

Abstract
To explore the role of both Aplysia cell adhesion molecule (ApCAM) and activity of specific protein kinase C (PKC) isoforms in the initial formation of sensory neuron synapses with specific postsynaptic targets (L7 but not L11), we examined presynaptic growth, initial synapse formation, and the expression of the presynaptic neuropeptide sensorin following cell-specific reduction of ApCAM or of a novel PKC activity. Synapse formation between sensory neurons and L7 begins by 3 h after plating and is accompanied by a rapid accumulation of a novel PKC to sites of synaptic interaction. Reducing ApCAM expression specifically from the surface of L7 blocks presynaptic growth and initial synapse formation, target-induced increase of sensorin in sensory neuron cell bodies and the rapid accumulation of the novel PKC to sites of interaction. Selective blockade of the novel PKC activity in L7, but not in sensory neurons, with injection of a dominant negative construct that interferes with the novel PKC activity, produces the same actions as downregulating ApCAM; blockade of presynaptic growth and initial synapse formation, and the target-induced increase of sensorin in sensory neuron cell bodies. The results indicate that signals initiated by postsynaptic cell adhesion molecule ApCAM coupled with the activation of a novel PKC in the appropriate postsynaptic neuron produce the retrograde signals required for presynaptic growth associated with initial synapse formation, and the target-induced expression of a presynaptic neuropeptide critical for synapse maturation.
Introduction
Many factors contribute to the formation of specific synapses: molecules that regulate the growth of axons and dendrites, expression of surface molecules or diffusible factors that influence the assembly of presynaptic and postsynaptic elements including the trafficking of receptors to sites apposed to presynaptic release sites, and activity-dependent modulation of synaptic contacts (Sanes and Yamagata, 2009). How do multiple cell surface and diffusible factors, activity and their respective signaling pathways orchestrate target selection and synapse formation?
The Ig family of cell adhesion molecules play critical roles in axon pathfinding and synapse formation (Goodman, 1996; Maness and Schachner, 2007), and in synaptic plasticity associated with learning and memory (Doherty et al., 1995; Dalva et al., 2007). One member of the Ig family, neural cell adhesion molecule (NCAM) in vertebrates, its homolog in Drosophila (fasciclin II) or Aplysia (ApCAM), contributes to neuritic growth, synapse formation and stabilization (Keller and Schacher, 1990; Davis et al., 1997; Goodman et al., 1997; Baines et al., 2002; Packard et al., 2003; Ashley et al., 2005; Kohsaka et al., 2007; Maness and Schachner, 2007), and long-term activity-dependent synaptic plasticity (Mayford et al., 1992; Lüthl et al., 1994; Zhu et al., 1995; Bukalo et al., 2004).
In Aplysia, ApCAM is a major membrane protein expressed at synaptic contacts (Keller and Schacher, 1990; Mayford et al., 1992). Changing ApCAM expression or transcripts for specific ApCAM splice variants in sensory or motor neurons accompany long-term plasticity at sensory neuron synapses (Mayford et al., 1992; Bailey et al., 1992, 1997; Wu and Schacher, 1994; Zhu et al., 1994, 1995; Schacher et al., 2000; Lee et al., 2007). Decreasing ApCAM on L7 regulates presynaptic growth of sensory neurons contacting L7 and disrupts synapse formation (Zhu et al., 1994). However, the potential roles of ApCAM in synapse specificity were not clear, since ApCAM levels expressed on the non-target L11 are comparable to that expressed on L7 (Schacher et al., 2000). This raises the possibility that down stream signaling initiated by cell surface molecules on L7, but not on L11, regulate synapse formation.
PKC activity is critical for initiating synapse formation and growth by sensory neurons contacting L7 (Hu et al., 2007b). Pharmacological inhibition of all PKC isoforms blocked growth and initial synapse formation by sensory neurons but did not interfere with sensory neuron growth contacting the non-target L11. Aplysia neurons express at least three PKC isoforms—classical (Apl I), novel (Apl II) and atypical (Apl III)—and their activation play important roles in both short- and long-term facilitation (LTF) of sensory neuron synapses (Manseau et al., 1998, 2001; Zhao et al., 2006; Sossin, 2007; Bougie et al., 2009; Villareal et al., 2009). Which PKC isoform is critical for initiating synapse formation and growth?
We report here that ApCAM on the surface of L7 and the activation of a novel PKC in L7 (but not L11) are required for the initial formation of synapses and synapse-associated growth by the presynaptic sensory neuron and target-induced increase in sensorin expression in sensory neurons.
Materials and Methods
Cell culture.
Sensory neurons were isolated from pleural ganglia dissected from adult animals (80 g) and motor neurons L7 and L11 were isolated from abdominal ganglia dissected from juvenile animals (2 g) and maintained in culture for varying times (see below) under conditions previously described (Rayport and Schacher, 1986; Hu et al., 2007b). Each cell body was isolated with a segment (300–600 μm) of its original axon as well as some fine dendritic-like branches extending from the axons of the motor neurons. Cocultures were prepared under the following conditions: (1) both presynaptic sensory neuron and postsynaptic target (L7 or L11) were plated together (∼2 h apart) with the axon stump of the sensory neuron touching the major axon of the target (SN-L7 or SN-L11), (2) postsynaptic target neurons (L7 or L11) were plated first and maintained overnight, and then a sensory neuron was added with its axon stump contacting the major axon of the target neuron (L7 + SN or L11 + SN), or (3) sensory neurons were plated first and maintained overnight, and then a postsynaptic target (L7 or L11) was added to a sensory neuron with the regenerated axons of the sensory neuron touching the major axon of the target (SN + L7 or SN + L11). Under these conditions, synapse formation is governed almost exclusively by both neuritic growth of the sensory neurons and the site-specific accumulation of glutamate receptors (Conrad et al., 1999; Li et al., 2009). Typically a single culture dish would contain one sensory neuron contacting L7 and another sensory neuron contacting L11. Sensory neurons plated alone (not contacting targets) were added to some dishes containing cocultures. In some cultures sensory neurons, L7 and L11 were plated alone. In some cultures, the PKC inhibitor chelerythrine (2 μm) was bath applied to cocultures and washed out at various times.
Electrophysiology.
Assays for synaptic connections were monitored with standard electrophysiological recordings (Hu et al., 2007b). Starting 2 h after onset of interaction between sensory neuron and L7 (or L11), an intracellular electrode was used to monitor EPSPs following action potentials evoked with an extracellular electrode placed near the cell body of the sensory neurons (Schacher and Montarolo, 1991; Hu et al., 2007b). The membrane potential of the motor neuron was held at −80 mV. No EPSP was recorded in L11 at any time point. Resting membrane potential and input resistance of L7 did not vary significantly from 2 h to 20 h in coculture.
Incubation with anti-ApCAM antibody.
L7 and L11 were plated alone in the same culture dish with or without isolated sensory neurons and 4 h later were incubated overnight (16 h) with 10 μg/ml monoclonal antibody 4E8 (Keller and Schacher, 1990) or mouse IgG. Previously we found that exposure to monoclonal antibody 4E8 leads to antibody-triggered endocyotsis of ApCAM from the surface of the cells (Keller and Schacher, 1990; Zhu et al., 1994). To test the degree of downregulation of ApCAM from the surface of the cells that were incubated overnight, cells were then fixed 3 h after washout of the antibody and assayed for the surface expression of ApCAM using immunocytochemistry of nonpermeabilized cells. To test consequences on synapse formation and growth, partner neurons (sensory neurons added to L7/L11 or L7/L11 added to sensory neurons) were added to the cultures immediately after antibody washout. Synapse formation was tested using electrophysiology, growth was assayed by imaging axons of sensory neurons following injection of the fluorescent dye 5,6-carboxyfluoroscein (Glanzman et al., 1989; Hu et al., 2007b), and sensorin expression was assayed with immunocytochemistry (Hu et al., 2007b).
Plasmid construction and microinjection of plasmid vectors.
The pNEX3 enhanced green fluorescent protein (eGFP) PKC Apl II as well as its dominant negative construct were described previously (Manseau et al., 2001). Briefly, each plasmid was injected using a pneumatic picopump (PV820, World Precision Instruments). Solutions of plasmids (∼0.3 μg/μl DNA in distilled water) containing 0.2% fast green were microinjected into L7 or sensory neurons cultured overnight. The tip of the micropipette was inserted into the cell nucleus, and short pressure pulses (10–50 ms duration; 20–40 psi) were delivered until the nucleus became uniformly green. The other member of the synaptic pair (12–16 h later for sensory neurons or L7) was added to form L7+SN or SN+L7 cocultures.
Imaging sensory neuron growth and varicosities.
After measuring the strength of the synapses, the fluorescent dye 5(6)-carboxyfluorescein (Invitrogen; 6% in 0.44 m KOH, pH = 7.0) was injected with 0.3–0.5 nA hyperpolarizing current pulses (500 ms at 1 Hz) for 3–5 min into some of the sensory neurons that were cocultured either with L7 or L11 (Glanzman et al., 1989; Hu et al., 2007b). Nomarski or phase contrast, and fluorescent images of the same view areas along the original axons of L7 or L11 were taken to map out the location of regenerated sensory neuron neurites and varicosities. Varicosities were defined as swellings along the sensory neuron neurites with diameters ≥ 1.5 μm. Growth cones at the leading tips of neurites and enlarged branch points were not counted as varicosities. Extent of growth from the axon stump of a sensory neuron was measured as the length along the original axons of the targets covered with regenerated axons of the sensory neurons. The cultures were viewed with a Nikon Diaphot microscope attached to a silicon-intensified target (SIT) (Dage 68; Dage-MTI) video camera. All images were captured and processed by the Microcomputer-Controlled Imaging Device (MCID) software package (Imaging Research).
Immunocytochemistry.
Immunocytochemistry was used to monitor expression of sensorin, ApCAM, and PKC Apl II (Keller and Schacher, 1990; Nakhost et al., 2002; Pepio et al., 2002; Hu et al., 2004a, 2007b). Cultures were rinsed briefly in artificial seawater and fixed in 4% paraformaldehyde and processed as described previously (Keller and Schacher;, 1990; Hu et al., 2004b, 2007b). The cells were exposed to rabbit polyclonal antibody specific for sensorin (1:1000) or PKC Apl II (1:250) diluted in 2% normal goat serum in 0.01 m PBS with 0.3% Triton X-100 at 4°C for 24 h. To evaluate surface expression of ApCAM, the fixed cultures were exposed to mouse monoclonal antibody 4E8 (1:500) diluted in the above buffer but without Triton X-100 at 4°C for 24 h. In contrast, if detergents are used for incubation significant intracellular staining was observed, especially after preincubation with 4E8 (data not shown; Zhu et al., 1994). Thus this technique measures mostly surface receptors. The incubated cultures were washed in 0.01 m PBS and incubated in FITC- or rhodamine-conjugated goat anti-rabbit IgG (1:200, Sigma) for sensorin or PKC Apl II detection or rhodamine-conjugated goat anti-mouse IgG (1:200, Sigma) for ApCAM detection at 4°C for 4 h. After washing in 0.01 m PBS, cultures were imaged directly with a filter set for detecting either FITC or rhodamine signal. To test the specificity of the primary antibody, controls were performed, including the substitution of normal rabbit serum for the primary antibody and omission of the primary antibody. To test specificity of the sensorin antibody, sensorin and its antibody at equimolar concentrations were preincubated for 30 min before their application to fixed cultures for immunostaining (Hu et al., 2004a,b). All controls showed little immunocytochemical reaction. The anti-ApCAM monoclonal antibody 4E8 recognizes the extracellular domain expressed in all splice variants of ApCAM (Keller and Schacher, 1990; Mayford et al., 1992). Illumination for detecting fluorescent signals was maintained at a constant setting for all treatments. Images were captured and processed as described above.
Quantification and data analysis.
All data are expressed as mean ± SEM produced by the indicated treatments. The averages for EPSP amplitude (including failures), axon growth and varicosities were measured in mV, μm and number, respectively. The intensity of sensorin immunostaining was tested by measuring average fluorescent intensity in the sensory neuron cell body, in the entire original axon and axon stump, and in the regenerated neurites and varicosities of sensory neurons contacting the original axons of L7 or L11. For most experiments staining intensities for sensorin for the various experimental treatments or target interactions were normalized to the average intensity measured for each cellular compartment in sensory neurons plated alone at the earliest time point (100% for each compartment). For monitoring staining for PKC Apl II at sites of interaction, the axon stump, neurites and varicosities for each sensory neuron were imaged following dye injection and their locations were mapped out and outlined on the surface of the original axons of L7 or L11 (see Zhu et al., 1994, 1995 for similar methods for monitoring site-specific expression of molecules at sensory neuron-target interaction). Staining intensities per unit area at sites of contact were then compared with staining intensity at adjacent areas of L7 or L11 that excluded any sensory neuron structure (normalized as 100%). Staining intensity for ApCAM on the membrane surface of the cells was measured by first mapping the contour of the cell surface (cell body, axon or neurite) and then measuring staining intensity per unit length. Staining intensity for ApCAM or PKC Apl II was normalized to the average staining detected for controls (see Results). ANOVA and Scheffe F test were used to gauge significant differences between treatments.
Results
Synapse formation is initiated by 3 h of cell–cell interaction
Sensory neurons form synapses with motor neuron targets by 6 h (Lyles et al., 2006; Hu et al., 2007b). Using an electrophysiological assay we examined a more exact time course for synapse formation using three types of cultures—simultaneous plating of a sensory neuron with L7 (SN-L7), L7 plated first then sensory neuron added the next day (L7 + SN), or sensory neuron plated first then L7 added the next day (SN + L7). We monitored EPSP amplitudes starting at 2 h after contact between sensory neurons and L7. We also monitored sensorin expression and the extent of sensory neuron growth starting either at 2 or 4 h after cell–cell contact.
Synapse formation begins by 3 h after contact and increases in strength thereafter (Fig. 1 A–C). No EPSP (<0.25 mV) was detected at 2 h, regardless of the culture procedure (data not shown). The time course for subsequent increases in synapse strength was dependent on the culture procedure. For SN-L7 cultures (simultaneous plating), 41% of cultures had detectable EPSPs (>0.25 mV) at 3 h (7/17; range of 0–1 mV) that increased to 83% (15/18; range of 0–6 mV) at 6 h. Average EPSP amplitudes increased from 0.4 ± 0.1 mV at 3 h to 3.4 ± 0.4 mV at 6 h. For L7 + SN cultures, synapse formation and rate of increase in EPSP amplitude was somewhat slower. At 3 h, only 25% of cultures (4/16; 0–1 mV) had detectable EPSPs with average amplitudes of 0.2 ± 0.1 mV. By 6 h the percentage of cultures with detectable EPSPs increased to 72% (13/18; range of 0–5 mV) with average EPSP amplitudes of 2.6 ± 0.4 mV. By 12 h all L7 + SN cultures had detectable EPSPs (see below). In contrast, synapse maturation was accelerated for SN + L7 cultures. At 3 h, 59% of cultures (10/17; range of 0–2 mV) had detectable EPSPs with average amplitudes of 1.0 ± 0.2 mV. By 6 h 100% of the cultures (20/20; range of 2–12 mV) had detectable EPSPs with average amplitudes of 7.2 ± 0.6 mV. Although synapse formation was initiated at approximately the same time, increase in synapse strength was accelerated when sensory neurons were plated first and motor neurons added the next day.
Figure 1.

Time course for synapse formation, sensorin expression, and axon growth of sensory neurons when interacting with L7. A–C, Time course for synapse formation. EPSPs evoked in L7 at 3–6 h of interaction for the three types of cell culture: simultaneous plating (SN-L7); L7 first, then sensory neuron added the next day (L7 + SN); and sensory neuron first and L7 added the next day (SN + L7). EPSP amplitudes increased with time (A). Calibration: Vertical bar, 10 mV; horizontal bar, 25 ms. Plating sensory neuron first leads to stronger synapses (B) and a higher proportion of cultures with detectable synapses (C). In B the height of each bar is the mean ± SEM of EPSP amplitude at each time point for the three culture conditions. ANOVA indicated a significant difference with condition and time (df = 4, 36; F = 4.032, p < 0.02). Individual comparisons (Scheffe F) indicated that EPSP amplitudes for SN + L7 cultures were significantly greater than the amplitudes in the other two culture conditions at each time point (p < 0.05 at 3 h and p < 0.01 at both 4 and 6 h). In addition to EPSP amplitudes, the proportion of cultures with synapses was higher for SN + L7 cultures (C). D, E, Sensorin expression in each cellular compartment increases over time for SN-L7 cultures. Phase contrast and corresponding epifluorescent images for sensorin immunostaining depict changes in sensorin expression at the various time points (D). Sensorin expression is low at 2 h (n = 6) but increases significantly at distal sites by 3 h (n = 8). At 4 h (n = 8), sensorin staining peaked in the axon, while peak intensity in the cell body occurred at 6 h (n = 10). Scale bar, 50 μm. In E the height of each bar is the mean ± SEM staining intensity for sensorin in each compartment at each time point after normalizing to the 2 h intensity values for each compartment (100% ± SEM). The peak intensity is first achieved in the distal neurites, while the peak intensity in the cell body is achieved last. ANOVA indicated a difference with time in each compartment (df = 2, 28, F = 78.785, p < 0.001). Individual comparisons indicated that staining intensity in the cell body increased with each time point (p < 0.01), staining intensity in the axon increased at each time point (p < 0.01) up to 4 h, and staining intensity in the distal neurites increased at 3 h (p < 0.01) and remained relatively constant afterward. F, Axon growth by sensory neurons increased continuously from 2 to 6 h. Height of each bar is the mean ± SEM extent of sensory neuron axon growth in contact with the major processes of L7 based on detectable sensorin immunochemistry in the distal neurites.
Target-induced regulation of sensorin expression follows initial synapse formation
By 20 h after plating, sensorin expression and secretion and regenerated growth were significantly higher when sensory neurons contact and form synapses with L7 compared with sensory neurons contacting L11 (Hu et al., 2004b, 2007b). When do these L7-dependent increases emerge? We first examined the time course for sensorin expression and growth in sensory neurons contacting L7 when the cells are plated simultaneously.
The time of peak expression for sensorin varied for cellular compartment, but the peaks were reached after synapse initiation (Fig. 1 D,E). Sensorin staining in all compartments was relatively low at 2 h (each normalized to 100%). Sensorin staining at the distal axon stump and newly regenerated neurites and varicosities increased to a peak level of intensity at 3–4 h that persisted at 6 h. Sensorin staining in the original axon reached peak values at 4 h that persisted at 6 h. In contrast, sensorin staining in sensory neuron cell bodies only reached peak levels of expression at 6 h.
New growth from sensory neurons accompanied the initial formation of synapses (Fig. 1 F). At 2 h, growth from the axon stump of the sensory neurons (SN-L7 cultures) was just beginning (42.2 ± 3.7 μm) and increased twofold (88.1 ± 4.0 μm) when synapses were first detected at 3 h. By 6 h sensory neuron growth along the major axons of L7 increased to 225.7 ± 7.5 μm. The results indicate that neuritic growth begins coincidentally with synapse formation, and peak levels of sensorin expression are reached after synapse initiation, in time to contribute to synapse maturation.
We next determined the time point when sensorin expression in sensory neurons begins to diverge as a result of specific target interaction. Sensorin staining at distal regenerated neurites, original axon, and cell bodies was independent of target interaction at 4 h (see supplemental Figs. 1–3, available at www.jneurosci.org as supplemental material), when most sensory neuron-L7 cultures had initiated synapses (40–75% for the three conditions; Fig. 1 C). Thus at the time of synapse initiation, no contact or initial contact with L11 produced the same level of sensorin expression throughout the sensory neurons as contact with L7.
Target-dependent differences in the expression of sensorin became evident after 6 h of cell interaction (supplemental Figs. 1–3, available at www.jneurosci.org as supplemental material). Sensorin staining in all compartments of sensory neurons contacting L7 were now significantly greater (from p < 0.04 to p < 0.01) than the staining in sensory neurons plated alone or sensory neurons contacting L11. Staining for sensorin in some of the compartments of the sensory neuron contacting L11 declined significantly (p < 0.05) by 6–8 h in SN-L11 (regenerated neurites), L11 + SN (original axon and regenerated neurites) and SN + L11 (regenerated neurites) cultures (supplemental Figs. 1B, 2B, 3B, available at www.jneurosci.org as supplemental material). Although sensorin expression was target-independent when synapse formation was initiated (by 4 h), continued target interaction (6–8 h) had both a positive effect on expression when sensory neurons contacted L7 and a negative effect on expression when sensory neurons contacted L11.
Target-dependent differences in neuritic growth by sensory neurons in SN-L7/L11 and L7/L11 + SN cultures also emerged by 6 h (see supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material; see below for growth in SN + L7 cultures compared with SN + L11 cultures). Neuritic growth from the sensory neurons plated alone or contacting different targets was not significantly different at 4 h (supplemental Figs. 1C, 2C, available at www.jneurosci.org as supplemental material). By 6–8 h target-dependent differences became more evident. Neuritic growth by sensory neurons contacting L7 in SN-L7 cultures (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material) was significantly greater than growth by sensory neurons alone (p < 0.05) or sensory neurons contacting L11 (p < 0.02). By 8 h, growth by sensory neurons contacting L7 in L7 + SN cultures (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material) was significantly greater than growth by sensory neurons contacting L11 (p < 0.05). Thus target-dependent differences in growth first emerge after the initial formation of synapses and during the early phase of synapse maturation.
In summary, regardless of culture procedure, synapse formation is initiated at a common time point (∼3 h), and initial sensorin expression in all compartments are similar to those observed for sensory neurons plated alone or for sensory neurons contacting L11. Target-dependent differences in sensorin expression and neuritic growth emerge at a later time point (6–8 h) and the action of the target had either a positive effect (L7) or a negative effect (L11) on sensorin expression. Specific retrograde signals from the targets must therefore influence the cellular events in the presynaptic neuron required for the initial formation of synapses and synapse-associated growth, and sensorin expression.
Reducing ApCAM on L7 interferes with synapse initiation, synapse-associated growth, and the increase in sensorin expression
Downregulation of ApCAM from the surfaces of neurites decreases fasciculation of neurites, but does not impact on axon elongation per se (Keller and Schacher, 1990; Peter et al., 1994). Since ApCAM expression on L7 is downregulated by preincubation with anti-ApCAM monoclonal antibody 4E8 (Keller and Schacher, 1990; Zhu et al., 1994, 1995), we examined changes in the expression of ApCAM on the surface of the target neurons, L7 and L11, and the presynaptic sensory neurons following an overnight incubation with anti-ApCAM antibody 4E8 (Fig. 2).
Figure 2.
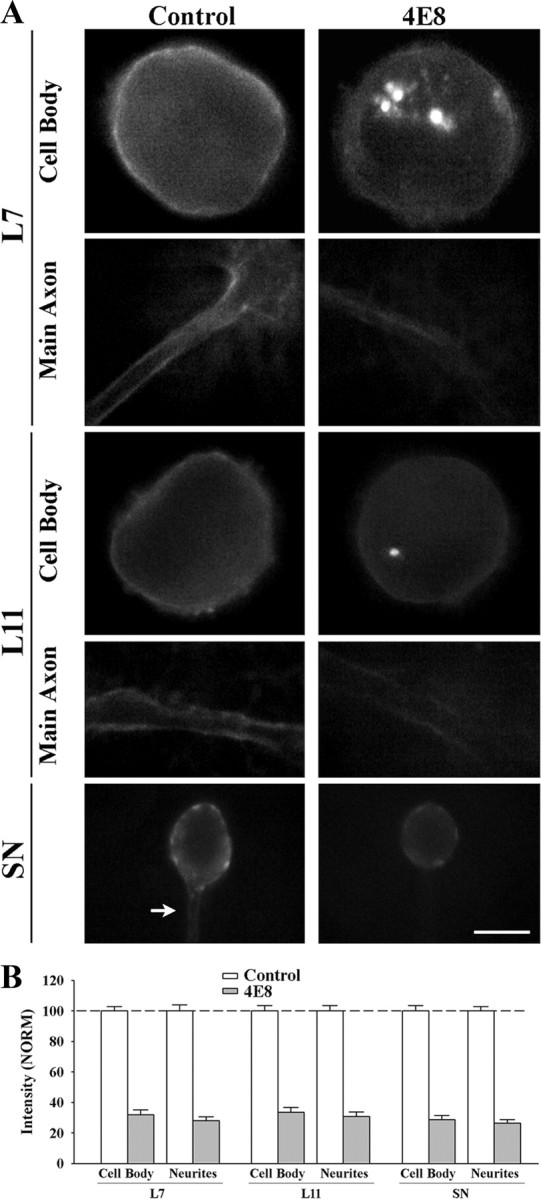
Treatment with anti-ApCAM antibody 4E8 downregulates ApCAM expression on the cell surface membrane of L7, L11, and sensory neurons. A, Epifluorescent images of ApCAM immunostaining of cell body and processes of L7 (n = 7 for each treatment), L11 (n = 7 for each treatment), or SN (n = 9 for each treatment) following overnight preincubation (14 h) with control or 4E8 (anti-ApCAM antibody). Midlevel focal planes (between substrate and top of each structure) are presented here. After antibody washout, cells were fixed 3 h later and processed for ApCAM immunochemistry of the surface membrane (no detergent added). Treatment with 4E8 significantly reduced surface membrane staining throughout the cells. Scale bar, 50 μm. B, Treatment with 4E8 reduced ApCAM on the surface of cells by ∼70%. The height of each bar is the mean ± SEM percentage for ApCAM staining on the surface of the cell body or processes following treatment with 4E8 normalized to the staining levels measured on the surface of each compartment following treatment with controls (100% ± SEM). ANOVA indicated a significant effect of treatment (df = 5, 40; F = 177.02, p < 0.001). Individual comparisons indicated that treatment with 4E8 significantly reduced ApCAM levels from the cell bodies (p < 0.01) and processes (p < 0.01) of all cells compared with controls. There were no significant differences in surface membrane staining for ApCAM for the control cells (see Results).
Preincubation with anti-ApCAM antibody 4E8 significantly reduced ApCAM from the surface membranes of all neurons (Fig. 2 A,B). Compared with expression of ApCAM on the surface of the cell body and distal processes of the neurons treated with control IgG (normalized to 100%; left panel in 2A), ApCAM expression on the surfaces of original axons and neurites of the neurons were significantly (p < 0.01) reduced 3 h after the termination of the incubation with anti-ApCAM antibody 4E8; 28.1 ± 2.6% for L7, 30.7 ± 3.1% for L11, and 26.4 ± 2.4% for sensory neurons. Expression on the surface membrane of the cell bodies of each neuron was also reduced significantly (p < 0.01) by similar amounts after incubation with anti-ApCAM antibody 4E8 (32.1 ± 3.2% in L7, 33.6 ± 3.2% in L11, and 28.7 ± 2.8% in sensory neurons). Staining intensity for ApCAM per unit area of surface membrane (cell body, and distal neurites) for all the neurons in the control-treated cultures was not significantly different from each other. When normalized to staining levels on L7 (100%), staining on the surface of the cell body and distal neurites of L11 was 94.0 + 4.6% and 95.7 + 3.4%. Staining on the surface of the cell body and distal neurites of sensory neurons was 96.3 + 5.5% and 93.7 + 3.7%. The reduction from the surface membranes of L7 and L11 treated with 4E8 was correlated with the presence of fluorescent staining in large granules in the cell bodies of L7 and L11 (Fig. 2 A). These granules likely contain high concentration of internalized ApCAM and/or 4E8 recognized by the secondary antibody even in the nonpermeabilized neurons. Such granules were not detected in control-treated L7 or L11. The significant reduction in ApCAM from the surfaces of the cells 3 h after incubation, a time point when synapse formation is initiated, allowed us to examine the consequences of cell-specific alterations in ApCAM expression on the surface of the target neurons on the initial formation of synapses, sensorin expression, and axon growth by sensory neurons. Motor neurons (L7 or L11) were maintained in culture overnight and incubated with 4E8 antibody or controls. Antibody was washed out just before adding the sensory neurons. Sensory neurons were plated such that their axon stumps contacted the major axons of the motor neurons.
Reducing ApCAM on L7 blocked initial synapse formation by sensory neurons (Fig. 3 A,B). After 6 h of contact with L7 motor neurons that were preincubated with anti-ApCAM antibody 4E8, the number of cultures with detectable EPSPs was reduced to 25% (4/16; range of 0–1 mV) with an average EPSP amplitude of 0.2 ± 0.1 mV compared with EPSPs detected in ∼85% of control cultures (11/13; range of 0–5 mV) with an average EPSP amplitude of 2.6 ± 0.4 mV (p < 0.01). Because the downregulation of ApCAM from the surface of L7 is reversed after washout of the antibody (Zhu et al., 1995), we expected that synapse formation would proceed with time. At 20 h, we reexamined the same cultures and found all L7 + SN cultures with 4E8 antibody pretreatment now had detectable EPSPs (range of 1–8 mV) with average EPSP amplitude of 3.5 ± 0.4 mV. All control cultures had detectable EPSPs (range of 6–15 mV) with higher average EPSP amplitudes of 8.3 ± 0.8 mV (p < 0.01). The transient blockade of synapse formation is consistent with the observation that the reduction in ApCAM expression on the surface of L7 following 4E8 treatment is reversed following antibody washout (Zhu et al., 1994, 1995). The postponement of synapse initiation is removed when ApCAM expression on the surface of L7 recovers.
Figure 3.

Downregulating ApCAM from the surfaces of L7 blocks initiation of synapse formation, L7-induced regulation of sensorin expression in the cell body of the sensory neurons and synapse-associated growth. A, Phase contrast and epifluorescent images of sensorin immunostaining in sensory neurons after 6 h of contact with L7 (n = 10 for control and n = 12 for 4E8) or L11 (n = 8 for each treatment) that were plated the previous day and preincubated with control or 4E8 antibody. Antibodies were washed out and sensory neurons plated ∼1 h later. Sensorin staining in the sensory neuron cell body was low following treatment of L7 with 4E8. Also note that there was no growth from the axon stump of the sensory neuron. Treatment of L11 with 4E8 did not interfere with sensorin expression in the cell body, axon or neurites of the sensory neurons. Scale bar, 50 μm. B, Downregulating ApCAM on L7 with 4E8 blocked synapse formation at 6 h, and significantly reduced synapse strength when EPSP were recorded again at 20 h. The height of each bar is the mean ± SEM of the EPSP amplitude measured at 6 and 20 h for control and 4E8 treatment of L7 before the addition of the sensory neuron. ANOVA indicated a significant effect of treatment (df = 1, 26; F = 7.979, p < 0.01). Individual comparisons indicated a significant reduction in EPSP with treatment with 4E8 at both 6 and 20 h (p < 0.01). C, Downregulating ApCAM on L7 with 4E8 blocked the increase in sensorin expression, especially in the cell body, while treatment of L11 with 4E8 did not interfere with sensorin expression throughout the sensory neuron. The height of each bar is the mean ± SEM staining intensity in the various compartments of the sensory cell normalized to the staining intensity in each compartment for control L7 + SN cultures (100%). Since no varicosities formed with pretreatment of L7 with 4E8, there is no bar for that compartment. ANOVA indicated a significant effect of treatment (df = 6, 72; F = 195.526, p < 0.001). Individual comparisons indicated that treatment with 4E8 resulted in a significant reduction in sensorin expression in the cell body of sensory neurons contacting L7 (p < 0.01), but had no effect on expression in sensory neuron axons. There was no significant difference in the treatment of L11 with 4E8; both 4E8 and control treatments did not alter the significant reduction (p < 0.05) in sensorin staining in the cell body and original axons. D, Downregulating ApCAM on the surface of L7 significantly blocked neuritic growth by sensory neurons, but downregulating ApCAM on the surface of L11 did not affect neuritic growth by sensory neurons. The height of each bar is the mean ± SEM in the extent of neuritic growth of sensory neurons in contact with L7 or L11 based on detectable sensorin immunochemistry in the distal neurites of the sensory neurons. Treatment of L7 with 4E8 blocked neuritic growth by sensory neurons. This was also verified by injection with carboxyfluorescein (n = 3, data not shown). ANOVA indicated a significant effect of treatment (df = 3, 36; F = 62.015, p < 0.001). Individual comparisons indicated that pretreatment of L7 with 4E8 significantly reduced growth (p < 0.01) while pretreatment of L11 with 4E8 did not affect growth by sensory neurons contacting L11.
Reducing ApCAM expression on the surface of L7 also blocked the target-dependent upregulation of sensorin expression in the cell body of the sensory neurons, but did not interfere with the upregulation of sensorin expression in the original axons of the sensory neurons (3A and 3C). Whereas sensorin expression was high in all compartments of sensory neurons contacting L7 pretreated with control IgG (each compartment normalized to 100%), reducing ApCAM expression on L7 resulted in the failure to upregulate sensorin expression in the cell body (57.5 ± 4.3%). Sensorin expression in the axon stump and original axon remained high (97.3 ± 3.6%). Because there was no growth (see below), staining in distal neurites and varicosities was indeterminate. In contrast, reducing ApCAM expression on L11 had no significant impact on sensorin expression. Compared with control L7 cultures (normalized to 100% for each compartment) sensory neurons contacting L11 had reduced sensorin expression in all compartments regardless of pretreatment of L11 with the 4E8 antibody. In sensory neurons contacting L11 treated with control antibody, sensorin expression was reduced in the cell body (55.3 ± 5.4%), axons (73.0 ± 5.2%) and varicosities (83.3 ± 4.2%). Treating L11 with 4E8 did not affect sensorin expression in the sensory neurons compared with L11 treated with control antibody; sensorin expression in the cell body (56.4 ± 5.8%), in the axons (71.9 ± 5.5%) and in the varicosities (81.1 ± 6.0%) was also reduced. Thus ApCAM on the surface of L7 contributes to the target-induced increase in sensorin, but ApCAM on L11 does not influence the target-induced decrease in sensorin when sensory neurons contact L11.
Reducing ApCAM levels on L7 also had a significant impact on growth by sensory neurons contacting L7, but reducing ApCAM levels on L11 had little or no consequence on axon growth by sensory neurons contacting L11 (Fig. 3 D). At 6 h, growth by sensory neurons contacting L7 pretreated with 4E8 was reduced significantly (p < 0.01) to 16.1 ± 2.6 μm compared with 210.5 ± 14.8 μm of growth for controls (Fig. 3 A,D). In contrast, reducing ApCAM expression on L11 had no significant impact on overall sensory neuron growth compared with control (Figs. 3 A–D). Neuritic growth by sensory neurons contacting L11 that were treated with 4E8 was 173.3 ± 13.1 μm compared with 168.5 ± 12.0 μm for growth by sensory neurons contacting L11 treated with control antibody. Thus synapse-associated growth and initial synapse formation by sensory neurons contacting L7 requires postsynaptic ApCAM, but growth along L11 was not affected by changes in ApCAM levels on the non-target L11.
ApCAM on sensory neurons is not critical for synapse initiation, synapse-associated growth, or target-induced changes in sensorin expression
To selectively reduce ApCAM expression on the sensory neurons, we cultured sensory neurons overnight in the presence of 4E8, washed out the 4E8 or control antibody and then plated L7 or L11 near the sensory neurons such that the stump and extending neurites of the sensory neurons were in contact with the original axons of the newly added target neurons. Images of the regenerated neurites of each sensory neuron were taken immediately before adding the target neuron. These neurites as well as any new growth contacting the targets were imaged again either after injection of fluorescent dye into the sensory neuron (Fig. 4) or after processing for sensorin immunochemistry (Fig. 5).
Figure 4.

Reducing ApCAM from the surface of the sensory neurons does not affect synapse formation and growth. Sensory neurons were plated and treated overnight with either control or anti-ApCAM antibody 4E8 and washed, and then axon growth was imaged before plating L7 or L11 (only SN + L7 cultures are shown here). After 6 h, EPSPs were recorded, and in some cultures, sensory neurons were injected with carboxyfluorescein to map out new growth by the sensory neurons. Other cultures were maintained overnight and EPSP amplitudes were reexamined. A, Phase contrast images (left) of axon growth from sensory neurons treated overnight with control IgG or 4E8. The dashed outline represents the position of the motor neuron L7 (axon hillock and original axon) that was plated with each sensory neuron after taking the images. Treatment with 4E8 does not impact axon growth by sensory neuron. On the right are epifluorescent images of neuritic growth following injection with carboxyfluorescein from the same sensory neurons after 6 h of interacting with the motor neurons. Arrowheads point to some of the varicosities and arrows point to some of the new branches that had grown over the 6 h of interaction. The white dashed lines represent the borders of the L7 motor neurons. Treatment with 4E8 failed to block new growth and formation of new varicosities. Scale bar, 75 μm. B, Reducing ApCAM from the surface of the sensory neurons did not interfere with synapse formation. The height of each bar is the mean ± SEM of the EPSP amplitudes recorded at 6 and 20 h. ANOVA indicated no significant effect of treatment with 4E8. At 20 h both control- and 4E8-treated cultures had formed strong synapses. C, Reducing ApCAM from the surface of the sensory neurons did not interfere with growth and formation of new varicosities. The height of each bar is the mean ± SEM of the total number of varicosities contacting L7 or L11 (left bars) and the length of new sensory neuron axon branches contacting L7 or L11 (right bars). For each treatment and culture (n = 6 for each treatment of SN + L7 or SN + L11 cultures), sensory neuron varicosities contacting each target were divided into two groups: old varicosities (O) on preexisting sensory neuron axon branches when the motor neuron was first plated, and new varicosities (N) formed by new axon growth. Regardless of target (L7 or L11), treatment with 4E8 had no impact on growth. Growth and varicosity number were higher for sensory neurons contacting L7 then sensory neurons contacting L11.
Figure 5.

Reducing ApCAM from the surface of the sensory neurons does not affect target-induced regulation of sensorin expression. Cultures were processed as described above, except at 6 h the cocultures were fixed and processed for sensorin immunochemistry. A, Phase contrast images (left) of axon growth from the stump of sensory neurons treated overnight with control IgG (n = 7 for SN + L7 or SN + L11) or 4E8 (n = 7 for SN + L7 or SN + L11). The dashed outline represents the position of the motor neuron L7 (axon hillock and major processes) that was plated with each sensory neuron after taking the images (only SN + L7 cultures are shown here). Treatment with 4E8 did not affect sensorin expression in the regenerated neurites, especially the new branches and varicosities. On the right are epifluorescent images of sensorin immunostaining from the same sensory neuron at 6 h of interacting with the motor neuron. In both images, arrowheads point to the location of some of the varicosities and arrows point to the location of some of the new branches that had grown over the 6 h of interaction. Scale bar, 40 μm. B, Reducing ApCAM from the surface of the sensory neurons did not block target-induced regulation of sensorin expression throughout the sensory neurons. The height of each bar is mean ± SEM of the staining intensity in the 4E8-treated cultures after normalization with the staining intensity for each compartment of sensory neurons treated with controls and interacting with L7 after 6 h. Treatment with 4E8 failed to block the strong staining in sensory neurons contacting L7, and failed to block the reduction in staining in each compartment for sensory neurons contacting L11.
Synapse formation and neuritic growth were not affected by reducing ApCAM expression on sensory neurons (Fig. 4). At 6 h, all cultures formed synapses both for sensory neurons preincubated with 4E8 (14/14) and cultures treated with control antibody (13/13). Average EPSP amplitude was 7.2 ± 1.0 mV in control cultures and 6.4 ± 1.0 mV in cultures treated with 4E8 (Fig. 4 B). When these cultures were reexamined at 20 h, synaptic strength was quite strong relative to the strength of synapses formed when cells are plated simultaneously (SN-L7) or L7 plated first then sensory neurons added the next day (L7 + SN). Regardless of treatment (4E8 or control), EPSP amplitudes increased significantly (p < 0.01) to 33.8 ± 3.1 mV or 37.2 ± 2.5 mV, respectively (Fig. 4 B). Both control- and 4E8-treated cultures exhibited additional neuritic growth by sensory neurons during the first 6 h of contact, including the formation of new varicosities (Fig. 4 A,C). In control cultures new growth contacting L7 extended 184.5 ± 14.7 μm with 5.0 ± 0.6 new varicosities. The total number of sensory neuron varicosities contacting L7 was 15.7 ± 2.2. Pretreatment of sensory neurons with 4E8 antibody did not impact on growth. Axon extension along L7 was 178.0 ± 13.6 μm with 4.7 ± 0.7 new varicosities. The total number of varicosities contacting L7 was 14.8 ± 2.0. Pattern of growth by sensory neurons in contact with L11 was also unaffected by pretreatment with 4E8 (Fig. 5 C). Axon extension was 131.3 ± 15.0 μm with 3.3 ± 0.7 new varicosities in controls and 129.2 ± 12.3 μm with 3.2 ± 0.5 new varicosities after pretreatment of sensory neurons with 4E8. The total number of sensory neuron varicosities contacting L11 was 10.5 ± 1.6 for controls and 9.7 ± 1.2 for cultures where sensory neurons were pretreated with 4E8. Thus reducing ApCAM on sensory neurons did not impact on synapse formation with L7 or neuritic growth along the surface of L7 or L11.
We next examined whether reducing ApCAM on the surface of the sensory neurons would impact on the target-induced changes in sensorin that emerge at 6 h after contact (Fig. 5). Sensorin expression was strong in all compartments of sensory neurons preincubated with control IgG and interacting with L7. Preincubation with 4E8 antibody did not affect the expression of sensorin in any of the compartments: 97.6 ± 4.0% in the cell body, 98.0 ± 4.6% in the original axon, and 99.7 ± 4.3% in regenerated neurites and varicosities. In both control- and 4E8-treated cultures, new branches and varicosities expressed high levels of sensorin (Fig. 5 A). Sensorin expression in sensory neurons interacting with L11 was significantly lower (p < 0.01) in all compartments than sensorin expression in sensory neurons contacting L7 when sensory neurons were preincubated either with IgG control or 4E8 antibody (Fig. 5 B). Thus reducing ApCAM on the sensory neurons did not interfere with the target-induced regulation of sensorin expression, synapse-associated growth or initial synapse formation while reducing ApCAM on L7 significantly affected these properties. Since L11 also expresses ApCAM on its surfaces, the down stream signaling by postsynaptic ApCAM or other cell surface molecule binding to a ligand on sensory neurons may be cell-specific and contribute to the target-dependent regulation of sensory neuron responses. We next explored the signaling cascades that might contribute to the target-specific regulation of initial synapse formation, growth and sensorin expression.
PKC activation is required for synapse initiation
Bath application of the general PKC inhibitor chelerythrine at the start of cell–cell interaction reversibly blocks synapse initiation and synapse-associated growth by sensory neurons, target-induced regulation of sensorin expression and synapse maturation (Hu et al., 2007b). The same conditions failed to alter growth by sensory neurons either plated alone or in contact with L11. We examined the time course of these changes produced by chelerythrine by examining the effects of the inhibitor when it was applied at various times after the initial interaction between a sensory neuron and L7 (Fig. 6, SN-L7 cultures).
Figure 6.

Blocking PKC activity in sensory neuron and L7 with chelerythrine interferes with initial synapse formation, synapse-associated growth, and sensorin expression. A–C, Initial synapse formation, growth, and sensorin expression were affected differently when chelerythrine was bath applied to SN-L7 cultures at the indicated times after plating both cells. Cocultures were imaged after 16 h of drug treatment (18–22 h in culture) after processing for sensorin immunoreactivity (A). Note that the time at which chelerythrine was added to the coculture had a significant impact on sensorin expression. The differences in synapse strength and sensorin expression in control cultures at 18 and 22 h were not significant. An overall ANOVA indicated a significant effect of treatment on sensorin expression in the different compartments (df = 4, 43, F = 82.193, p < 0.001). Individual comparisons indicated that expression in the axon and distal sites was reduced only when inhibitor was added at 2 h (F = 3.825 p < 0.05). Expression in the cell body remained suppressed when inhibitor was added at 3 and 4 h (F = 32.145, p < 0.01, and F = 30.762, p < 0.01) but reached control levels at 6 h. EPSP amplitude detected after drug treatment was significantly affected (B). Compared with control, adding drug at each time point significantly reduced EPSPs (F = 19.894–38.278; p < 0.01). Applying drug after 2 h blocked synapse formation, while adding drug after 6 h allowed cultures to form stronger synapses than those detected when drug was added at 3 or 4 h (p < 0.05). Extent of growth after drug treatment was also influenced by the time of application (C). For each time point, growth was significantly reduced compared with control (F = 13.802–92.526; p < 0.01).
Blocking PKC activities in SN-L7 cocultures starting at each time point (2 h-6 h) blocked all subsequent changes normally produced by target interaction. Starting the 16 h incubation with the PKC inhibitor at 2 h of cell–cell contact blocked synapse formation (10/10 cultures failed to form synapses) and synapse-associated growth by sensory neurons compared with control (Fig. 6 A–C). Sensorin expression in axons and cell body were significantly reduced (p < 0.01 for cell body and p < 0.05 for original axon + stump) compared with control. Onset of PKC inhibition at subsequent time points (3 and 4 h) allowed the initial formation of synapses (7/12 cultures when inhibitor was added at 3 h and 8/10 cultures when added at 4 h) but halted the expected increase in synapse strength measured 16 h later (0–2 mV compared with 13.8 ± 1.3 mV for controls; Fig. 6 B). This is consistent with results in Figure 1 indicating that synapse formation is initiated at 3 h. Adding the PKC inhibitor at 3–4 h also blocked further sensory neuron growth (Fig. 6 C), and the increase in sensorin expression in the cell body (Fig. 6 A; reduced by 69.0 + 3.7% at 3 h and 64.5 + 4.8% at 4 h compared with control; p < 0.01). Intensities of sensorin expression at the few distal neurites and varicosities were not significantly different from controls. The increase in sensorin expression in the cell body was detected at control levels when the PKC inhibitor was added at 6 h. Although all cultures (10/10) formed synapses, the increase in synapse strength and growth was blocked when the PKC inhibitor was added at 6 h, since PKC activity is required for sensorin secretion that regulates synapse maturation (Hu et al., 2007a,b). Thus PKC activation in sensory neurons and/or L7 during the first 6 h is required for the initial formation of synapses, synapse-associated growth by sensory neurons, and the increase in sensorin expression in the sensory neuron.
Downregulating ApCAM from the surface of L7 interferes with the accumulation of PKC Apl II at sites of contact with sensory neurons
The novel PKC isoform, PKC Apl II, is expressed in neurons of Aplysia (both sensory neurons and motor neurons), is activated by serotonin (5-HT), and is required for some forms of short-term synaptic plasticity (Manseau et al., 2001; Farah et al., 2009). Injecting plasmids expressing a mutated form of this PKC into sensory neurons blocks activation of the kinase and some forms of short-term synaptic plasticity of sensory neuron synapses. Does this novel PKC participate in the initial formation of sensory neuron synapses? We first examined, with immunochemistry, the distribution of the novel PKC during the early phase of synapse formation or when sensory neurons contact L11, and whether downregulating ApCAM from the surface of L7 affects that distribution.
Expression of the novel PKC is enhanced at sites of sensory neuron contact with L7, but not at sensory neuron contact sites with L11, at 6–8 h in L7 + SN cultures (Fig. 7 A,B). Before fixation and processing for immunochemistry, sensory neurons were injected with fluorescent dye and sites of contact with L7 or L11 were imaged (Fig. 7 A). The same view areas and focal planes were then reimaged for immunostaining of the novel PKC. Intensity of the staining near the original axon, axon stumps, varicosities and regenerated neurites of the sensory neurons in contact with L7 and L11 were measured and compared with adjacent sites on L7 or L11 without any sensory neuron contacts (see the general areas outline by the dashed lines in Fig. 7 A; see Materials and Methods for details). Contact sites between sensory neuron and L7 (Fig. 7 A, within areas designated as C) had significantly higher (p < 0.01) staining intensities (149.2 ± 7.4%) than non-contact sites (Fig. 7 A, within areas designated as NC) at neighboring surfaces of L7 (normalized to 100%). In contrast, sites of contact between the original axon, axon stump, varicosities and regenerated neurites of sensory neurons and L11 showed no difference in staining intensity compared with neighboring sites on the surface of L11 without sensory neuron contacts (98.1 ± 7.3% compared with 95.9 ± 8.6% normalized to the intensity of staining at non-contact sites on L7) and were not significantly different from the staining detected at non-contact sites of L7 + SN cultures. The enhanced accumulation at sensory neuron-L7 contact sites is not a result of simply the sum of the staining from the sensory neuron plus the target. Thus interaction with sensory neurons induces a target-specific (L7 but not L11) accumulation of the novel PKC at or near sites where the sensory neuron contacts L7.
Figure 7.

ApCAM on the surface of L7 is required for local targeting of PKC Apl II to sites of sensory neuron contact. A, B, PKC Apl II staining intensity is enhanced at contact sites of sensory neuron with L7 (n = 12), but not with L11 (n = 8). In A, phase contrast (left), epifluorescent images of PKC Apl II immunostaining (red; middle), and combined epifluorescent images of PKC Apl II staining and dye-filled neurites and varicosities of the sensory neuron (green; right) contacting either L7 (L7 + SN) or L11 (L11 + SN) are compared. Note the enhanced staining at the contact sites (within the large dashed zone in the middle micrographs) between sensory neuron and L7 compared with nearby sites on L7 without contacts (such as areas within the NC zone). Contact sites on the surface of L11 showed little or no differences in staining compared with neighboring NC sites. In B, staining intensity at sites of contact with the target by the axon stump, neurites, and varicosities of sensory neurons are normalized to neighboring non-contact sites on the surface of L7 (normalized to 100%). An overall ANOVA indicated a significant effect of the target on staining intensity at contact sites (df = 3, 36, F = 14.516, p < 0.001). Individual comparisons indicated that staining intensity at sensory neuron contact sites (C) with L7 were significantly greater than that in neighboring non-contact sites (NC) on L7 (F = 9.645, p < 0.01), and contact sites between sensory neurons and L11 (F = 13.461, p < 0.01). Intensity at contact sites between sensory neurons and L11 were not significantly different from intensity at the non-contact sites on L11. C, D, Reducing ApCAM on the surface of L7 by incubating with anti-ApCAM antibody 4E8 abolishes the increase in PKC Apl II at L7 contact sites with sensory neurons. Preplated L7 was incubated overnight either with control (n = 16) or 4E8 antibodies (n = 16). After antibody washout, sensory neurons were added to some of the preplated L7 (n = 10 per treatment). In C, epifluorescent views of PKC Apl II immunostaining for both L7 + SN cultures and L7-alone cultures (n = 6 per treatment) treated with both antibodies are compared. In the two upper right panels, dye-filled staining of the sensory axon, stump, and neurites (green) is superimposed on PKC Apl II immunostaining (red) for control- and 4E8-treated L7 and compared. We also compared the overall staining of sections of L7's axon that were not contacted by sensory neurons (200–300 μm sections) in L7 + SN cultures (C, top left) to the overall staining of sections of L7 axons plated alone (C, bottom left). No significant difference was detected in the immunostaining for the novel PKC. In D, staining intensities at sites of contact with L7 by axon, axon stump, and varicosities of the sensory neuron are compared with normalized staining for the PKC Apl II on the surface of control-treated L7 axon (300 μm portion of the axon normalized to 100%). An overall ANOVA indicated a significant effect of antibody treatment on staining at contact sites (df = 3, 28, F = 33.371, p < 0.001). Individual comparisons indicated that sensory neuron contact with L7 increased staining for PKC Apl II at contact sites compared with overall staining along the axon of control L7 plated alone (F = 19.944, p < 0.01). Treatment with the anti-ApCAM antibody abolished the increase in PKC Apl II staining at contact sites in L7 + SN cultures compared with control-treated L7 + SN cultures (F = 10.447, p < 0.01). Staining at contact sites between sensory neurons and L7 when ApCAM was downregulated from L7 was not significantly different from overall staining for L7 plated alone. Treatment of L7 alone with anti-ApCAM 4E8 did not change significantly overall staining for PKC Apl II compared with control-treated L7 alone.
Downregulating ApCAM from the surface of L7, which interferes with initial synapse formation (see above), blocked the targeting of the novel PKC to sites of sensory neuron contact (Fig. 7 C,D). Motor neurons were incubated overnight with 4E8 or control antibody, and after washout sensory neurons were added to some of the treated neurons. At 6–8 h, sensory neurons were injected with fluorescent dye, imaged and then processed for PKC Apl II immunochemistry. Staining intensity on the axon surface of L7 plated alone plus treated with control antibody was normalized to 100%. As noted above, staining was significantly higher at sites of contact between the sensory neuron axon, axon stump, regenerated neurites and varicosities and L7 treated with the control antibody compared with the overall staining on the surface of L7 plated alone and in the same culture dish (Fig. 7 D). Overall staining of L7 in areas that had no sensory neuron contact was not significantly different from the overall staining of L7 plated alone. Treatment of L7 with 4E8 blocked most growth by the sensory neuron and blocked any accumulation of staining for the novel PKC even at sites of contact between the original axon and axon stump of the sensory neuron and the treated L7. When L7 is plated alone, treatment with 4E8 did not impact on basal level of staining for the novel PKC. Thus one down stream consequence of ApCAM on the surface of L7 may be to signal the local accumulation of the novel PKC to sites of sensory neuron contact. Since the resolution was not sufficient to determine whether the accumulation of the novel PKC was presynaptic or postsynaptic, we next examined the consequences of cell-specific reduction in the activity of the novel PKC on initiating synapse formation, growth and sensorin expression.
Blocking activity of the novel PKC in L7, but not in sensory neurons, blocked synapse formation, sensory neuron growth, and the increase in sensorin expression
Each cell partner was plated alone and the nucleus was injected using intracellular electrodes containing: fast green (0.2%; vehicle); plasmid with eGFP linked to the normal PKC Apl II gene; or plasmid with eGFP linked to a mutated form of the PKC Apl II that acts as a dominant negative inhibitor of activation for the novel PKC (Manseau et al., 2001; Zhao et al., 2006). After waiting 12–16 h to allow protein expression in the injected cell, a synaptic partner (sensory neuron added to injected L7, or L7 added to injected sensory neuron) was added to each culture. To allow 100% of all control cultures to form a synapse, EPSPs were monitored at 12 h in L7 + SN cultures (Fig. 8)and 6–8 h in SN + L7 cultures (Fig. 9). Growth and sensorin expression were measured using sensorin immunochemistry (Fig. 8) for L7 + SN cultures, while growth in SN + L7 cultures was measured by imaging eGFP expression before and 6–8 h after plating the L7. For sensory neurons injected with fast green, new sensory neuron growth was measured from images following fluorescent dye injection (see Fig. 6 above).
Figure 8.

Downregulating activity of PKC Apl II in the motor neuron L7 by injecting a dominant negative construct blocks initial synapse formation, synapse-associated growth, and increased expression of sensorin. A, Phase contrast (left), rhodamine epifluorescent images of sensorin immunostaining in sensory neuron cell bodies, axons, and varicosities (red; middle) and double label for sensorin immunostaining (red) in sensory neurons and eGFP-expression (green) epifluorescent images indicating expression of the construct in L7 (right panel) of the same view areas showing sensory neuron interaction with L7. Growth of sensory neuron and sensorin expression, especially in the cell body, are significantly reduced following injection and expression of the dominant negative PKC (PKC Apl II dn eGFP; bottom) compared with the normal PKC Apl II (PKC Apl II eGFP; top). B, Initial synapse formation is blocked following injection of the dominant negative construct. An ANOVA indicated a significant effect of construct injection (df = 2, 28, F = 19.968, p < 0.001). Individual comparisons indicated that injection of the dominant negative construct (n = 11) significantly reduced EPSP amplitude compared with fast green (F = 15.356, p < 0.01; n = 10) or to injections with the control construct (F = 14.096, p < 0.01; n = 10). Injection of the normal gene into L7 did not affect synapse formation compared with injection of fast green alone. C, Sensory neuron growth was significantly reduced when L7 was injected with the dominant negative construct. An ANOVA indicated a significant effect of construct injection (df = 2, 28, F = 47.128, p < 0.001). Individual comparisons indicated that injection with the dominant negative construct significantly reduced growth compared with fast green injection (F = 37.016, p < 0.01) or injection of construct for the normal gene (F = 32.415, p < 0.01). Growth following injection of construct for the normal gene was not different from the growth after fast green injection. D, Sensorin expression, particularly in the cell body of the sensory neuron, was significantly reduced by injection of the dominant negative construct. An overall ANOVA indicated a significant effect of treatment (df = 4, 56, F = 20.703, p < 0.001). Individual comparisons indicated that injection of the dominant negative construct in L7 significantly reduced sensorin expression in the cell body of the sensory neuron compared with injection of fast green (F = 15.119, p < 0.01) or the normal gene (F = 13.008, p < 0.01). Sensorin staining in the axons was not affected by injections. Since there were no stained varicosities after injection of the dominant negative construct, differences in growth were indeterminate. There were no significant differences in the staining in all compartments between injection of fast green and injection of the construct for the normal gene.
Figure 9.

Downregulating activity of PKC Apl II in the sensory neuron by injecting a dominant negative construct does not interfere with initial synapse formation, and new synapse-associated growth. A, Injection of the dominant negative construct into the sensory neuron (green) does not interfere with growth of new sensory neuron branches that form new varicosities. The left images are epifluorescent views of eGFP expression in the preplated sensory neuron before adding L7 (Pre). The eventual locations of some of the new branches (yellow arrows) and new varicosities (red arrowheads) are indicated. The right images (Post) show epifluorescent views of the same area after 8 h of interaction with L7 (n = 8 per treatment). Arrows and arrowheads indicate some new branches and varicosities, respectively. Note that expression of the dominant negative construct does not interfere with new growth and varicosity formation. The right image is a phase contrast view of the same area 8 h after adding L7. Arrows point to the axon of the SN and the major axon of L7. B, EPSP amplitude (top), formation of new varicosities (middle), and growth of new branches (bottom) are unaffected by injection of the dominant negative construct. An overall ANOVA indicated no significant effect of treatment.
Expression of the dominant negative form of the novel PKC in L7 (Fig. 8 A) blocked synapse formation (Fig. 8 B), synapse-associated growth by the sensory neuron (Fig. 8 C) and target-induced increase in sensorin expression in the cell body (Fig. 8 D). Over 50% of the cultures expressing the dominant negative inhibitor (PKC Apl II dn eGFP) failed to form a synapse by 12 h (6/11 cultures) and the range of EPSP amplitudes was 0–2 mV (0.7 ± 0.3 mV). In contrast 100% of the cultures in the other two groups (fast green and PKC Apl II eGFP; n = 10 each) formed synapses with a range of EPSP amplitudes of 2–10 mV (5.5 ± 0.8 mV) for fast green and a range of 3–9 mV (5.3 ± 0.7 mV) for cultures overexpressing PKC Apl II eGFP. Growth from the sensory neuron stump was significantly reduced by the expression of the dominant negative PKC Apl II compared with the other two groups (78.2 ± 7.2 μm compared with 268.2 ± 19.2 μm for fast green and 255.9 ± 18.1 μm for PKC Apl II eGFP). As was the case for downregulating ApCAM from the surface of L7, expressing the dominant negative PKC Apl II in L7 blocked the target-induced upregulation of sensorin expression in the cell body (when normalized to 100% for fast green injection); sensorin staining was reduced to 48.9 ± 7.1%. Thus blocking activation of the novel PKC in L7 had the same consequences on sensory neuron function as downregulating ApCAM from L7.
Overexpression of PKC Apl II eGFP in the motor neuron or sensory neuron did not enhance synapse strength, sensory neuron growth or sensorin expression. Because the activity of this kinase is not constitutive, it was not surprising that overexpression alone without additional stimulation did not produce a change in phenotype. The PKC Apl II eGFP did not accumulate at sensory neuron contacts (Figs. 8 A, 9 A; data not shown) probably due to overexpression.
Expression of the dominant negative form of the novel PKC in sensory neurons (Fig. 9 A) had no effect on initial synapse formation and new growth (Fig. 9 B) (see also Manseau et al., 2001). Detectable EPSPs were observed in 100% of cultures (n = 8 cultures per treatment) with comparable EPSP amplitudes (5.6 ± 0.6 mV for PKC Apl II dn eGFP, 5.8 ± 0.8 mV for PKC Apl II eGFP and 6.0 ± 0.9 mV for fast green). New growth and varicosities formed by the new branches were not affected by the expression of the dominant negative construct (range of 7.5 ± 0.8 to 8.1 ± 0.9 new varicosities and 165.0 ± 5.7 μm to 178.5 ± 9.2 μm for new branch growth). Thus activation of the novel PKC in sensory neurons does not contribute to initial synapse formation and presynaptic growth, while its activation in L7 is required for synapse formation, growth and the increase in sensorin expression.
Discussion
ApCAM on the surface of L7 and activation of a novel PKC in L7 are both required for initial synapse formation, synapse-associated growth and the increase in sensorin expression by sensory neurons. Growth by sensory neurons contacting a non-target (L11) requires neither ApCAM nor the activation of PKC. ApCAM expression and activation of the novel PKC in sensory neurons did not affect the initial formation of synapses or synapse-associated growth by sensory neurons, suggesting ApCAM on L7 interacts with a heterophilic ligand/receptor on sensory neurons leading to the cellular responses required for initial synapse formation and synapse-associated growth by sensory neurons. Although L11 expresses the same splice variants of ApCAM (Schacher et al., 2000), its potential interaction with the putative heterophilic ligand/receptor on the sensory neuron failed to initiate downstream signaling required for synapse formation. Activity of the novel PKC in L7 is required to produce the presynaptic cellular responses including the increased expression of the presynaptic neuropeptide sensorin whose secretion is required for subsequent synapse maturation.
Common features of synapse formation
Regardless of culture procedure, initial synapse formation and the emergence of target-dependent differences generally followed similar time courses. One interesting difference was the more rapid rate of synapse maturation when sensory neurons regenerated overnight before adding L7 (SN + L7). After 20 h, the synapses were stronger than those formed in SN-L7 or L7 + SN cultures. The overall expression of sensorin in sensory neurons of SN + L7 cultures at 6 h was higher than sensorin expression in sensory neurons in the other cocultures (∼50%, data not shown). This level of expression might result in higher levels of secreted sensorin at a critical early phase of synapse maturation and lead to stronger mature synapses (Bank and Schacher, 1992; Zhu et al., 1994; Hu et al., 2004a,b, 2006, 2007b). Thus variations in synaptic weights within a circuit could arise when a timely event - a secreted factor or elevated calcium signaling with activity - impacts developing synapses at different stages of formation or maturation.
ApCAM and novel PKC activation contribute to initial synapse formation and growth
How does reducing ApCAM from the surface of L7, but not sensory neurons or L11, interfere with synapse-associated growth by sensory neurons and the initial formation of synapses? Although reducing ApCAM on the surface of L7 would lead to the defasciculation of new neuritic growth, such alterations do not affect the structure of preexisting L7 neurites (Keller and Schacher, 1990) where the sensory neurons generally form the initial synapses. We did not explore whether reducing ApCAM affected other postsynaptic properties of L7. This general decrease in adhesion following the downregulation of ApCAM on the surface of L7 may also reduce the signaling by other potential surface molecules critical for evoking appropriate responses. However, more likely is that ApCAM on L7 acts as a receptor/ligand interacting with a heterophilic receptor/ligand on the sensory neurons leading to bidirectional consequences on synapse-associated growth by the sensory neurons and synapse formation. This ligand is unlikely to be the homologous receptor ApCAM, since downregulating ApCAM from sensory neurons failed to interfere with synapse formation or synapse-associated growth. The receptor on the sensory neuron could be another member of the Ig superfamily or other receptor families that interact with members of the Ig superfamily (Maurel et al., 1994; Grumet, 1997; Storms and Rutishauser, 1998; Ashley et al., 2005; Nielsen et al., 2010). DsCAM is an Ig member that contributes to aspects of synapse formation between sensory neurons and L7 (Li et al., 2009) and may also contribute to the bidirectional signaling along with postsynaptic ApCAM to initiate synapse assembly.
One potential signaling cascade initiated by ApCAM is the activation of PKC (Maness and Schachner, 2007; Ditlevsen et al., 2008). Blocking all PKC activity, blocking the novel PKC activity only in L7, or reducing ApCAM on the surface of L7 all block initial synapse formation and synapse-associated growth by sensory neurons. In addition, reducing ApCAM on the surface of L7 interferes with the sensory neuron-induced accumulation of the novel PKC at sites of contact. While it is likely that the ApCAM-dependent accumulation of the novel PKC Apl II at contact sites is due to activation (Sossin, 2007), this was not shown directly. Thus ApCAM on L7 may activate signaling cascades both in sensory neurons via the heterophilic receptor and in L7 to influence the expression and trafficking of molecules including receptors required for the assembly of functional synapses (Conrad et al., 1999; Washbourne et al., 2004; Sytnyk et al., 2006; Kohsaka et al., 2007; Cai et al., 2008; Li et al., 2009; Villareal et al., 2009).
Although contact with L11 produces comparable axon growth by the sensory neuron during the first 4 h, that growth is not associated with the formation of synapses, does not lead to the accumulation of the novel PKC, and is independent of ApCAM expression or PKC activation. The failure of sensory neurons to produce accumulation of the novel PKC on L11 could result either from subtle differences in the properties of ApCAM expressed on L11 (different sugar side chains), or the absence of appropriate downstream adaptors in L11 to mediate signaling by ApCAM. Other factors may influence responses when sensory neurons contact L11. By 6 h, target-dependent differences emerge in sensory neuron growth when contacting L7 compared with L11 (Glanzman et al., 1989; Hu et al., 2004b, 2007b). The decline in axon growth when sensory neurons contact L11 relative to contact with L7 is independent of ApCAM expression on L11. Other retrograde signals emanating from L11, such as secretion of the L11 neuropeptide (Taussig et al., 1984), may influence these presynaptic responses. Thus several cell-specific factors might coordinate presynaptic and postsynaptic behavior in the formation and maturation of specific synapses (Fox et al., 2007).
The requirement for ApCAM expression on L7, but not sensory neurons, for synapse-associated growth and synapse formation parallels ApCAM's role in structural plasticity accompanying long-term facilitation (LTF) and long-term depression (LTD) of sensory neuron synapses (Mayford et al., 1992; Bailey et al., 1992, 1997; Zhu et al., 1994, 1995; Schacher et al., 2000; Lee et al., 2007). With LTF, ApCAM reduction from sensory neurons precedes the growth of new sensory neuron branches and the formation of new varicosities at sites where ApCAM expression on L7 is high (Mayford et al., 1992; Bailey et al., 1992, 1997; Zhu et al., 1995). In addition to the retrograde signals reaching the cell body of the sensory neuron produced by ApCAM removal (Lee et al., 2007), the structural plasticity initiated soon after the loss of ApCAM from the surface of the sensory neurons may be mediated by a local increase in the interaction between ApCAM expressed on L7 and the heterophilic receptor/ligand expressed on the sensory neurons that we suggest may drive initial synapse formation and synapse-associated growth. With LTD, ApCAM reduction from L7 precedes retraction of sensory neuron branches and the loss of synaptic contacts (Zhu et al., 1995). These presynaptic changes produced by the loss of ApCAM on L7 parallel the consequences on synapse formation and synapse-associated growth when ApCAM on L7 is reduced experimentally with pretreatment with antibody 4E8.
ApCAM and novel PKC activation in L7 regulate presynaptic sensorin expression
Multiple factors regulate the expression of sensorin in sensory neurons. Early upregulation at distal sites is independent of target interaction and may arise from other external cues that influence the local translation of sensorin mRNA that accumulates at distal sites (Schacher et al., 1999; Hu et al., 2002; Lyles et al., 2006). Blocking PKC activity in both cells before initial synapse formation blocked upregulation of sensorin even in the distal axon (Hu et al., 2007b) (Fig. 2 A). The activity of the novel PKC in L7 is required to signal a significant increase in the expression of sensorin in the sensory neuron cell body at 6 h. PKC activity in L7 may signal a retrograde response in the presynaptic terminal that reaches the cell body of the sensory neuron with a short delay to increase sensorin primarily by translational regulation (see Hu et al., 2002, their Fig. 1). With time, expression of sensorin in the distal processes and cell bodies of sensory neurons interacting with L7 remains high, while expression in sensory neurons interacting with L11 declines. The failure of ApCAM in L11 to signal the accumulation and the potential activation of the novel PKC may explain why sensorin levels fail to increase in the sensory neuron. This coincides with the emergence of target-dependent differences in the accumulation of sensorin mRNA at distal sites of the sensory neurons that might be produced by the degradation of the transported mRNA (Hu et al., 2002; Lyles et al., 2006). The signal from L11 also appears to influence sensorin expression in the cell body, since levels of sensorin declines in that compartment starting at 6–8 h.
In summary, our results identified two critical factors in a specific postsynaptic target—a member of the Ig family of cell adhesion molecules and activation of a novel PKC—that regulate the initial formation of specific synapses. Interactions by the surface protein and the activation of a novel PKC in the specific postsynaptic target influence both synapse-associated growth and the increased expression of a presynaptic neuropeptide required for synapse maturation.
Footnotes
The research was supported by National Institutes of Health (NIH) Grants MH 60387 and NS 42159 to S.S. and Canadian Institute of Health Research Grant MOP12046 to W.S.S. Animals were provided by the National Center for Research Resources for Aplysia at the University of Florida in Miami, which is supported by NIH Grant RR-10294. We thank Drs. John Koester and Amir Levine for comments on earlier drafts of the manuscript.
References
- Ashley J, Packard M, Ataman B, Budnik V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci. 2005;25:5943–5955. doi: 10.1523/JNEUROSCI.1144-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Chen M, Keller F, Kandel ER. Serotonin-mediated endocytosis of ApCAM: an early step of learning-related synaptic growth in Aplysia . Science. 1992;256:645–649. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kaang BK, Chen M, Martin KC, Lim CS, Casadio A, Kandel ER. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of ApCAM in Aplysia sensory neurons. Neuron. 1997;18:913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- Baines RA, Seugnet L, Thompson A, Salvaterra PM, Bate M. Regulation of synaptic connectivity: levels of Fasciclin II influence synaptic growth in the Drosophila CNS. J Neurosci. 2002;22:6587–6595. doi: 10.1523/JNEUROSCI.22-15-06587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank M, Schacher S. Segregation of presynaptic inputs on an identified target neuron in vitro: structural remodeling visualized over time. J Neurosci. 1992;12:2960–2972. doi: 10.1523/JNEUROSCI.12-08-02960.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougie JK, Lim T, Farah CA, Manjunath V, Nagakura I, Ferraro GB, Sossin WS. The atypical protein kinase C in Aplysia can form a protein kinase M by cleavage. J Neurochem. 2009;109:1129–1143. doi: 10.1111/j.1471-4159.2009.06045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O, Fentrop N, Lee AY, Salmen B, Law JW, Wotjak CT, Schweizer M, Dityatev A, Schachner M. Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J Neurosci. 2004;24:1565–1577. doi: 10.1523/JNEUROSCI.3298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Chen S, Glanzman DL. Postsynaptic regulation of long-term facilitation in Aplysia . Curr Biol. 2008;18:920–925. doi: 10.1016/j.cub.2008.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad P, Wu F, Schacher S. Changes in functional glutamate receptors on a postsynaptic neuron accompany formation and maturation of an identified synapse. J Neurobiol. 1999;39:237–248. [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signaling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic analysis of the mechanisms controlling target selection: target-derived Fasciclin II regulates the pattern of synapse formation. Neuron. 1997;19:561–573. doi: 10.1016/s0896-6273(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Ditlevsen DK, Povlsen GK, Berezin V, Bock E. NCAM-induced intracellular signaling revisited. J Neurosci Res. 2008;86:727–743. doi: 10.1002/jnr.21551. [DOI] [PubMed] [Google Scholar]
- Doherty P, Fazeli MS, Walsh FS. The neural cell adhesion molecule and synaptic plasticity. J Neurobiol. 1995;26:437–446. doi: 10.1002/neu.480260315. [DOI] [PubMed] [Google Scholar]
- Farah CA, Weatherill D, Dunn TW, Sossin WS. PKC differentially translocates during spaced and massed training in Aplysia . J Neurosci. 2009;29:10281–10286. doi: 10.1523/JNEUROSCI.1533-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fässler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, Rheault MN, Sado Y, Segal Y, Werle MJ, Umemori H. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Glanzman DL, Kandel ER, Schacher S. Identified target motor neuron regulates neurite outgrowth and synapse formation of Aplysia sensory neurons in vitro. Neuron. 1989;3:441–450. doi: 10.1016/0896-6273(89)90203-1. [DOI] [PubMed] [Google Scholar]
- Goodman CS. Mechanisms and molecules that control growth cone guidance. Annu Rev Neurosci. 1996;19:341–377. doi: 10.1146/annurev.ne.19.030196.002013. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Davis GW, Zito K. The many faces of fasciclin II: genetic analysis reveals multiple roles for a cell adhesion molecule during the generation of neuronal specificity. Cold Spring Harbor Symp Quant Biol. 1997;62:479–491. [PubMed] [Google Scholar]
- Grumet M. Nr-CAM: a cell adhesion molecule with ligand and receptor functions. Cell Tissue Res. 1997;290:423–428. doi: 10.1007/s004410050949. [DOI] [PubMed] [Google Scholar]
- Hu JY, Meng X, Schacher S. Target interaction regulates distribution and stability of specific mRNAs. J Neurosci. 2002;22:2669–2678. doi: 10.1523/JNEUROSCI.22-07-02669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Glickman L, Wu F, Schacher S. Serotonin regulates the secretion and autocrine action of a neuropeptide to activate MAPK required for long-term facilitation in Aplysia . Neuron. 2004a;43:373–385. doi: 10.1016/j.neuron.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Hu JY, Goldman J, Wu F, Schacher S. Target-dependent release of a presynaptic neuropeptide regulates the formation and maturation of specific synapses in Aplysia . J Neurosci. 2004b;24:9933–9943. doi: 10.1523/JNEUROSCI.3329-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Wu F, Schacher S. Two signaling pathways regulate the expression and secretion of a neuropeptide required for long-term facilitation in Aplysia . J Neurosci. 2006;26:1026–1035. doi: 10.1523/JNEUROSCI.4258-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Chen Y, Schacher S. Protein kinase C regulates local synthesis and secretion of a neuropeptide required for activity-dependent long-term synaptic plasticity. J Neurosci. 2007a;27:8927–8939. doi: 10.1523/JNEUROSCI.2322-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Chen Y, Schacher S. Multifunctional role of protein kinase C in regulating the formation and maturation of specific synapses. J Neurosci. 2007b;27:11712–11724. doi: 10.1523/JNEUROSCI.3305-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller F, Schacher S. Neuron-specific membrane glycoproteins promoting neurite fasciculation in Aplysia californica . J Cell Biol. 1990;111:2637–2650. doi: 10.1083/jcb.111.6.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka H, Takasu E, Nose A. In vivo induction of postsynaptic molecular assembly by the cell adhesion molecule Fasciclin II. J Cell Biol. 2007;179:1289–1300. doi: 10.1083/jcb.200705154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lim CS, Park H, Lee JA, Han JH, Kim H, Cheang YH, Lee SH, Lee YS, Ko HG, Jang DH, Kim H, Miniaci MC, Bartsch D, Kim E, Bailey CH, Kandel ER, Kaang BK. Nuclear translocation of CAM-associated protein activates transcription for long-term facilitation in Aplysia . Cell. 2007;129:801–812. doi: 10.1016/j.cell.2007.03.041. [DOI] [PubMed] [Google Scholar]
- Li HL, Huang BS, Vishwasrao H, Sutedja N, Chen W, Jin I, Hawkins RD, Bailey CH, Kandel ER. Dscam mediates remodeling of glutamate receptors in Aplysia during de novo and learning-related synaptic plasticity. Neuron. 2009;61:527–540. doi: 10.1016/j.neuron.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthl A, Laurent JP, Figurov A, Muller D, Schachner M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature. 1994;372:777–779. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- Lyles V, Zhao Y, Martin KC. Synapse formation and mRNA localization in cultured Aplysia neurons. Neuron. 2006;49:349–356. doi: 10.1016/j.neuron.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Manseau F, Sossin WS, Castellucci VF. Long-term changes in excitability induced by protein kinase C activation in Aplysia sensory neurons. J Neurophysiol. 1998;79:1210–1218. doi: 10.1152/jn.1998.79.3.1210. [DOI] [PubMed] [Google Scholar]
- Manseau F, Fan X, Hueftlein T, Sossin W, Castellucci VF. Ca2+-independent protein kinase C Apl II mediates the serotonin-induced facilitation at depressed Aplysia sensorimotor synapses. J Neurosci. 2001;21:1247–1256. doi: 10.1523/JNEUROSCI.21-04-01247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel P, Rauch U, Flad M, Margolis RK, Margolis RU. Phosphacan, a chondroitin sulfate proteoglycan of brain that interacts with neurons and neural cell-adhesion molecules, is an extracellular variant of a receptor-type protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 1994;91:2512–2516. doi: 10.1073/pnas.91.7.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Barzilai A, Keller F, Schacher S, Kandel ER. Modulation of an NCAM-related adhesion molecule with long-term synaptic plasticity in Aplysia . Science. 1992;256:638–644. doi: 10.1126/science.1585176. [DOI] [PubMed] [Google Scholar]
- Nakhost A, Kabir N, Forscher P, Sossin WS. Protein kinase C isoforms are translocated to microtubules in neurons. J Biol Chem. 2002;277:40633–40639. doi: 10.1074/jbc.M205099200. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Kulahin N, Walmod PS. Extracellular protein interactions mediated by the neural cell adhesion molecule NCAM: heterophilic interactions between NCAM and cell adhesion molecules, extracellular matrix protein and viruses. Adv Exp Med Biol. 2010;663:23–53. doi: 10.1007/978-1-4419-1170-4_2. [DOI] [PubMed] [Google Scholar]
- Packard M, Mathew D, Budnik V. FASt remodeling of synapses in Drosophila . Curr Opin Neurobiol. 2003;13:527–534. doi: 10.1016/j.conb.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Pepio AM, Thibault GL, Sossin WS. Phosphoinositide-dependent kinase phosphorylation of protein kinase C Apl II increases during intermediate facilitation in Aplysia . J Biol Chem. 2002;277:37116–37123. doi: 10.1074/jbc.M202264200. [DOI] [PubMed] [Google Scholar]
- Peter N, Aronoff B, Wu F, Schacher S. Decrease in growth cone-neurite fasciculation by sensory or motor cells in vitro accompanies downregulation of Aplysia cell adhesion molecules by neurotransmitters. J Neurosci. 1994;14:1413–1421. doi: 10.1523/JNEUROSCI.14-03-01413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport SG, Schacher S. Synaptic plasticity in vitro: cell culture of identified Aplysia neurons mediating short-term habituation and sensitization. J Neurosci. 1986;6:759–763. doi: 10.1523/JNEUROSCI.06-03-00759.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M. Many paths to synaptic specificity. Annu Rev Cell Dev Biol. 2009;25:161–195. doi: 10.1146/annurev.cellbio.24.110707.175402. [DOI] [PubMed] [Google Scholar]
- Schacher S, Montarolo PG. Target-dependent structural changes in sensory neurons of Aplysia accompany long-term heterosynaptic inhibition. Neuron. 1991;6:679–690. doi: 10.1016/0896-6273(91)90166-w. [DOI] [PubMed] [Google Scholar]
- Schacher S, Wu F, Panyko JD, Sun ZY, Wang D. Expression and branch-specific export of mRNA are regulated by synapse formation and interaction with specific postsynaptic targets. J Neurosci. 1999;19:6338–6347. doi: 10.1523/JNEUROSCI.19-15-06338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacher S, Wu F, Sun ZY, Wang D. Cell-specific changes in expression of mRNAs encoding splice variants of Aplysia cell adhesion molecule accompany long-term synaptic plasticity. J Neurobiol. 2000;45:152–161. doi: 10.1002/1097-4695(20001115)45:3<152::aid-neu3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Sossin WS. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn Mem. 2007;14:236–246. doi: 10.1101/lm.469707. [DOI] [PubMed] [Google Scholar]
- Storms SD, Rutishauser U. A role for polysialic acid in neural cell adhesion molecule heterophilic binding to proteoglycans. J Biol Chem. 1998;273:27124–27129. doi: 10.1074/jbc.273.42.27124. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Nikonenko AG, Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J Cell Biol. 2006;174:1071–1085. doi: 10.1083/jcb.200604145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig R, Kaldany RR, Scheller RH. A cDNA clone encoding neuropeptides isolated from Aplysia neuron L11. Proc Natl Acad Sci U S A. 1984;81:4988–4992. doi: 10.1073/pnas.81.15.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villareal G, Li Q, Cai D, Fink AE, Lim T, Bougie JK, Sossin WS, Glanzman DL. Role of protein kinase C in the induction and maintenance of serotonin-dependent enhancement of the glutamate response in isolated siphon motor neurons of Aplysia californica . J Neurosci. 2009;29:5100–5107. doi: 10.1523/JNEUROSCI.4149-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG, McAllister AK. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci. 2004;24:8253–8264. doi: 10.1523/JNEUROSCI.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Schacher S. Pre- and postsynaptic changes mediated by two second messengers contribute to expression of Aplysia long-term heterosynaptic inhibition. Neuron. 1994;12:407–421. doi: 10.1016/0896-6273(94)90281-x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Leal K, Abi-Farah C, Martin KC, Sossin WS, Klein M. Isoform specificity of PKC translocation in living Aplysia sensory neurons and a role for Ca2+-dependent PKC APL I in the induction of intermediate-term facilitation. J Neurosci. 2006;26:8847–8856. doi: 10.1523/JNEUROSCI.1919-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu F, Schacher S. Aplysia cell adhesion molecules and serotonin regulate sensory cell-motor cell interactions during early stages of synapse formation in vitro . J Neurosci. 1994;14:6886–6900. doi: 10.1523/JNEUROSCI.14-11-06886.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu F, Schacher S. Changes in expression and distribution of Aplysia cell adhesion molecules can influence synapse formation and elimination in vitro . J Neurosci. 1995;15:4173–4183. doi: 10.1523/JNEUROSCI.15-06-04173.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]