

Abstract
Converting one enzyme into another is challenging due to the uneven distribution of important amino acids for function in both protein sequence and structure. We report a strategy for protein engineering allowing an organized mixing and matching of genetic material that leverages lower throughput with increased quality of screens. Our approach successfully tested the contribution of each surface exposed loop in the trypsin fold alone and the cooperativity of their combinations towards building the substrate selectivity and Na+-dependent allosteric activation of the protease domain of human coagulation factor Xa into a bacterial trypsin. As the created proteases lack additional protein domains and protein co-factor activation mechanism requisite for the complexity of blood coagulation, they are stepping-stones towards further understanding and engineering of artificial clotting factors.
Keywords: cooperativity, allostery, protein engineering, substrate selectivity, blood coagulation, protease, mutagenesis
Introduction
Trypsin-like serine proteases, like many eukaryotic proteins, are challenging bioengineering targets despite their well-documented structure-function relationships and evolutionary success. 1; 2; 3; 4 The biological utility of the trypsin fold is evident by the varied roles played by these proteases in hemostasis, fibrinolysis, immunity, development, apoptosis, and homeostasis – processes requiring both catalytic efficiency and substrate selectivity. Pioneering work in protein redesign showed alteration of the primary specificity of trypsin into chymotrypsin 5 or elastase 6 involved an unexpected degree of difficulty and a number of amino acid substitutions outside those directly contacting the substrate. Indeed, such necessity for significant change in protein sequence to build the function of one protease into another appears to be a general feature of the trypsin family.
Trypsin-like serine proteases are comprised of two six-stranded Greek key β-barrels lying perpendicular to and on top of one another with the active site cleft between them (Figure 1A).7 Eight surface exposed loops circle the active site in the trypsin fold and are potentially involved in macromolecular interactions and allosteric activation by mono- or divalent cations or protein co-factors.8 Sequence analysis of the trypsin family reveals a high correlation between regions of sequence variability and positions where these loops are located - a feature common to all protein families.9 In contrast, the inner hydrophobic core is highly conserved. For the example protease pair used in this study, the core of both human coagulation factor Xa (FXa) and the bacterial Streptomyces griseus trypsin (SGT) are highly similar (Figure 1B).11; 12; 13 However, the surface differences between these two proteins are sufficiently extensive to warrant the use of a color-coded pie diagram as a notation system rather than writing out the 54 individual point mutations (Figure 1C). The considerable evolutionary distance between these two proteins are only one problem amongst many for interconverting their properties.
Figure 1. The problem addressed by combinatorial design.

(A) The trypsin fold is comprised of two Greek-key β-barrels with eight of the eleven surface exposed loops encircling the catalytic triad and numbered according to the chymotrypsin convention. (B) Like most S1A peptidases, the target FXa (PDB 2BOK) and bacterial trypsin (PDB 1OS8) share a common hydrophobic core. 12; 33 However, the eight loops that surround the active site differ completely between them (see Figure 2). (C) A color coded pie digram is a useful representation of fragment combinations as the changes introduced into SGT are extensive and involve 4 to 15 amino acid substitutions in each loop.
Issues generally encountered upon attempting to build eukaryotic proteins and other restraints peculiar to the trypsin family have restricted progress in their engineering. We have been engaged in converting a bacterial trypsin into an artificial blood coagulation factor 11; 12; 13 and hindered by many of these difficulties that limit throughput of any design strategy. In particular, trypsin-like proteases are rarely found in bacterial genomes and not readily expressed in soluble form by bacteria.4 The inability of these proteases to spontaneously fold is due, in part, to the presence of propeptides that assist protein folding yet whose presence restricts activity analysis of recombinant material directly without zymogen activation.14 High-throughput screening for protease variants with beneficial properties is also difficult because the enzymes start out with better kinetic parameters - higher kcat and lower KM - than the target for all possible substrates owing to their digestive function.15 In our pursuit, the changes to build functionality are presumably considerable as clotting factors exhibit cooperative energetic between distal sites whose origins and connectivity are poorly defined.16 For example, alanine-scanning mutagenesis of clotting factors illustrates that many residues spread throughout the protease domain are involved in function.17; 18; 19; 20 Moreover, clotting factors are multidomain proteins whose N-terminal domains play vital roles in phospholipid binding and co-factor recognition,21 which are aspects not examined in this study yet whose contributions towards coagulation can be more thoroughly investigated by the protease domains built using our approach. To minimize the impact of the technical limitations and enable progress, we devised a new method for protein engineering that allows grafting surface loops at multiple sites in a polypeptide to simplify the search for gain-of-function mutations. The method is generally applicable to any gene, easy to perform, works with all plasmids and expression systems and can be modified for different goals.
Our combinatorial design strategy efficiently manipulates segments of genetic material at one or more positions in a gene and tests their combination. In contrast to other approaches, the strategy adopts a lower throughput and focuses upon increasing the quality of that screened. By testing combinations of sequence fragments both additive and cooperative relationships between sites in a protein are probed. Our strategy enabled us to test the contribution of each surface exposed loop alone or in different combinations in the trypsin fold towards substrate selectivity and Na+-dependent allosteric activation. In turn, the approach created an FXa-like protease domain from the bacterial homolog SGT. Importantly, gains in function are shown to arise from the cooperativity of three distal sites in the folded protein encoded in five loops and are not solely due to protein-protein interfaces involved directly in substrate binding. These mutations would therefore be difficult to find using other protein engineering approaches. Despite the significant advances engineered into the scaffold protein, the resulting protease lacks the coagulation factor Va and phospholipid binding and activation mechanism, which is provided, at minimum, by the associated N-terminal EGF and γ-carboxylation domains of FXa and the molecular basis for these processes remain poorly understood.21 Our designs are therefore stepping-stones on the road to the creation of more complex entities.
Results
Definition of Sequence Fragments
Sequence fragments were defined by the absence of common polypeptide backbone architecture using the available structures of both SGT and FXa.22; 23 Changes in sequence were chosen to not include the hydrophobic core of the bacterial protein in an effort to maintain spontaneous folding of the modified SGT in the bacterial expression system used. As observed with most trypsin-like proteases, the protease domain of FXa was not able to be produced in soluble active form in this expression system to enable direct comparison. Eight fragments were designed from the sequence of human FXa and encoded into oligonucleotides where both the 5′ and 3′ ends were of sufficient length and composition to prime PCR-based amplification of the SGT gene and enable bidirectional amplification during PCR (Table 1). Our engineering problem was therefore reduced into eight components separated fairly evenly across the SGT gene (Figure 2).
Table 1.
Oligonucleotide primer design for the eight fragments to build FXa into a bacterial trypsin. Changes in sequence are shown in bold and are flanked by 5′ and 3′ ends of sufficient length and composition to enable PCR-based amplification. Two primers were necessary for the 70-loop fragment due to its proximity to the 60-loop. Two generic primers iSGT-f and iSGT-r designed from the beginning and end of the SGT were also used.
| Name | Sequence |
|---|---|
| FX35-f | PFMVRLINEENEGFCGGALY 5′-CCCTTCATGGTCCGGCTCATCAATGAAGAAA ATGAAGGTTTTTGCGGCGGCGCCCTCTAC-3′ |
| FX60-f | TAAHCVYQAKSITATG 5′-ACCGCCGCCCACTGCGTGTATCAAGCGAAGT CGATCACGGCCACCGGC-3′ |
| FX70-1-f | NTSITARVGDRNTEQEEGGEVKARST 5′-AACACCTCGATCACGGCCAGAGTCGGAGATA GAAACACTGAGCAAGAGGAGGGTGGTGAGGTCAA GGCCCGCTCCACC-3′ |
| FX70-2-f |
AKSITARVGDRNTEQEEGGEVKARST 5′-GCGAAGTCGATCACGGCCAGAGTCGGAGATA GAAACACTGAGCAAGAGGAGGGTGGTGAGGTCAA GGCCCGCTCCACC-3′ |
| FX99-f | VLQAPGFTKETYGKDWALI 5′-GTCCTCCAGGCCCCCGGCTTCACTAAGGAGA CCTACGGCAAGGACTGGGCGCTCATC-3′ |
| FX145-r | TVAGWGRTHEKGSQQRY 5′-GTAGCGCTGCTGGCTGCCCTTCTCATGAGTT CTGCCCCAGCCGGCGACGGT-3′ |
| FX170-r | DAACRSSSSFILVANEM 5′-CATCTCGTTGGCCACGAGGATGAAGCTGGAC GAGGAGCGGCAGGCGGCGTC-3′ |
| FX186-r | MICAGYDTKQEDTCQGD 5′-GTCACCCTGGCAGGTGTCCTCCTGCTTGGT GTCGTATCCGGCGCAGATCAT-3′ |
| FX220-r | GEGCARKGKYGVYTEV 5′-GACCTCGGTGTAGACACCATATTTGCCCTT CCGGGCGCAGCCCTCGCC-3′ |
| iSGT-f | PFMVRLS 5′-CCCCTTCATGGTCCGGCTCT-3′ |
| iSGT-r | TFASAI 5′-GATGGCGGAAGCGAAGGT-3′ |
Figure 2. Sequence alignment of the protease bearing all eight fragments (SGX) with the SGT starting scaffold and the target protease FXa.

Positions indicated are based upon the chymotrypsin numbering system. Combinatorial enzyme design manipulates 54 amino acid substitutions throughout the entire SGT gene and significantly alters loop length and composition. Although the fully substituted protease was produced in small amounts and was not studied in detail, the discrete and distributed nature of the changes introduced into the SGT gene highlights the utility of combinatorial enzyme design.
Combinatorial Enzyme Design
To graft sequence loops in a parsimonious manner, we were inspired by the mechanisms of natural competence in gram-positive bacteria whose preference for long linear strands of DNA contrasts the prevailing use of circular plasmids (Figure 3A).24 Oligonucleotide primers where one or both contain a mutagenic segment first generate a PCR amplicon, which is purified and added in excess to a second reaction containing plasmid bearing the target gene previously linearized by a restriction endonuclease. Importantly, the initial PCR product spans the endonuclease cleavage site used to linearize the plasmid (Figure 3B). Thermocycling with a long and accurate DNA polymerase merges and amplifies the amplicon and plasmid into larger concatamers as the oligonucleotides used in the first amplification step enable bidirectional expansion during the reaction, yet still through 3′ nucleotide addition by the polymerase. The second PCR reaction directly transforms competent bacteria. Following uptake and outgrowth the mutated plasmid is repaired to circular form by the bacteria and then replicates through normal mechanisms. To maximize efficiency, both the linearized plasmids and PCR amplicons bearing the desired substitutions are retained at each step to allow rapid and iterative combination of loop fragments in a cost-effective manner. For soluble expression of active recombinant protein we used naturally competent Bacillus subtilis as expression host, yet found the concatamers could transform chemically induced competent states of E. coli likely owing to the generality of the mechanism of plasmid replication. Hence, the method may be useful for pre-processing of a gene before use in other expression systems. Without need for additional reagents or processing steps the DNA generated can be used iteratively to move efficiently through the sequence space between scaffold and target protein.
Figure 3. Combinatorial enzyme design.

(A) One or more fragment-bearing primers are used to create a PCR product that subsequently drives plasmid concatamerization when provided a linearized plasmid template. The long linear product is capable of directly transforming competent bacteria, which repair the plasmid to circular form based on the mechanism of plasmid replication. Genetic material at each step is retained for iterative re-use to create many fragment combinations quickly. (B) In the design of FXa, eight fragments corresponding to surface exposed loops are encoded in oligonucleotide primers (Table 1). These primers span the entire SGT gene whose protein sequence is numbered from 16 to 245 according to the chymotrypsin convention. A variety of naturally occurring unique restriction endonuclease sites can be used to linearize the plasmid bearing the SGT gene for concatamerization. (C) The eight primers combine to generate single and double fragment substituted proteases (I) whose plasmids can then be purified, linearized with a restriction enzyme and the material generated from the first twenty four PCR reactions used to build double, triple and quadruple fragment substituted constructs (II). Importantly, all eight sites are tested in a variety of combinations to identify important intermediates and their cooperativity. These sites can be investigated with further fragment exchange in a third round thus simplifying the navigation of sequence space.
Our combinatorial approach generates extensive targeted diversity in a short period of time. In this example, the eight loops can be combined in 256 different ways, yet the method allows one to navigate sequence space with sufficient dexterity to require sampling less of these possibilities as intermediates hinted at the importance of key sites. In full diversification mode, two rounds of mutagenesis can use the eight primers in this example to produce 8 single, 28 double, 48 triple and 36 quadruple fragment substitutions (Figure 3C). These 120 combinations occur at eight different positions spread throughout the target gene and can be generated in a short period of time. These intermediates can be used in a further round to build in all eight sites or more thoroughly investigate combinations that appear beneficial. We have also found that more extensive variation can be generated more rapidly by multiplexing the PCR reaction in the first step or using more forward and reverse primers, including ones bearing sequence degeneracy (data not shown).
Combinatorial enzyme design efficiently maps the contribution of each surface loop toward substrate selectivity and Na+-dependent allosteric activation in FXa when grafted on the SGT scaffold (Figure 4A). Forty four constructs bearing various fragment combinations were sorted according to their hydrolysis of a representative pair of chromogenic paranitroanilide (pNA) derivatives of tripeptide substrates (H-D-Arg-Gly-Arg-pNA and H-D-Phe-Gly-Lys-pNA) in the presence or absence of the allosteric activator Na+. An additional ten constructs were also built but could not be examined due to very low or absent levels of expression, which is not surprising given the general difficulty of producing active trypsin-like proteases in bacteria. The ratio of activity towards the two substrates was measured directly from the culture supernatant using equal volumes to account for differences in expression levels between mutant proteases and guide our choice of what proteases to purify and characterize in more detail.
Figure 4. Properties of fragment substituted proteases.

(A) Iteration produces a library whose organized diversity at multiple sites could not be achieved as readily by conventional approaches. Of eight possible loop fragments incorporated into the SGT scaffold, three (170, 186 and 220) create Na+ dependent allostery (top panel shows the ratio of initial velocities in NaCl relative to LiCl at 250 mM) and five (35, 99, 170, 186 and 220) maximize substrate selectivity between the primary substrate binding pocket and extended active site as determined by the two substrates d-Arg-Gly-Arg-pNA and d-Phe-Gly-Lys-pNA that differ at P1 and P3 (middle panel illustrates the ratio of initial rates of hydrolysis of each substrate at 50 μM). The fragments responsible overlap and these properties do not arise from other fragment combinations (bottom panel shows the product of the two ratios). (B) When mapped into two dimensions the simplicity of the output illustrates the utility of combinatorial enzyme design for navigating the sequence space between two distantly related enzymes. Although there are many possible combinations of fragments, most are not beneficial and only two pathways are observed for gain of function. In the first, Na+-dependent allosteric activation emerges from the 170/186/220 combination leading to intermediates with very low activity 11 and dramatic gains in selectivity and activity following mutagenesis of the 99-loop 13. In the second, the 220 and 35 fragment combination builds selectivity iteratively with maximal selectivity achieved with the late emergence of Na+ activation.
Selectivity and Allostery of Engineered Proteases
Mutagenesis of trypsin active site cleft residues does not generate substrate selectivity but requires a defined set of additional fragments from multiple places in the gene. On the basis of known structural data, loops at positions 60, 99 and 170 are the only ones capable of directly contacting small chromogenic substrates in all trypsin-like proteases but their inclusion alone or with most other fragments produce proteases that are neither selective nor particularly active and fall into the lowest of three selectivity strata observed in the library of proteases.25 Notably, lowered selectivity of the 99/170 construct can be considered a threshold for promiscuity, which is known to be an important barrier to cross in protein redesign.26 Adding the 220-loop fragment to create 99/170/220 improves substrate selectivity 15-fold relative to the promiscuous 99/170 mutant suggesting an important role for the composition of the 220-loop. With respect to allosteric activation, the 170/186/220 loop combination is the minimal fragment set to engineer Na+-dependent activation. This finding agrees with our previous observations showing the need for absence of large hydrophobic residues at position 172 provided a proper Na+ binding environment and excludes contributions from other loops such as the 145-loop on the other side of the cation binding site.11 Selectivity of the active site cleft and Na+-site fragment-bearing proteases becomes maximized 185-fold more than the promiscuous 99/170 mutant by addition of the 35-loop fragment to build 35/99/170/186/220 even in the absence of loops that would directly link these substitutions on the surface of the folded protein. The observed gain in selectivity is notable as the 35-loop is on the opposite side of the molecule in exosite I and does not interact with the chromogenic substrates used in the assay. Moreover, the beneficial effect only appears when the 220-loop fragment is also present with the 35-loop and maximized with the presence of Na+-dependent allosteric activation. Hence, the gain of function is the result of cooperativity between distal sites in the engineered protease.
Five fragments encoding for the 35-, 99-, 170-, 186- and 220-loops of FXa are necessary for and contributed to maximal selectivity towards tripeptide substrates. Addition of the 70-loop to this five fragment set reduced selectivity for reasons that are unclear, yet this loop plays a role in co-factor and substrate recognition in some trypsin family members.7 Importantly, simple addition of the loops “above the active site” in the standard view of the protease domain – 35, 60, 70, 99, and 170 – that are known to interact with small and larger substrates and co-factors does not generate substrate selectivity. Changes introduced follow two pathways for gain of function with a converging endpoint bearing 25 of the 54 possible amino acid substitutions (Figure 4B). The Na+-binding site and the 220-loop in particular is crucial for the communication between all five fragments whose positions span the entire gene sequence and at three clusters separated 25 to 30 Å from one another in the folded protein. Separation between the five loops in both sequence and structure and their cooperativity explains why mutagenesis of the active site cleft alone or by larger chimeras fails to introduce selectivity and activity of FXa into homologous proteases.27; 28; 29; 30 Designs including fragments adjacent to these regions increase neither selectivity nor activity, reduced the yield of recombinant protein and impaired their analysis. Guided by the preliminary assessment of selectivity using chromogenic tripeptide substrates, we purified and characterized several proteases bearing combinations of the five fragments and compared them with FXa to determine if the observed cooperativity extends to the recognition of larger substrates and inhibitors.
Macromolecular Selectivity of Engineered Proteases
Properties of fragment-exchanged proteases towards chromogenic substrates are mirrored and more dramatic with larger substrates and inhibitors. Only the engineered protease bearing all five fragments is selective towards oligopeptide substrates 25 to 30 amino acids in length derived from natural substrates of clotting factor proteases. Substrates tested include the extracellular region of protease-activated receptors 1, 2, 3 and 4 and the three sites required to activate coagulation factor V (Figure 5). These peptides are common diagnostics of naturally occurring clotting factors and are stringent measures of selectivity given their lack of structure. Overnight digestion of these peptides is a useful, if qualitative, overview of the selectivity introduced. FXa cleaves after two of the potential twenty five arginine or lysine C-terminal peptide bonds in the seven oligopeptides. The five-fragment protease cleaved, and more importantly did not cleave, these oligopeptide substrates in a fashion identical to FXa, including clipping the two factor V peptides at the same site to a similar extent.
Figure 5. Properties of fragment-engineered proteases towards larger substrates compared to FXa.
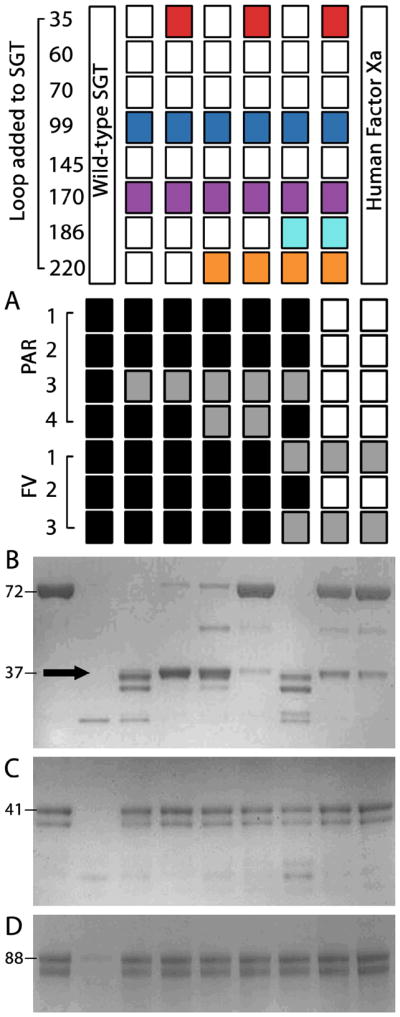
The top panel indicates the loop added to the SGT scaffold with the rightmost lane resulting from the use of human FXa. All analyses were conducted at equal concentrations of protease and excess substrate. (A) Summary of the extensive hydrolysis of seven representative oligopeptide substrates. Black boxes indicate cleavage at two or more sites, grey boxes note single peptide bond hydrolysis and empty boxes denote no noticeable cleavage. Each protease (10 nM) was incubated with oligopeptide (10 μM) for 24 hrs then resolved by reverse phase HPLC. The wild-type trypsin readily cleaves after any arginine or lysine side chain. Addition of the five fragments except in any combination improves this selectivity, yet only when all are present does the selectivity profile of FXa become recapitulated. For the two factor V (FV) peptides, FXa and the five fragment substituted protease cleaved at the same site and near identical rate. Limited proteolysis of (B) prothrombin, (C) protein C, and (D) plasminogen by engineered proteases compared to the starting scaffold and human FXa. The left lane of each SDS-PAGE gel shows the undigested zymogen for comparison and their molecular weights in kDa. Equal concentration of proteases (100 nM, not visible on the gel) were allowed to hydrolyze each zymogen (2 μg) for 1 hour at 37°C in 25 mM Tris pH 8, 10 mM CaCl2, 200 mM NaCl, and 0.1% PEG 8000 before SDS-PAGE analysis. Only the 35/99/170/186/220 fragment-substituted protease mimics the activity and selectivity of FXa and liberates prethrombin-2 (black arrow) with an equivalent rate.
Loop substitution had a dramatic and unexpected effect on the inhibition by antithrombin. Unlike FXa, the engineered proteases are remarkably resistant to inhibition by this serpin (Figure 6A). The five fragment substituted protease is 625-fold less reactive to antithrombin than SGT and 40-fold less reactive than FXa. The discrepancy between the magnitude of gain in selectivity and inhibition hints at the evolutionary pressure for balanced inhibition in vivo for proper blood coagulation that was not part of our screening strategy and did not emerge from the changes made to the trypsin scaffold. Time-dependent hydrolysis of zymogen protein precursors further confirms the necessity for multisite engineering to create selectivity similar to FXa.
Figure 6. Inhibition and activity of engineered proteases compared to FXa.

The left panel indicates the loop added to the SGT scaffold with the bottom line showing the values derived using human FXa. (A) Rates of association of the fragment engineered proteases with the serpin antithrombin III are influenced upon loop addition. However, the engineered proteases are remarkably resistant to inhibition to an extent more than FXa. (B) Activity of constructs towards the representative FXa substrate d-Arg-Gly-Arg-pNA. Despite introduction of various degrees of 25 amino acid substitutions at five sites, the proteases retain activity (kcat) similar to both the wild-type scaffold and FXa.
Limited proteolysis of the zymogens plasminogen, protein C, which are not FXa substrates, and prothrombin, a substrate of FXa, illustrates the five fragment protease is a FXa-like protease devoid of co-factor activation (Figure 5B–D). The wild-type SGT quickly destroyed these proteins at multiple sites and did not produce their active forms. In contrast and surprisingly, the loop additions identified through the analysis of small chromogenic substrates readily conferred an inability to cut non-natural FXa targets in a reasonable timeframe. The inability to cut these proteins when combined with the pattern of oligopeptide hydrolysis illustrates that the digestive nature of the trypsin has been restricted.
Prothrombin activation to thrombin requires cleavage of the two peptide bonds after Arg271 and Arg320 whose differential hydrolysis by FXa is a challenging problem for protein engineering. In the absence of factor Va and a phospholipid surface, FXa selectively cleaves the Arg271 site of prothrombin over Arg320 by several orders of magnitude and yields the inactive precursor prethrombin-2 31. In contrast, FXa in the prothrombinase complex flips the prothrombin site preference of FXa and makes Arg320 the preferred site by improving the rate of this hydrolysis reaction several million-fold. Such discrimination by FXa between these two sites is striking as the Ile-Asp/Glu-Gly-Arg-Thr sequence is nearly identical at both sites. Indeed, the similarity between these two sites in prothrombin would presumably exclude the use of short chromogenic substrates as the sole selection criteria for FXa-like activity using directed evolution or other protein engineering strategies.
Similar to observations made from oligopeptide hydrolysis, the five fragment protease (35/99/170/186/220) selectively cleaved prothrombin at Arg271 in a manner identical to FXa (Figure 5B). Other design intermediates also produced prethrombin-2 yet with different rates than FXa and also continued processing prothombin into non-natural fragments. Like FXa, none of the designs activated prothrombin into active thrombin with any efficiency. Selective production of prethrombin-2 with a rate comparable to FXa suggests a difficult to define fidelity in the recapitulation of substrate selectivity has been attained. Importantly, gains in selectivity are not the result of differences in maximal activity of the engineered enzymes as each protease presents kcat values towards chromogenic substrates not significantly different than FXa (Figure 6B). The addition of an appropriate membrane surface and factor Va to potentially form the prothombinase complex using the engineered proteases did not significantly change the cleavage rates or pattern nor produce active thrombin (data not shown) suggesting the additional protein domains of FXa and other changes to the protease domain are required for protein and phospholipid co-factor binding and activation.32 The five fragments introduced into the trypsin scaffold therefore act cooperatively to build selective recognition of macromolecular substrates similar to FXa in the absence of its natural cofactors.
Structural Properties of Engineered Proteases
Crystal structures of the non-selective (35/99/170) and the most selective (35/99/170/186/2220) fragment exchanged proteases explain the basis for the observed properties. Both structures show the enzymes in a catalytically competent state with unchanged regions nearly identical to the starting scaffold. In the absence of the fragments responsible for recognition of the allosteric activator Na+, the active site cleft of the protease bearing the 35-, 99-, and 170- loops of FXa adopts a conformation unlike either the initial scaffold or target (Figure 7A). The 99-loop is flipped away from the active site cleft and the Phe174 of the added 170-loop buries into the core of the protein. In these incorrect states the two loops provide little for molecular discrimination in the active site cleft. Burial of an introduced Phe174 was observed upon similar protein engineering in both rat anionic and bovine trypsins suggesting a general importance for distal site cooperativity and Na+ activation to build function similar to FXa.27; 28; 29
Figure 7. Comparison of the crystal structures of two fragment-engineered proteases to FXa.

The top panel shows the molecular surface of each protein (wheat) with the changes introduced to the protein colored according to the schematic in Figure 1. The lower panel compares the active site cleft of two structures compared to FXa (PDB 2BOK, light purple). 33 (A) In the 35/99/170-loop mutant protease both the 99- and 170-loops adopt an unconventional state with Phe174 buried into the core of the protein underneath Trp215 – a critical and highly conserved determinant of the extended substrate site pocket of all trypsin-like proteases – and the 99-loop is flipped upwards with Tyr99 unable to contribute to substrate discrimination. (B) In contrast, the high selectivity mutant bearing the 35/99/170/186/220-loops of FXa bound to the small molecule inhibitor benzamidine and Na+ presents an identical active site cleft to FXa with Tyr99 and Phe174 correctly positioned with well defined electron density (σ = 1.0, green).
In contrast to the low selectivity protease, the structure of the FXa-like protease bearing the same three loop fragments and the two Na+ binding loops bound to the allosteric activator Na+ (35/99/170/186/220) presents an active site cleft nearly identical to FXa with both the 99- and 170-loops properly positioned (Figure 7B). Such an architecture was not observed in the previously documented 99/170/186/220 construct whose structure presented the 99-loop incorrectly and flipped upwards as in the 35/99/170 construct.13 The Na+ binding site establishes active site cleft architecture by polarizing the cavity where Phe174 is buried in the other structure through polar contacts between surface residues in the remainder of the 170-loop and formation of an electrostatic interaction between Glu217 and Lys224, which is observed in FXa and our previous construct 99/170/186/220.13; 33 Both of the new structures present the 35-loop in a conformation unlike FXa due to crystal packing between adjacent molecules and the mechanism by which this loop confers gains on the enzymatic properties and rectifies the conformation of the 99-loop remains unclear. Nevertheless, the two structures and biochemical data illustrate the utility of combinatorial enzyme design for creating distal site cooperativity in a protein domain and provide a useful starting point for more complex designs of clotting factor proteases.
Discussion
As documented in previous attempts to design and modify trypsins and coagulation factor Xa in particular, simple substitution of residues directly contacting the desired substrate does not build selectivity suggesting an elusive requirement for distal site contributions for function.27; 28; 29 Absence of such knowledge limits our understanding of trypsin biology – a notable issue given their abundance in our genome and emerging importance in complex processes that are in stark contrast to the digestive function typically ascribed to members of this family.34; 35; 36; 37 We previously introduced Na+-dependent allostery similar to FXa into a trypsin and found structural overlap with determinants of primary specificity.11 Improving our initial construct required optimization of residues near a critical Y172S substitution. However, this fourteen amino acid substituted protease was 200-fold less active than the wild-type trypsin. In the absence of obvious paths to improve the activity of this enzyme, we further engineered the adjacent 99-loop and observed a dramatic and unexpected increase in both activity and selectivity. The crystal structure of this protease revealed an identical Na+ binding site and extended active site cleft as FXa yet an incorrect flipped conformation of the added 99-loop in the active site cleft.13 As shown here, the properties of this engineered protease (99/170/186/220) are not similar to FXa even though we had introduced nineteen amino acid substitutions at four different loops in the protease including changes to their length. The large number of substitutions and their distributed positions throughout the SGT gene suggested dramatic changes were necessary for further progress.
Obvious mutations to improve our design would engineer the residues responsible for exosite recognition into the previous high activity construct. However, the exosite of factor Xa is poorly defined and further complicated by documented energetic cooperativity between this loosely defined region and both the active site and Na+ binding site suggesting a degree of complexity.38; 39 We therefore did not know whether to add the 35- and/or the 70-loop in exosite I or their neighbors in the folded protein - the 60- and 145-loops - or any combination thereof. Additionally, we did not know if the effect of the Na+-activation mechanism could be replaced by inclusion of other loops on the surface of the protein or if these considerable changes would need to be made with subtle changes in the hydrophobic core of the folded protein. As described above, multiple constraints limited our assessment of these issues and were made more problematic by the 103-fold lower plasmid DNA transformation efficiency of the B. subtilis expression host. Combinatorial enzyme design minimizes these limitations and offers a number of useful advantages for protein engineering.
Here we have converted a broad selectivity bacterial trypsin into a simplified human blood clotting factor protease using a new method for grafting surface loops of one enzyme onto another. The twenty-five amino acid substitutions introduced into SGT are less than 12% of total protein and the key changes could only have been obtained by substituting the near entirety of the protease domain of FXa into SGT using other protein engineering strategies that rely on larger segmental exchange of genetic information - an obvious and uninformative outcome. It is unclear whether strategies involving more subtle randomized mutagenesis with activity screening for FXa-like selectivity using chromogenic tetrapeptide substrates would have created the selectivity to produce prethrombin-2 given near identical sequence composition of the two cleavage sites in prothrombin. Indeed, the active site cleft of FXa is documented to exhibit low selectivity towards chromogenic substrates.40
Sites identified by combinatorial enzyme design overlay regions of known importance in trypsin-like proteases and appear obvious on the basis of coagulation factor biochemistry yet the minimal set of substitutions to generate their effects in a bacterial protein could not have been predicted a priori. The critical role for the 220-loop, which includes the P225Y substitution, for transducing the effect of the composition of the 35-loop to the active site cleft on the opposite side of the molecule finds remarkable parallel with the evolutionary pressure documented at this region in naturally occurring trypsin sequences.41 Sequence dichotomy of residue 225 between proline and tyrosine, whose codons do not interconvert by single base substitution, partitions the family tree of S1A peptidases and the Tyr225-containing branch of proteases are responsible for development, coagulation, and immunity. Combinatorial enzyme design therefore appears to capture broader trends in the evolutionary separation between two distantly related proteins.
Despite the observed gain in desired properties, the most selective protease produced in this study requires, at minimum, additional protein domains to be considered a true coagulation factor as these modules provide the proper geometry, co-factor binding, and phospholipid binding necessary for efficient blood clotting.21; 32 The modules created here will be useful to probe the relationship between protease domain and N-terminal protein domains toward co-factor binding and activation, which is a poorly understood process for FXa when it becomes engaged in the prothrombinase complex. Our data demonstrates the vital role of the allosteric Na+-binding site for orchestrating the contribution of exosite I and the active site cleft in FXa, which is distinct from the activating effect ascribed to this allostery. Whether this relationship extends to other Na+-activated coagulation factors, such as thrombin, activated protein C or Factor IXa, and how this integrates with the other modes of co-factor activation in these proteases remains to be determined and requires analysis using multidomain protein constructs. Combinatorial enzyme design provides a general strategy for protein engineering of mechanisms requiring distal site cooperativity and will have particular utility in the creation protease domain modules to dissect the biological pathways involving clotting factors and their many homologs in the human genome.
Materials & Methods
The initial genetic construct of wild-type SGT cloned into plasmid pWB980 in B. subtilis was manipulated as described previously.11; 12; 13 In this system, SGT and its mutants are constitutively expressed in their active form and secreted directly into the culture supernatant. Combinatorial enzyme design was performed using purified plasmid DNA obtained from recombinant clones using the QIAprep Miniprep Spin kit (Qiagen, Valencia, CA). Initial PCR amplicons were generated using either one or two mutagenic oligonucleotides encoding fragment substitutions and an appropriate generic primer if necessary. We designed and applied four forward oligonucleotide primers for the loops in the N-terminal β-barrel and four reverse primers for those of the C-terminal β-barrel and a pair of forward and reverse generic primers from the ends of the SGT gene (Table 1). An additional forward primer was also needed due to the proximity of the 60- and 70-loops. PCR reactions (250 μL) were prepared using Deep Vent polymerase using standard conditions for amplification using plasmid DNA templates (New England Biolabs, Beverly, MA). The initial PCR reaction was ethanol precipitated prior to agarose gel electrophoresis. Amplicons were excised from the gel and purified using the QIAquick Gel Extraction Kit according to the manual and a final elution volume of 50 μL (Qiagen, Valencia, CA), which removes non-specific products and unused oligonucleotides primers.
Initial amplicons (12 μL of the spin column eluate) were combined with wild-type SGT in plasmid pWB980 (4 μL, 100 ng/μL) linearized with AhdI, EcoNI, NaeI, BamHI or EagI depending on the oligonucleotide pair used in the first step. Mutated plasmids were similarly linearized allowing combinatorial exchange of fragments. Each restriction endonuclease used cut the plasmid once within the SGT gene and not in the remainder of the plasmid. The amplicon-plasmid mixture was amplified using KlenTaqLA (DNA Polymerase Technology, St. Louis, MO) in 50 μL total volumes. Other long and accurate thermophilic polymerases were also capable of building concatamers. After an initial denaturation of 2 minutes at 95°C, the amplicon and linearized plasmid were subjected to 32 cycles of 95°C for 45 s, 58°C for 1 minutes and 72°C for 8 minutes. In most instances, the entire reaction product was used to directly transform B. subtilis according to the method of Spizizen.42 During validation of the method 10 μL of the reaction was run on an agarose gel and appeared as in Figure 3A. Restriction digest mapping and sequence analysis of purified plasmids validated recombinant clones. Typically more than 80% of the clones generated contained the desired fragment combination and double substitution designs were rarely observed to contain only one fragment. Measures to reduce background plasmid due to multicopy carryover into the final clone did not change the kinetic properties estimated from crude culture supernatants such as using plasmids with inactivated genes that were repaired, longer outgrowth of the transformation reaction prior to plating and antibiotic selection, and secondary transformation of purified plasmid after sequence analysis. The lack of significant background may derive from the degradation of long linear DNA upon entering competent cells and mechanism of plasmid replication.
Protein Purification
Recombinant proteases were purified as described previously using a three-step process involving affinity capture with soybean trypsin inhibitor agarose, ion exchange with SP Sephadex and then gel filtration using Sephadex G-75. 11; 12; 13 Final protein was 99% pure as assessed by active site titration using the irreversible chloromethylketone inhibitor PPACK.
Determination of Substrate Specificity and Inhibitor Affinity
Chromogenic tripeptide substrates bearing a C-terminal paranitroanilide (pNA) leaving group and a D-enantiomer at the N-terminus (d-Arg-Gly-Arg-pNA and d-Phe-Gly-Lys-pNA) were used to probe active site substrate selectivity (Midwest Biotech, Indianapolis, IN). The substrates differ at two sites to explicitly test subsite cooperativity in the engineered proteases. Initial screening of protease selectivity and Na+-activation was measured from crude culture supernatant directly to take advantage of the available chromogenic petide substrates and done simultaneously to account for differences in protein expression levels. In the absence of the SGT-bearing plasmid B. subtilis cultured supernatant does not present activity towards these substrates nor does it contain inhibitors of trypsin activity. Selectivity was further confirmed in notable enzyme design intermediates using purified enzymes. For the initial rate data presented in Figure 4, monovalent cation dependent activity and substrate selectivity was determined in 250 mM NaCl or LiCl with 50 μM of either substrate in 25 mM Tris pH 8.0 using a SpectraMax microplate reader. Activity measurements required at maximum 1/10th reaction volume of culture supernatant. Protein C, plasminogen and prothrombin from Haematologic Technologies (Essex Junction, VT) were cleaved under similar reaction conditions then denatured under reducing conditions, resolved by SDS-PAGE using NuPAGE 4-12% Bis-Tris gels (Invitrogen, Carlsbad, CA) and visualized with Coomassie staining.
Oligopeptides synthesized by the W.M. Keck Peptide Facility (Yale University) were designed to encompass at minimum seven residues on the N-terminal side and 16 residues on the C-terminal side of the scissile bond in the naturally occurring human proteins and whose composition is provided in Table 2. Substrate selectivity was assessed by the incubation of 10 μM of each peptide substrate with 10 nM of recombinant protease or human factor Xa for 24 hrs. Reactions were separated using a C18 Waters Novapak column (4.6 × 250 mm, 4 μm) and a sodium phosphate buffer/acetonitrile linear gradient over 30 min at a flow rate of 1 mL/min on a Shimadzu Prominence system and the extent of oligopeptide hydrolysis quantified by the peak area from the absorbance 205 nm relative to uncleaved peptide. Discontinuous assays were used to measure the inhibition of each protease by antithrombin III. Protease at 20 to 40 nM was incubated with 0.05 to 8 μM of antithrombin III and at various time points the residual activity towards 500 μM d-Arg-Pro-Arg-pNA measured. Initial velocity as a function of time was fit to a hyperbolic decay function to determine the second order rate constant of association.
Table 2.
Oligopeptides used to assess substrate of fragment engineered proteases. Of the twenty-five peptide bonds following positively charged amino acids arginine and lysine in the seven peptides, only the two denoted in bold are cleaved by FXa and the five fragment protease 35/99/170/186/220 with significant and equal rates.
| Oligopeptide | Composition |
|---|---|
| PAR1 | ATNATLDPRSFLLRNPNDKYEPFWEDEEKN |
| PAR2 | TNRSSKGRSLIGKVDGTSHVTGKGVTVETV |
| PAR3 | AKPTLPIKTFRGAPPNSFEEFPFSALEGWT |
| PAR4 | TPSILPAPRGYPGQVCANDSDTLELPDSSRA |
| FV-709 | QNRLAAALGIRSFRNSSLNQEEEEFNLTALALE |
| FV-1018 | KHTHHAPLSPRTFHPLRSEAYNTFSERRLKHSL |
| FV-1545 | DPDNIAAWYLRSNNGNRRNYYIAAEEISWDYSE |
Protein Crystallization and Structure Determination
Hanging-drop crystallization was used to obtain diffraction quality crystals. The mutant of SGT bearing the 35/99/170/186/220-loops of FXa (15 mg/mL) in 10 mM calcium acetate, pH 6.0 was mixed with an equal volume of reservoir solution containing 2.0 M ammonium sulphate, 0.1 M CAPS pH 10.5, 75 mM NaCl and 25 mM benzamidine. Crystals (0.4 × 0.3 × 0.2 mm) appeared after three weeks of incubation at 21°C. For the mutant of SGT bearing the 35/99/170-loops of FXa in similar preparation buffer, crystals were obtained from reservoir containing 10 mM calcium acetate, pH 6.0 and 1.5 M ammonium sulphate in the absence of small molecule inhibitors. Small, but diffraction quality, crystals (0.05 × 0.04 × 0.04 mm) grew within an hour at room temperature. Crystals were soaked briefly in artificial mother liquor containing 15 then 30% glucose as cryoprotectant for a few minutes immediately prior to cryofreezing and data collection. Data was collected on an ADSC Quantum 315 CCD at Beamline 14-BM-C at BIOCARS (Argonne, IL) for the three fragment protease and a Mar345 image-plate detector for the five fragment substituted protease. Integration and scaling of diffraction data were carried out with HKL-2000 43. Structures were solved by molecular replacement using the CCP4 suite.44 Initial positional and temperature factor refinement was done in REFMAC with alternating cycles of refinement and model building with COOT performed until the final models were produced.45 Statistics of the models are summarized in Table 3. Greater than 98% of geometry was in the preferred or allowed region of the Ramachandran plot for both models.
Table 3.
Data collection and refinement statistics. Highest resolution shell is shown in parenthesis.
| 35/99/170 (PDB ID 3I77) | 35/99/170/186/220 (PDB ID 3I78) | |
|---|---|---|
| Data collection | ||
| Space group | I422 | P3221 |
| Cell dimensions | ||
| A, b, c (Å) | 138.3, 138.3, 81.4 | 113.7, 113.7, 52.6 |
| σ, β, α (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 120.00 |
| Resolution (Å) | 35.0 - 2.10 (2.16-2.10) | 36.0 - 3.00 (3.08-3.00) |
| Rmerge | 0.162 (0.624) | 0.105 (0.323) |
| I/σI | 14.4 (3.8) | 11.8 (2.6) |
| Completeness (%) | 98.9 (99.5) | 97.6 (94.7) |
| Redundancy | 10.2 (10.5) | 5.8 (3.4) |
| Refinement | ||
| Resolution (Å) | 35 – 2.10 | 36 -3.00 |
| No. reflections | 21800 | 7515 |
| Rwork/Rfree | 0.162/0.195 | 0.208/0.275 |
| No. atoms | ||
| Protein | 1693 | 1698 |
| Ligands/ions | 15 | 30 |
| Water | 296 | 0 |
| B-factors | ||
| Protein | 17.4 | 56.8 |
| Ligand/ion | 56.0 | 66.9 |
| Water | 34.8 | n/a |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.017 | 0.013 |
| Bond angles (°) | 1.4 | 1.6 |
Protein Data Bank Accession Numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org, as PDB ID codes 3I77 and 3I78.
Acknowledgments
This work was supported by American Heart Association Scientist Development Grant 09SDG2260722 (to M.J.P.) and by NIH research grants HL49413, HL58141, HL73813 and HL95315 (to E.D.C.).
Abbreviations used
- RGR
H-D-Arg-Gly-Arg-p-nitroanilide
- FGK
H-D-Phe-Gly-Lys-p-nitroanilide
- PPACK
H-D-Phe-Pro-Arg-CH2Cl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–24. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3:509–19. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 3.Page MJ, Di Cera E. Serine peptidases: Classification, structure and function. Cell Mol Life Sci. 2008;65:1220–36. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Page MJ, Di Cera E. Evolution of peptidase diversity. J Biol Chem. 2008;283:30010–4. doi: 10.1074/jbc.M804650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–53. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 6.Hung SH, Hedstrom L. Converting trypsin to elastase: substitution of the S1 site and adjacent loops reconstitutes esterase specificity but not amidase activity. Protein Eng. 1998;11:669–73. doi: 10.1093/protein/11.8.669. [DOI] [PubMed] [Google Scholar]
- 7.Page MJ, Macgillivray RT, Di Cera E. Determinants of specificity in coagulation proteases. J Thromb Haemost. 2005;3:2401–8. doi: 10.1111/j.1538-7836.2005.01456.x. [DOI] [PubMed] [Google Scholar]
- 8.Page MJ, Di Cera E. Role of Na+ and K+ in enzyme function. Physiol Rev. 2006;86:1049–92. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 9.Pawlowski K, Godzik A. Surface map comparison: studying function diversity of homologous proteins. J Mol Biol. 2001;309:793–806. doi: 10.1006/jmbi.2001.4630. [DOI] [PubMed] [Google Scholar]
- 10.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 11.Page MJ, Bleackley MR, Wong S, MacGillivray RT, Di Cera E. Conversion of trypsin into a Na(+)-activated enzyme. Biochemistry. 2006;45:2987–93. doi: 10.1021/bi052481a. [DOI] [PubMed] [Google Scholar]
- 12.Page MJ, Wong SL, Hewitt J, Strynadka NC, MacGillivray RT. Engineering the primary substrate specificity of Streptomyces griseus trypsin. Biochemistry. 2003;42:9060–6. doi: 10.1021/bi0344230. [DOI] [PubMed] [Google Scholar]
- 13.Page MJ, Carrell CJ, Di Cera E. Engineering protein allostery: 1.05 A resolution structure and enzymatic properties of a Na+-activated trypsin. J Mol Biol. 2008;378:666–72. doi: 10.1016/j.jmb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venekei I, Graf L, Rutter WJ. Expression of rat chymotrypsinogen in yeast: a study on the structural and functional significance of the chymotrypsinogen propeptide. FEBS Lett. 1996;379:139–42. doi: 10.1016/0014-5793(95)01483-7. [DOI] [PubMed] [Google Scholar]
- 15.Stosova T, Sebela M, Rehulka P, Sedo O, Havlis J, Zdrahal Z. Evaluation of the possible proteomic application of trypsin from Streptomyces griseus. Anal Biochem. 2008;376:94–102. doi: 10.1016/j.ab.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Bock PE, Panizzi P, Verhamme IM. Exosites in the substrate specificity of blood coagulation reactions. J Thromb Haemost. 2007;5(Suppl 1):81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–25. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 18.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–53. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 19.Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270:16854–63. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- 20.Rezaie AR, Kittur FS. The critical role of the 185-189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J Biol Chem. 2004;279:48262–9. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 21.Mann KG. Thrombin formation. Chest. 2003;124:4S–10S. doi: 10.1378/chest.124.3_suppl.4s. [DOI] [PubMed] [Google Scholar]
- 22.Read RJ, James MN. Refined crystal structure of Streptomyces griseus trypsin at 1.7 A resolution. J Mol Biol. 1988;200:523–51. doi: 10.1016/0022-2836(88)90541-4. [DOI] [PubMed] [Google Scholar]
- 23.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. Structure of human des(1-45) factor Xa at 2.2 A resolution. J Mol Biol. 1993;232:947–66. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 24.Chen I, Dubnau D. DNA uptake during bacterial transformation. Nat Rev Microbiol. 2004;2:241–9. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- 25.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–71. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Rauh D, Klebe G, Stubbs MT. Understanding protein-ligand interactions: the price of protein flexibility. J Mol Biol. 2004;335:1325–41. doi: 10.1016/j.jmb.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 28.Rauh D, Klebe G, Sturzebecher J, Stubbs MT. ZZ made EZ: influence of inhibitor configuration on enzyme selectivity. J Mol Biol. 2003;330:761–70. doi: 10.1016/s0022-2836(03)00617-x. [DOI] [PubMed] [Google Scholar]
- 29.Reyda S, Sohn C, Klebe G, Rall K, Ullmann D, Jakubke HD, Stubbs MT. Reconstructing the binding site of factor Xa in trypsin reveals ligand-induced structural plasticity. J Mol Biol. 2003;325:963–77. doi: 10.1016/s0022-2836(02)01337-2. [DOI] [PubMed] [Google Scholar]
- 30.Hopfner KP, Kopetzki E, Kresse GB, Bode W, Huber R, Engh RA. New enzyme lineages by subdomain shuffling. Proc Natl Acad Sci U S A. 1998;95:9813–8. doi: 10.1073/pnas.95.17.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orcutt SJ, Krishnaswamy S. Binding of substrate in two conformations to human prothrombinase drives consecutive cleavage at two sites in prothrombin. J Biol Chem. 2004 doi: 10.1074/jbc.M410866200. [DOI] [PubMed] [Google Scholar]
- 32.Myles T, Yun TH, Hall SW, Leung LL. An extensive interaction interface between thrombin and factor V is required for factor V activation. J Biol Chem. 2001;276:25143–9. doi: 10.1074/jbc.M011324200. [DOI] [PubMed] [Google Scholar]
- 33.Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, Benz J, Stihle M, Diederich F. Quantification of cation-pi interactions in protein-ligand complexes: crystal-structure analysis of Factor Xa bound to a quaternary ammonium ion ligand. Angew Chem Int Ed Engl. 2005;44:4400–4. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- 34.Puente XS, Sanchez LM, Gutierrez-Fernandez A, Velasco G, Lopez-Otin C. A genomic view of the complexity of mammalian proteolytic systems. Biochem Soc Trans. 2005;33:331–4. doi: 10.1042/BST0330331. [DOI] [PubMed] [Google Scholar]
- 35.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, Antalis TM. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22:237–58. doi: 10.1023/a:1023003616848. [DOI] [PubMed] [Google Scholar]
- 36.Ovaere P, Lippens S, Vandenabeele P, Declercq W. The emerging roles of serine protease cascades in the epidermis. Trends Biochem Sci. 2009;34:453–63. doi: 10.1016/j.tibs.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Choi SY, Bertram S, Glowacka I, Park YW, Pohlmann S. Type II transmembrane serine proteases in cancer and viral infections. Trends Mol Med. 2009;15:303–12. doi: 10.1016/j.molmed.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Bianchini EP, Pike RN, Le Bonniec BF. The elusive role of the potential factor X cation-binding exosite-1 in substrate and inhibitor interactions. J Biol Chem. 2004;279:3671–9. doi: 10.1074/jbc.M309691200. [DOI] [PubMed] [Google Scholar]
- 39.Underwood MC, Zhong D, Mathur A, Heyduk T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor xa. Studies on catalytic efficiency and inhibitor binding. J Biol Chem. 2000;275:36876–84. doi: 10.1074/jbc.M001386200. [DOI] [PubMed] [Google Scholar]
- 40.Bianchini EP, Louvain VB, Marque PE, Juliano MA, Juliano L, Le Bonniec BF. Mapping of the catalytic groove preferences of factor Xa reveals an inadequate selectivity for its macromolecule substrates. J Biol Chem. 2002;277:20527–34. doi: 10.1074/jbc.M201139200. [DOI] [PubMed] [Google Scholar]
- 41.Krem MM, Di Cera E. Molecular markers of serine protease evolution. Embo J. 2001;20:3036–45. doi: 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spizizen J. Transformation of Biochemically Deficient Strains of Bacillus Subtilis by Deoxyribonucleate. Proc Natl Acad Sci U S A. 1958;44:1072–8. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]