

Summary
Agouti (ASIP) and Agouti-related protein (AgRP) are endogenous antagonists of melanocortin receptors that play critical roles in the regulation of pigmentation and energy balance, respectively, and which arose from a common ancestral gene early in vertebrate evolution. The N-terminal domain of ASIP facilitates antagonism by binding to an accessory receptor, but here we show that the N-terminal domain of AgRP has the opposite effect and acts as a prodomain that negatively regulates antagonist function. Computational analysis reveals similar patterns of evolutionary constraint in the ASIP and AgRP C-terminal domains, but fundamental differences between the N-terminal domains. These studies shed light on the relationships between regulation of pigmentation and body weight, and they illustrate how evolutionary structure function analysis can reveal both unique and common mechanisms of action for paralogous gene products.
Introduction
The Agouti-melanocortin system plays a critical role in the regulation of pigmentation and energy balance in a wide variety of vertebrate species. Agouti protein (ASIP) and Agouti-related protein (AgRP) are paracrine-signaling molecules; ASIP is normally produced in the skin, where it promotes the synthesis of reddish-yellow pigment by hair follicle melanocytes, while AgRP is normally produced in the hypothalamus, where it promotes increased feeding and decreased energy expenditure. ASIP and AgRP act, respectively, via the melanocortin 1 receptor (MC1R) expressed on melanocytes and the MC3R and MC4R expressed in the brain to decrease receptor coupling to adenylate cyclase. From a pharmacologic perspective, AgRP and ASIP are inverse agonists, preventing receptor activation by small melanocortin peptides such as α-melanocyte-stimulating hormone (α-MSH) and, in addition, decreasing basal receptor activity in the absence of α-MSH.
Orthologs of ASIP, AgRP, MC1R, and MC4R have been identified in mammalian, teleost fish, and avian genomes, but not in invertebrate genomes, which suggests that the Agouti-melanocortin system evolved by gene duplication from individual ligand and receptor genes in the last 500 million years. Indeed, the specialized expression patterns of ASIP and AgRP, and their ability to crossreact with the other’s receptor in vitro, are consistent with the view that distinct physiologic functions of ASIP and AgRP have arisen through socalled “subfunctionalization,” such that the current expression pattern and function of each molecule represents a subset of an ancestral gene that existed early in vertebrate evolution.
Biophysical and pharmacologic studies of ASIP and AgRP are consistent with this view, at least with respect to the C-terminal domains of the two proteins. ASIP and AgRP are, respectively, 109 and 112 amino acids in length (after signal peptide cleavage), and they have 40 and 46 residue C-terminal domains that, in cell culture, are sufficient for potent antagonist function at their cognate melanocortin receptors [1, 2]. The C-terminal domains of ASIP and AgRP have nearly identical spacing of 10 key cysteine residues; homonuclear 1H Nuclear Magnetic Resonance (NMR) and biophysical studies demonstrate that the domains have very similar protein folds with an unusual inhibitor cystine knot (ICK) motif stabilized by 5 disulfide bonds.
By contrast, the N-terminal domains of ASIP and AgRP, while similar in length and exon structure, exhibit little primary sequence similarity, and their physiological roles are less clear. In the case of ASIP, the N-terminal domain is required for interaction with Attractin, a large single-transmembrane-spanning domain protein that is required for ASIP signaling in vivo, and is thought to act as an accessory receptor for ASIP-mediated antagonism of MC1R. An analogous role has been suggested for AgRP, whereby the N terminus would interact with syndecan-3, a CNS-specific cell surface proteoglycan, and facilitate the ability of AgRP to antagonize MC4R function [3–5]. While this hypothesis was supported by initial genetic studies [3], recent work by White and colleagues [6, 7] provides strong evidence that the biologically active form of AgRP is its C-terminal fragment, AgRP(83–132), produced by the action of a proprotein convertase (PC). This result is particularly surprising given that the biologically active form of ASIP consists of the full-length protein (after signal peptidase cleavage) [8], ASIP(23–131), and that the genes that encode AgRP and ASIP exhibit the same size and exon structure, pointing to a single and conserved evolutionary origin for the entire protein-coding region.
To gain further insight into the biochemistry and evolution of the Agouti-melanocortin system, we have carried out structural and pharmacologic studies on full-length AgRP, and we evaluated these observations from the perspective of a comparative genomic analysis based on measuring local evolutionary rates of AgRP and ASIP. Our structural and pharmacologic observations confirm that the N-terminal domain of AgRP acts to suppress antagonist activity of the biologically active C-terminal domain, and that it does so in a way that is independent of C-terminal domain structure. Genomic analysis further indicates that the N-terminal domains of the two proteins exhibit distinct patterns of evolutionary constraint, revealing functional divergence. Taken together, our results suggest that the different roles of the N-terminal domains of AgRP and ASIP—a prodomain and a ligand for an accessory receptor, respectively—reflect distinct physiologic functions acquired after duplication of an ancestral melanocortin antagonist early in vertebrate evolution. These observations highlight an interesting mechanism for evolution of paralogous genes whereby subfunctionalization due to complementary expression patterns occurs together with coding sequence changes in distinct structural modules, and they illustrate how bioinformatic structure function analysis can reveal both unique and common mechanisms of action for paralogous gene products.
Results
Our previous structural studies of C-terminal AgRP and C-terminal ASIP were based on synthetic peptides of 46 and 40 residues, respectively. However, the size of full-length AgRP is too large for efficient chemical synthesis; therefore, we used a bacterial expression system based on the work of Rosenfeld et al. [9], in which Met and Lys replace the first 5 amino acids (after signal sequence cleavage) to yield a 109 residue protein, MKd5-AgRP (referred to in what follows as full-length AgRP), with the following sequence, (the cysteine-rich C-terminal domain is underlined):
| 24–50 | MKAPMEGIRRPDQALLPELPGLGLRAP |
| 51–80 | LKKTTAEQAEEDLLQEAQALAEVLDLQDRE |
| 81–110 | PRSSRRCVRLHESCLGQQVPCCDPCATCYC |
| 111–132 | RFFNAFCYCRKLGTAMNPCSRT |
15N uniformly labeled full-length AgRP was expressed and purified as described in Experimental Procedures by making use of a His tag, which was then removed by enterokinase cleavage. Folding of native protein under oxidizing conditions was monitored by reverse-phase high-performance liquid chromatography (HPLC) and mass spectrometry; after 4 hr of folding, we recovered a single HPLC peak that eluted at an earlier retention time than the fully reduced protein, and that showed the expected loss of 10 Da corresponding to five disul- fide bridges.
Agonist binding to melanocortin receptors leads to the production of cAMP; AgRP functions to suppress this activity at MC3R and MC4R. To evaluate the functional consequences of AgRP’s N-terminal domain, we examined the ability of full-length AgRP to inhibit NDP-MSH (a potent analog of α-MSH) at MC4R. As shown in Figure 1A, full-length AgRP causes a dose-dependent rightward shift, consistent with competitive antagonism, and a standard Schild analysis gives an inhibition constant (Ki) of 3.6 nM. This value is ~10-fold greater than reported values for the C-terminal domain alone [2, 10] (see the Supplemental Data available with this article online), thus demonstrating significantly reduced antagonist function. To ensure that the reduced activity was not due to misfolded protein, full-length AgRP was first folded and then proteolyzed with Factor Xa, which normally cleaves after exposed Arg residues, thus liberating AgRP(83–132). Cleavage was carried out at pH 5.0 to avoid disulfide exchange. AgRP(83–132), prepared in this manner, was then directly compared to full-length AgRP and commercially available AgRP(86–132) under identical conditions. Measurements performed in triplicate at twoprotein concentrations are shown in Figure 1B. Full-length AgRP, AgRP(83–132), and AgRP(86–132) were evaluated for their ability to suppress α-MSH-stimulated cAMP production. AgRP(83–132) exhibited equivalent activity to AgRP(86–132) (Ki = 0.35 nM, see Supplemental Data). However, full-length AgRP was significantly less potent than the two forms of C-terminal AgRP, showing significant inhibition only when its concentration was 10-fold greater than that of α-MSH. Binding displacement studies with the radioligand 125I-NDP-MSH were performed to compare the affinities of full-length and C-terminal AgRP. IC50 values are 18.1 ± 0.4 nM for full-length AgRP and 1.6 ± 0.2 nM for AgRP(86–132) at MC4R. These data argue that the decrease in function of the full-length protein, relative to its cleaved C-terminal domain, arises from an approximate 10-fold reduction in affinity.
Figure 1. Pharmacology of Full-Length MKd5-AgRP versus C-Terminal AgRP(83–132).
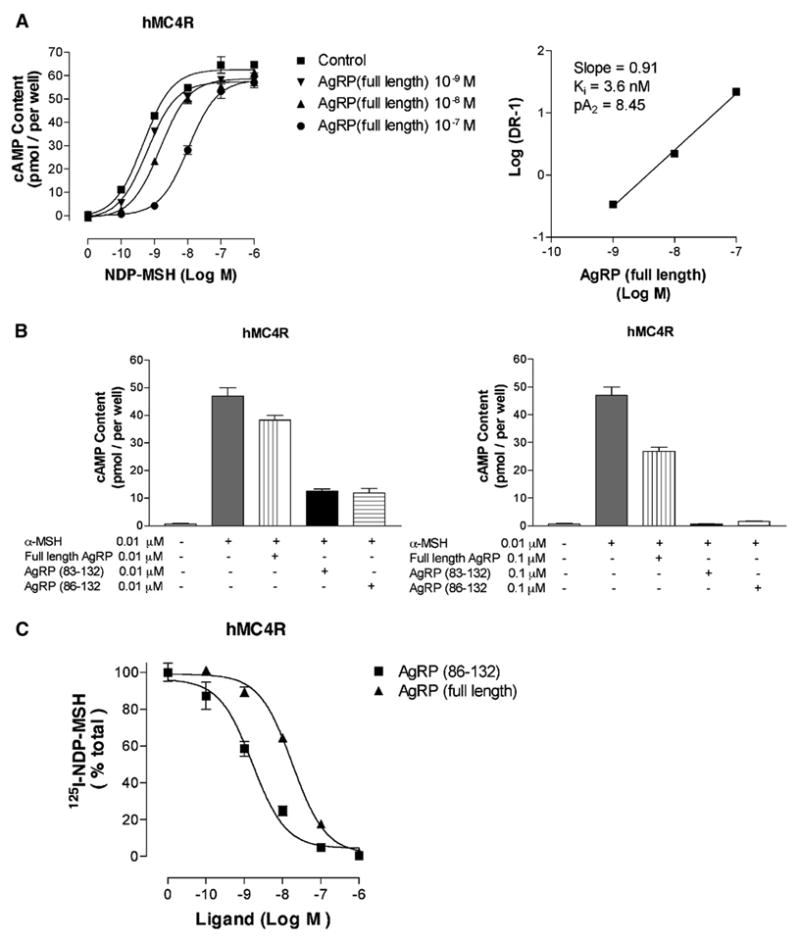
AgRP(83–132) is obtained by cleavage of the oxidatively folded full-length protein.
(A) Inhibition of NDP-MSH-stimulated cAMP generation at hMC4R by full-length AgRP, and corresponding Schild analysis, reveals competitive antagonism with a dissociation constant (Ki) of 3.6 nM.
(B) Both full-length AgRP and AgRP(83–132) inhibit MC4R cAMP production (measured at antagonist concentrations of 0.01 μM and 0.1 μM) stimulated by α-MSH, but AgRP(83–132) is substantially more potent and, to within experimental error, equivalent to AgRP(86–132) with a Ki of 0.36 nM.
(C) Displacement of the radioligand 125I-NDP-MSH from MC4R by full-length AgRP and C-terminal AgRP, giving respective IC50 values of 18.1 ± 0.4 nM and 1.6 ± 0.2 nM.
Error bars represent standard deviations determined from triplicate measurements.
To directly compare the NMR structures of C-terminal and full-length AgRP, we produced 15N-labeled AgRP(83–132) by cleavage of folded, full-length AgRP with Factor Xa (vide supra). Spin systems for the 15N-labeled C-terminal AgRP(83–132) were assigned by using 3D nuclear Overhauser spectroscopy-heteronuclear single quantum coherence (NOESY-HSQC) and 3D total correlation spectroscopy-heteronuclear single quantum coherence (TOCSY-HSQC), and the 1H chemical shifts were found to be equivalent to those previously reported. 1H chemical shifts are well dispersed and exhibit considerable variation from random coil values, consistent with a folded domain (Figures 2A and 2B) [11, 12]. (15N chemical shifts are also well dispersed; see Supplemental Data). 3D NMR spectra were used to assign the spin systems of the C-terminal domain residues in full-length AgRP. Subtraction of the conformationally sensitive 1H chemical shift values of AgRP(83–132) from the corresponding values of the C-terminal domain within full-length AgRP reveal little variation (Figure 2B; C-terminal 15N chemical shifts are also consistent between full-length and AgRP(83–132).; see Supplemental Data). Along with the observation of long-range nuclear Overhauser enhancements (NOEs) that identify AgRP’s β sheet [13] (Supplemental Data), these data demonstrate that structural characteristics of the C-terminal domain in AgRP are affected very little by the presence of the N-terminal residues.
Figure 2. Analysis of AgRP(83–132) 1H Chemical Shifts Derived from Both the Full-Length Protein and the Isolated C-Terminal Domain.

(A) The measured 1H chemical shifts from AgRP(83–132), minus the consensus random coil chemical shifts. The significant variation is consistent with a folded domain. Circles represent measured values; squares represent proline, which is not observable in the HSQC spectra. The arrows show the locations of β strands adjacent to the RFF triplet (residues 111–113).
(B) The difference in chemical shifts, full-length minus C-terminal, for residues 83–132. Note that the vertical axis is expanded by a factor of three compared to that in (A). The limited scatter demonstrates that the HSQC from residues 83–132 in full-length AgRP is essentially equivalent to that of the isolated C-terminal domain, AgRP(83–132). The 2 residues that do show significant 1H chemical shift variations are Arg89 and His91, as indicated.
Display of the full-length AgRP HSQC arising from the backbone NH groups shows that essentially all of the well-dispersed peaks are due to residues in the C-terminal domain; the N-terminal residues show little or no dispersion. Consequently, we were not able to obtain sequential assignment of the N-terminal residues below position 83. The lack of chemical shift dispersion for the N-terminal domain is consistent with a polypeptide segment that lacks well-defined secondary or tertiary structure [14].
Individual 1H linewidths from the HSQC spectrum are narrower for the N-terminal domain (18 Hz) than for the C-terminal domain (27 Hz), indicative of N-terminal flexibility [15, 16]. To directly evaluate protein backbone dynamics, 15N[1H]-NOEs were recorded for full-length AgRP (Figure 3) [17, 18]. Nearly all of the positive peaks correspond to C-terminal residues, demonstrating that the C-terminal residues are ordered with respect to each other, and that they tumble in solution as a domain unit with a rotational correlation time, τc, ≫1 ns. Within the C-terminal domain, only the last 2 residues (Arg131 and Thr132) showed either weak or negative heteronuclear NOE peaks, and these follow the last Cys residue [13]. In contrast, nearly all N-terminal residues give either no observable NOE or a strong negative crosspeak consistent with backbone flexibility and τc ≪ 1 ns, including residues 83–86, between the C-terminal side of the putative PC cleavage site and the first ICK cysteine (residue 87). These data demonstrate that the N-terminal residues of full-length AgRP are flexible and highly dynamic.
Figure 3. NMR Spectra of Full-Length AgRP.

(A) [15N, 1H]-HSQC spectrum of full-length MKd5-AgRP. Assigned peaks are from residues 83–132.
(B) 15N [1H]-NOE spectrum of full-length MKd5-AgRP. Positive NOEs are red, and negative NOEs are green. Almost all N-terminal residues are either negative or unobservable, demonstrating the flexibility of this segment.
While the structure of the C-terminal domain of MKd5-AgRP is mostly unaffected by the N-terminal segment, Arg89 and His91 do show small, but significant (>0.03 ppm), 1H chemical shift differences (Figure 2B). Interestingly, these residues lie in a key loop of the ICK domain that docks directly to melanocortin receptors, and which is critical for high-affinity binding [13, 19–21]. Taken together, these data suggest that increased antagonist potency of C-terminal relative to full-length AgRP (Figure 1) is not caused by a change in protein folding, but by the ability of the flexible N-terminal domain to hinder accessibility to the C-terminal domain. Structural findings are summarized in a model of full-length AgRP (Figure 4A), based on the previously determined C-terminal domain structure [13], and an N-terminal domain that was energy minimized without distance restraints.
Figure 4. Structural Features of AgRP and How They Compare with Those of ASIP.
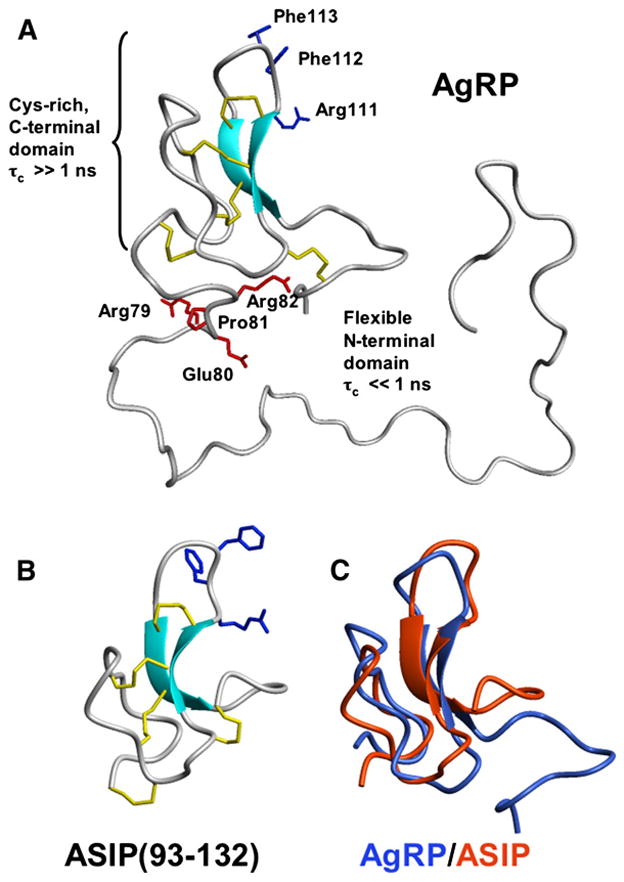
(A) Representation of full-length AgRP. The structure of the Cys-rich C-terminal domain was determined previously by 1H NMR (PDB code: 1HYK) and is preserved in the full-length protein. Positive 15N[1H]-NOEs from residues 87–130 (Figure 3B) show that this domain tumbles with a correlation time (τc) greater than 1 ns, consistent with a structured unit. By contrast, the N-terminal domain is flexible and has a backbone τc ≪ 1 ns. Cysteine residues involved in disulfide bonds are yellow; residues in the RFF triplet, required for high-affinity MCR interactions, are blue. Residues in the putative proprotein convertase recognition site are red. Cleavage takes place after Arg82. The demonstrated flexibility of this segment is consistent with PC cleavage in vivo. CYANA calculations [32] produced a broad ensemble of possible N-terminal structures (500 total were calculated); for clarity, only 1 of the several lowest energy structures is shown.
(B) The NMR structure of the C-terminal domain of ASIP.
(C) Structural alignment of ASIP with the corresponding domain of AgRP [33].
From an evolutionary perspective, these data present a paradox. The C-terminal domains of AgRP and ASIP are almost interchangeable from a structural and biochemical perspective (Figures 4B and 4C); however, the N-terminal domain of ASIP is required for receptor antagonism, whereas, as indicated above, the N-terminal domain of AgRP apparently inhibits receptor antagonism. To further investigate the basis of this difference, we used evolution structure function (ESF) analysis [22] to measure and compare local evolutionary rates for both proteins. This method uses statistically rigorous multiple sequence alignments and phylogenetic trees to reveal evolutionarily constrained regions, which correspond to the structurally and functionally most important regions of the proteins.
There are nine ASIP and eight AgRP orthologs annotated in publicly available databases; we identified an additional three ASIP orthologs (Zebrafish, Fugu, Tetraodon) and four AgRP orthologs (Goldfish, Zebrafish, Fugu, Tetraodon) by sequence similarity searches. ESF constraint profiles for seven mammalian homologs of ASIP and AgRP (human, dog, cow, pig, mouse, rat, and opossum) reveal large regions of evolutionary constraint in the C-terminal portion of both proteins that correspond to the cysteine-rich domains (Figure 5, highlighted in red). An additional evolutionarily constrained region is apparent in the N-terminal portion of ASIP, but not AgRP (Figure 5, highlighted in blue), indicating that an N-terminal domain of ASIP has evolved at a rate similar to that of the C-terminal domain. Strong evolutionary constraint of the ASIP N-terminal domain is not surprising given the requirement for this domain—serving as a ligand for the accessory receptor Attractin—that has previously been demonstrated in vivo and in vitro. However, the relative lack of constraint observed for theAgRP N-terminal domain indicates that its ability to function as a prodomain has few requirements in terms of protein sequence or structure. This observation is consistent with theNMR studies described above and suggests, additionally, that the AgRP N-terminal domain does not serve as a ligand for other receptors.
Figure 5. Evolution Structure Function Analysis of ASIP and AgRP.

Relative evolutionary constraint for each group of indicated proteins was carried out as described in Experimental Procedures; the multiple sequence alignments are indicated below each plot. Areas highlighted in blue and red indicate regions of high local constraint that correspond to the N-terminal domain of ASIP or the C-terminal domains of both proteins, respectively. Accession numbers for ASIP orthologs (and their species and source) are P42127 (human, UniProt), Q5UK76 (dog, UniProt), Q29414 (cow, UniProt), Q6ZYM3 (pig, UniProt), Q03288 (mouse, UniProt), Q99JA2 (rat, Uniprot), ENSMODP00000003361 (opossum, Ensembl), ENSGALP00000034003 (chicken, Ensembl), and Q5CC35 (gold-fish, Uniprot). Accession numbers for AgRP orthologs (and their species and source) are O00253 (human, UniProt), 73957497 (dog, GenBank), P56413 (cow, Uniprot), Q9TU18 (pig, UniProt), P56473 (mouse, UniProt), 62665164 (rat, GenBank), ENSMODP00000007302 (opossum, Ensembl), and ENSGALP00000003505 (chicken, Ensembl).
We extended the ESF analysis to include five additional homologs from nonmammalian vertebrates (Chicken, Goldfish, Zebrafish, Fugu, and Tetraodon). The resulting profiles are qualitatively similar to those obtained for mammalian homologs (Figure 5), which suggests that evolutionary constraint of the ASIP N-terminal domain has an origin similar to that of ASIP and AgRP themselves, early during vertebrate evolution.
Discussion
Several aspects of melanocortin receptor biology are based on comparative analysis of paralogous genes. In particular, recognition and characterization of the AgRP-MC3R and -MC4R pathways was based on hypotheses and/or DNA sequences that emerged from studying the ASIP-MC1R pathway; the ability of ASIP to antagonize the MC4R serves as the basis for the still widespread use of animals that ubiquitously express ASIP (as in the Ay or Avy mutations) as an obesity model. Nonetheless, there is a fundamental difference between the action of AgRP and ASIP; the N-terminal domain of AgRP functions as a prodomain [6], while the N-terminal domain of ASIP serves as a ligand for an accessory receptor [8, 23]. Our work provides both biophysical and evolutionary insight into this difference.
Clues that full-length AgRP might represent a proprotein with reduced activity compared to a C-terminal fragment were apparent from initial studies of recombinant AgRP produced by insect cells [2]; shorter forms generally exhibited greater antagonist activity than longer forms when tested on Xenopus melanophores. However, these [2, 23] and additional studies [1, 6, 9, 10, 24] are complicated by the use of heterogeneous mixtures of partially purified recombinant protein [2, 23], and/or by comparing full-length AgRP and C-terminal AgRP prepared and folded as separate polypeptides, often from different sources. The current approach, in which we first folded full-length AgRP and then released its C-terminal domain by proteolysis under conditions that quench any disulfide bond rearrangement, controls for any differences in peptide origin or preparation and therefore demonstrates unequivocally that the N-terminal domain in full-length AgRP inhibits the potency of the MCR-binding domain AgRP(83–132). The NMR experiments reported here show that the entire N-terminal domain of AgRP up through residue 86 is flexible and unstructured and thus certainly susceptible to proteolytic cleavage. The mechanism of N-terminal inhibition prior to proteolysis is likely to involve changes in the local environment of positively charged Arg89 and His91, probably via a transient collapse of a negatively charged segment of the N-terminal domain (residues 57–78) against the C-terminal surface involved in MCR docking, which, in turn, interferes with receptor binding. Thus, a primary biological role of the AgRP N-terminal domain is to negatively regulate its activity such that potent melanocortin antagonism is uncovered only after proteolytic processing.
This conclusion provides a new aspect to our understanding of melanocortin biology, in which the function of AgRP and ASIP in energy balance and pigmentation, respectively, represent not only different patterns of gene expression, but also different biochemical mechanisms mediated by paralogous N-terminal domains. Several considerations suggest that both the regulatory and the structural differences between AgRP and ASIP reflect so-called “subfunctionalization,” in which duplicated genes are preserved during evolution due to the partitioning of different functions between the duplicates [25, 26]. Both melanocortin receptors and their antagonistic ligands must have arisen early during vertebrate evolution, since orthologs of ASIP and AgRP, and of MC1R and MC4R, are found within vertebrate, but not protochordate, genomes. Function of the ASIP N-terminal domain as a ligand for an accessory receptor is likely to have a similar origin, since the patterns of evolutionary constraint among mammalian and vertebrate orthologs are nearly identical.
Taken together, these data suggest a model in which a single (antagonistic) ligand and its cognate receptor originally served to control both pigmentation and energy balance in an ancestral vertebrate several hundred million years ago, perhaps as a means to coordinately regulate metabolic rate and body temperature by means of radiant energy. For example, inhibition of CNS melanocortin signaling in a modern poikilotherm would probably decrease thyroid and reproductive function, while inhibition of pigment melanocortin signaling would probably decrease the absorption of radiant energy, causing a reduction of body temperature. Both the behavioral and the thermoregulatory responses would be adaptive during starvation and could help to explain the somewhat surprising connection between pigmentation and energy balance observed today.
Significance
Agouti (ASIP) and Agouti-related protein (AgRP) function as ligands for melanocortin receptors that control pigmentation (MC1R) and body weight (MC3R and MC4R), respectively, and diverged from a common ancestor early in vertebrate evolution. Our previous work demonstrated that the carboxy-terminal regions of ASIP and AgRP, which bind directly to melanocortin receptors, have nearly identical three-dimensional structures. A major challenge in the field has been to understand the biochemical and biophysical basis for differences in ASIP and AgRP action. We demonstrate that the amino-terminal region of AgRP acts as a prodomain that both inhibits function and is easily cleaved from the full-length protein, and we provide a biophysical explanation as to the nature of the inhibition. By contrast, the homologous region of ASIP plays a completely different role, serving as a ligand for an accessory receptor that is required for in vivo activity. We interpret our findings in the context of molecular evolution, demonstrating that the carboxy-terminal domains of both proteins have similar levels of constraint while the amino-terminal domains differ dramatically. These observations contradict the long-held view that biochemical mechanisms of action for ASIP and AgRP are identical, and they have direct implications both for interpreting previous work [3] and for current approaches to obesity drug development.
Experimental Procedures
Bacterial Expression, Purification, and Folding of Full-Length AgRP
The MKd5-AgRP(25–132) cDNA was created from the AgRP(1–132) plasmid in the bluescript vector with a pair of synthetic oligonucleotides, ACTGTAAGCTCATATGAAAGCCCCCATGGAGGGC and CGC TCTAGAACTAGTGGATCCTGTTGG, which encode a BAM-H1 and NDE1 cleavage site and Met Lys residues on the N terminus, respectively, and amplify AgRP(25–132). To include a Factor Xa or enterokinase- cleavable 10x His tag, the MKd5-AgRP cDNA was ligated into either the Pet 16B or Pet 19a cloning vectors, respectively (Invitrogen, San Diego, CA). The expression vectors were used to transform BL21 E. coli. Cells were grown to an optical density of 0.6 in LB media with ampicilin at 37°C and were then induced with 50 mM IPTG. The cell pellet was collected by centrifugation and was suspended in lysis buffer (20 mM Tris [pH 7.5], 500 mM NaCl, 0.1% Triton, 10% glycerol) and sonicated for 4 min. The suspension was centrifuged at 15,000 × g, and both the soluble and insoluble fractions were tested for the presence of AgRP by using PAGE gels. 10xHis-MKd5-AgRP was found exclusively in inclusion bodies. These inclusion bodies were solubilized in 6Mguanidine hydrochloride, 10 mM Tris, 1 mM DTT, and 15 mM imidazole, at pH 8.0. Proteins were purified by using a Ni-NTA superflow column, were washed with 50mMimidazole, and were eluted with 500mMimidazole, followed by reverse-phase HPLC on a C18 column. His tag removal was carried out with recombinant enterokinase or Factor Xa (Invitrogen) in a modified cleavage buffer consisting of 20 mM MES, 50 mM NaCl, and 2 mM CaCl2, at pH 6.4 over a 4 hr period. To fold MKd5-AgRP, 1 mg lyophilized material was dissolved in 100 μl DMSO, followed by the addition of 10 ml folding buffer, which contained 2.5 M guanidine hydrochloride, 100 mM Tris, 0.2 mM oxidized glutathione, and 1mMreduced glutathione, at pH 8.0. Folding was usually complete after 2 hr. HPLC showed the disappearance of the peak corresponding to the fully reduced protein, followed by the gradual emergence of a single HPLC peak that eluted at an earlier retention time than the reduced material. Mass spectrometry of the HPLC peak corresponding to folded MKd5-AgRP showed the expected loss of 10 Da relative to the fully reduced material, due to the formation of five disulfides bridges.
cAMP Assays
cAMP assays (performed with the Amersham Pharmacia Biotech cAMP assay kit TRK 432) and 125I-NDP-MSH displacements were performed as previously described [1, 27]. HEK293 cells stably transfected with the human melanocortin receptor hMC4R were used for these studies. All experiments were performed in triplicate, and data were analyzed with Graphpad Prism (Graphpad Software, San Diego, CA). AgRP(86–132) was purchased from Peptide Institute, Inc.
NMR Sample Preparation
The structure of C-terminal AgRP(87–132) was previously solved at pH 5.0, which produces the greatest solubility (>1.0 mM) [13]. However, under these conditions, full-length AgRP exhibits linewidths and T1 and T2 relaxation times inconsistent with monomeric protein, suggesting that the protein exists as soluble oligomers. Screening a wide variety of conditions (pH, temperature, salt, cosolvents) by NMR revealed that the protein was monomeric only when small amounts of organic solvent were added, specifically 4% acetonitrile w/v and 3% DMSO w/v at 37°C.
The NMR sample for all spectra acquired for full-length AgRP contained ~200 μM protein in 50 mM acetate-d5 buffer (pH 5.0) with 10% D2O, 4% w/v acetonitrile-d3, and 3% w/v DMSO-d6. NMR spectra for the C-terminal domain of AgRP contained the same buffer conditions as the full-length protein with 50 μM protein concentration. All solutions contained 0.005% sodium azide to inhibit bacterial growth. All isotopically enriched reagents were purchased from Cambridge Isotope Laboratories (Cambridge, MA).
NMR Spectroscopy
Spectra were obtained on Varian 600 MHz spectrometers, with and without a cryoprobe. NMR data were acquired at 37°C. Resonance assignments were made by using the following spectra: 2D 15NHSQC, 3D 15N-(NOESY)-HSQC with 100 ms mixing time, and 15N-(TOCSY)-HSQC with 80 ms mixing time. Spectra were acquired by using spectral widths of w1 = 2200, w2 = 10000, and, when applicable, w3 = 10000. All experiments used sensitivity enhanced gradient coherence selection. Heteronuclear 15N[1H]-NOE experiments were performed under the same conditions as those described above and with a relaxation delay of 3.0 s and a 3.0 s proton saturation.
Structure Calculations
The model of full-length AgRP was made by using the distance restraints previously determined for the C-terminal domain [13]. The N-terminal residues were left with no distance restraints. 500 CYANA structures were calculated, and, of these, one of the lowest-energy structures was selected for the model. All structure representations were developed with the aid of MOLMOL [28].
Evolution Structure Function Analysis
Local evolutionary rates over ASIP and AgRP were estimated by first generating a multiple sequence alignment with Probcons (http://probcons.stanford.edu/) [29] and deriving a phylogenetic tree by using SEMPHY (http://compbio.cs.huji.ac.il/semphy/) [30], and then, while holding the tree topology fixed, calculating substitutions per site by using PROTPARS (http://evolution.genetics.washington.edu/phylip.html) [31]. These single-site rate values were smoothed by using sliding-windows weighted averaging: in each 17-position-wide window, the relative weight was highest for the value at the center position, and it decreased linearly on either side to the edge of the window. The resulting value was assigned to the position in the protein corresponding to the center of the window. The rate values were then converted to relative constraint by normalizing to a range between 0 and 1, and subtracting from 1.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant DK64265 (G.L.M) and by instrumentation grants from the National Science Foundation (CHE-0342912) and NIH (RR19918) in support of the University of California, Santa Cruz (UCSC) 600 MHz NMR. The authors thank Professors Thomas James and Mark Kelly of University of California, San Francisco, for generously providing access to their 600 MHz cryoprobe NMR. Darren Thompson is gratefully acknowledged for advice and helpful comments on the manuscript.
Footnotes
Supplemental Data include AgRP(86–132)/MC4R pharmacological data, analysis of 15N chemical shifts, HPLC retention times of AgRP constructs, C-terminal NOEs from full-length AgRP consistent with the β sheet observed in AgRP(87–132), and 15N chemical shifts and are available at http://www.chembiol.com/cgi/content/full/13/12/1297/DC1/.
References
- 1.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SB, Gantz I. Characterization of Agouti-related protein binding to melanocortin receptors. Mol Endocrinol. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 2.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 3.Reizes O, Lincecum J, Wang Z, Goldberger O, Huang L, Kaksonen M, Ahima R, Hinkes MT, Barsh GS, Rauvala H, et al. Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell. 2001;106:105–116. doi: 10.1016/s0092-8674(01)00415-9. [DOI] [PubMed] [Google Scholar]
- 4.Reizes O, Clegg DJ, Strader AD, Benoit SC. A role for syndecan-3 in the melanocortin regulation of energy balance. Peptides. 2005;27:274–280. doi: 10.1016/j.peptides.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 5.Strader AD, Reizes O, Woods SC, Benoit SC, Seeley RJ. Mice lacking the syndecan-3 gene are resistant to diet-induced obesity. J Clin Invest. 2004;114:1354–1360. doi: 10.1172/JCI20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creemers JW, Pritchard LE, Gyte A, Le Rouzic P, Meulemans S, Wardlaw SL, Zhu X, Steiner DF, Davies N, Armstrong D, et al. Agouti-related protein is post-translationally cleaved by proprotein convertase 1 to generate AGRP83–132: interaction between AGRP83–132 and melanocortin receptors cannot be influenced by syndecan 3. Endocrinology. 2006;147:1621–1631. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- 7.Pritchard LE, White A. Agouti-related protein: more than a melanocortin-4 receptor antagonist? Peptides. 2005;26:1759–1770. doi: 10.1016/j.peptides.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 8.He L, Gunn TM, Bouley DM, Lu XY, Watson SJ, Schlossman SF, Duke-Cohan JS, Barsh GS. A biochemical function for attractin in agouti-induced pigmentation and obesity. Nat Genet. 2001;27:40–47. doi: 10.1038/83741. [DOI] [PubMed] [Google Scholar]
- 9.Rosenfeld RD, Zeni L, Welcher AA, Narhi LO, Hale C, Marasco J, Delaney J, Gleason T, Philo JS, Katta V, et al. Biochemical, biophysical, and pharmacological characterization of bacterially expressed human agouti-related protein. Biochemistry. 1998;37:16041–16052. doi: 10.1021/bi981027m. [DOI] [PubMed] [Google Scholar]
- 10.Quillan JM, Sadee W, Wei ET, Jimenez C, Ji L, Chang JK. A synthetic human Agouti-related protein-(83–132)-NH2 fragment is a potent inhibitor of melanocortin receptor function. FEBS Lett. 1998;428:59–62. doi: 10.1016/s0014-5793(98)00487-6. [DOI] [PubMed] [Google Scholar]
- 11.Mielke SP, Krishnan VV. An evaluation of chemical shift index-based secondary structure determination in proteins: influence of random coil chemical shifts. J Biomol NMR. 2004;30:143–153. doi: 10.1023/b:jnmr.0000048940.51331.49. [DOI] [PubMed] [Google Scholar]
- 12.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 13.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High resolution NMR structure of the chemically-synthesized melanocortin receptor binding domain AGRP(87–132) of the Agouti-related protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 14.Snyder DA, Chen Y, Denissova NG, Acton T, Aramini JM, Ciano M, Karlin R, Liu J, Manor P, Rajan PA, et al. Comparisons ofNMR spectral quality and success in crystallization demonstrate that NMR and X-ray crystallography are complementary methods for small protein structure determination. J Am Chem Soc. 2005;127:16505–16511. doi: 10.1021/ja053564h. [DOI] [PubMed] [Google Scholar]
- 15.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 16.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 17.Dayie KT, Wagner G. Relaxation-rate measurements for 15N-1H groups with pulsed-field gradients and preservation of coherence pathways. J Magn Reson A. 1994;111:121–126. [Google Scholar]
- 18.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 19.Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SB, Gantz I, Millhauser GL. NMR structure of a minimized human agouti related protein prepared by total chemical synthesis. FEBS Lett. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- 20.Millhauser GL, McNulty JC, Jackson PJ, Thompson DA, Barsh GS, Gantz I. Loops and links: structural insights into the remarkable function of the agouti-related protein. Ann N Y Acad Sci. 2003;994:27–35. doi: 10.1111/j.1749-6632.2003.tb03159.x. [DOI] [PubMed] [Google Scholar]
- 21.Wilczynski A, Wang XS, Joseph CG, Xiang Z, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, et al. Identification of putative agouti-related protein(87–132)-melanocortin-4 receptor interactions by homology molecular modeling and validation using chimeric peptide ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 22.Simon AL, Stone EA, Sidow A. Inference of functional regions in proteins by quantification of evolutionary constraints. Proc Natl Acad Sci USA. 2002;99:2912–2917. doi: 10.1073/pnas.042692299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ollmann MM, Barsh GS. Down-regulation of melanocortin receptor signaling mediated by the amino terminus of Agouti protein in Xenopus melanophores. J Biol Chem. 1999;274:15837–15846. doi: 10.1074/jbc.274.22.15837. [DOI] [PubMed] [Google Scholar]
- 24.de Rijke CE, Jackson PJ, Garner KM, van Rozen RJ, Douglas NR, Kas MJ, Millhauser GL, Adan RA. Functional analysis of the Ala67Thr polymorphism in agouti related protein associated with anorexia nervosa and leanness. Biochem Pharmacol. 2005;70:308–316. doi: 10.1016/j.bcp.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 25.Li WH, Yang J, Gu X. Expression divergence between duplicate genes. Trends Genet. 2005;21:602–607. doi: 10.1016/j.tig.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Massingham T, Davies LJ, Lio P. Analysing gene function after duplication. Bioessays. 2001;23:873–876. doi: 10.1002/bies.1128. [DOI] [PubMed] [Google Scholar]
- 27.Jackson PJ, McNulty JC, Yang YK, Thompson DA, Chai B, Gantz I, Barsh GS, Millhauser GL. Design, pharmacology and NMR structure of a minimized cystine knot with Agouti-related protein activity. Biochemistry. 2002;41:7565–7572. doi: 10.1021/bi012000x. [DOI] [PubMed] [Google Scholar]
- 28.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 29.Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman N, Ninio M, Pe’er I, Pupko T. A structural EM algorithm for phylogenetic inference. J Comput Biol. 2002;9:331–353. doi: 10.1089/10665270252935494. [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J. PHYLIP. 2005 ( http://evolution.genetics.washington.edu/phylip.html)
- 32.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 33.McNulty JC, Jackson PJ, Thompson DA, Chai B, Gantz I, Barsh GS, Dawson PE, Millhauser GL. Structures of the agouti signaling protein. J Mol Biol. 2005;346:1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.