOI] [PubMed] [Google Scholar]
- 36.Weisel J.W., Medved L. The structure and function of the alpha C domains of fibrinogen. Ann. N.Y. Acad. Sci. 2001;936:312–327. doi: 10.1111/j.1749-6632.2001.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 37.Toscano A., Santore M.M. Fibrinogen adsorption on three silica-based surfaces: conformation and kinetics. Langmuir. 2006;22:2588–2597. doi: 10.1021/la051641g. [DOI] [PubMed] [Google Scholar]
- 38.Marchin K.L., Berrie C.L. Conformational changes in the plasma protein fibrinogen upon adsorption to graphite and mica investigated by atomic force microscopy. Langmuir. 2003;19:9883–9888. [Google Scholar]
- 39.Agnihotri A., Siedlecki C.A. Time-dependent conformational changes in fibrinogen measured by atomic force microscopy. Langmuir. 2004;20:8846–8852. doi: 10.1021/la049239+. [DOI] [PubMed] [Google Scholar]
- 40.Ohta R., Saito N., Ishizaki T., Takai O. Visualization of human plasma fibrinogen adsorbed on highly oriented pyrolytic graphite by scanning probe microscopy. Surf. Sci. 2006;600:1674–1678. [Google Scholar]
- 41.Alkjaersig N., Fletcher A.P. Formation of soluble fibrin oligomers in purified systems and in plasma. Biochem. J. 1983;213:75–83. doi: 10.1042/bj2130075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hantgan R.R., Hermans J. Assembly of fibrin. A light scattering study. J. Biol. Chem. 1979;254:11272–11281. [PubMed] [Google Scholar]
- 43.Hunziker E.B., Straub P.W., Haeberli A. Molecular morphology of fibrin monomers and early oligomers during fibrin polymerization. J. Ultrastruct. Mol. Struct. Res. 1988;98:60–70. doi: 10.1016/s0889-1605(88)80934-0. [DOI] [PubMed] [Google Scholar]
- 44.Weisel J.W., Veklich Y., Gorkun O. The sequence of cleavage of fibrinopeptides from fibrinogen is important for protofibril formation and enhancement of lateral aggregation in fibrin clots. J. Mol. Biol. 1993;232:285–297. doi: 10.1006/jmbi.1993.1382. [DOI] [PubMed] [Google Scholar]
- 45.Bernocco S., Ferri F., Profumo A., Cuniberti C., Rocco M. Polymerization of rod-like macromolecular monomers studied by stopped-flow, multiangle light scattering: set-up, data processing, and application to fibrin formation. Biophys. J. 2000;79:561–583. doi: 10.1016/S0006-3495(00)76317-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mosesson M.W., Siebenlist K.R., Hainfeld J.F., Wall J.S. The covalent structure of factor XIIIa crosslinked fibrinogen fibrils. J. Struct. Biol. 1995;115:88–101. doi: 10.1006/jsbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- 47.Mosesson M.W., Siebenlist K.R., Diorio J.P., Matsuda M., Hainfeld J.F. The role of fibrinogen-D domain intermolecular association sites in the polymerization of fibrin and fibrinogen Tokyo-II (gamma-275 Arg->Cys) J. Clin. Invest. 1995;96:1053–1058. doi: 10.1172/JCI118091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pechik I., Yakovlev S., Mosesson M.W., Gilliland G.L., Medved L. Structural basis for sequential cleavage of fibrinopeptides upon fibrin assembly. Biochemistry. 2006;45:3588–3597. doi: 10.1021/bi0525369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spraggon G., Everse S.J., Doolittle R.F. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 50.Geer C.B., Tripathy A., Schoenfisch M.H., Lord S.T., Gorkun O.V. Role of ‘B-b’ knob-hole interactions in fibrin binding to adsorbed fibrinogen. J. Thromb. Haemost. 2007;5:2344–2351. doi: 10.1111/j.1538-7836.2007.02774.x. [DOI] [PubMed] [Google Scholar]
- 51.Okumura N., Terasawa F., Haneishi A., Fujihara N., Hirota-Kawadobora M. B:b interactions are essential for polymerization of variant fibrinogens with impaired holes ‘a’. J. Thromb. Haemost. 2007;5:2352–2359. doi: 10.1111/j.1538-7836.2007.02793.x. [DOI] [PubMed] [Google Scholar]
- 52.Weisel J.W. Which knobs fit into which holes in fibrin polymerization? J. Thromb. Haemost. 2007;5:2340–2343. doi: 10.1111/j.1538-7836.2007.02794.x. [DOI] [PubMed] [Google Scholar]
- 53.Blomback B., Hessel B., Hogg D., Therkildsen L. 2-Step fibrinogen-fibrin transition in blood-coagulation. Nature. 1978;275:501–505. doi: 10.1038/275501a0. [DOI] [PubMed] [Google Scholar]
- 54.Weisel J.W. Fibrin assembly—lateral aggregation and the role of the 2 pairs of fibrinopeptides. Biophys. J. 1986;50:1079–1093. doi: 10.1016/S0006-3495(86)83552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collet J.P., Moen J.L., Veklich Y.I., Gorkun O.V., Lord S.T. The alphaC domains of fibrinogen affect the structure of the fibrin clot, its physical properties, and its susceptibility to fibrinolysis. Blood. 2005;106:3824–3830. doi: 10.1182/blood-2005-05-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Litvinov R.I., Yakovlev S., Tsurupa G., Gorkun O.V., Medved L. Direct evidence for specific interactions of the fibrinogen alpha C-domains with the central E region and with each other. Biochemistry. 2007;46:9133–9142. doi: 10.1021/bi700944j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Binnie C.G., Lord S.T. A synthetic analog of fibrinogen-alpha-27–50 is an inhibitor of thrombin. J. Thromb. Haemost. 1991;65:165–168. [PubMed] [Google Scholar]
- 58.Carr M.E., Jr., Hermans J. Size and density of fibrin fibers from turbidity. Macromolecules. 1978;11:46–50. doi: 10.1021/ma60061a009. [DOI] [PubMed] [Google Scholar]
- 59.Collet J.P., Woodhead J.L., Soria J., Soria C., Mirshahi M. Fibrinogen Dusart: electron microscopy of molecules, fibers and clots, and viscoelastic properties of clots. Biophys. J. 1996;70:500–510. doi: 10.1016/S0006-3495(96)79596-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collet J.P., Shuman H., Ledger R.E., Lee S., Weisel J.W. The elasticity of an individual fibrin fiber in a clot. Proc. Natl. Acad. Sci. USA. 2005;102:9133–9137. doi: 10.1073/pnas.0504120102. [DOI] [PMC free article] [PubMed] [Google Scholar]