

Abstract
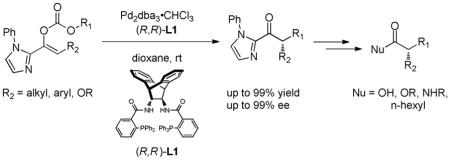
A broad range of highly enantioenriched 2-acylimidazoles are synthesized by palladium-catalyzed decarboxylative asymmetric allylic alkylation (DAAA) of 2-imidazolo-substituted enol carbonates. The enantioenriched 2-acylimidazole products can easily be converted to the corresponding carboxylic acid, ester, amide, and ketone derivatives with complete retention of the enantiopurity. The synthetic utility of this new method is demonstrated in the short, efficient synthesis of cetiedil.
The stereocontrolled alkylation of carbonyl compounds is a staple reaction in organic synthesis for the construction of highly enantioenriched materials. In particular, the alkylation of ester enolate equivalents has found widespread use due to the versatility of the resulting ester or amide products.1 Success in the field of ester enolate alkylations has relied on the use of chiral auxiliaries to direct the stereochemical outcome of the alkylation.2 Catalytic asymmetric ester enolate alkylations, although more desirable, are rare.3,4 Andrus and coworkers have reported the alkylation of a single 2-acylimidazole derivative (1-(1-methyl-1H-imidazol-2-yl)-2-(naphthalen-2-ylmethoxy)ethanone)5 with allylic and benzylic bromides that gives products with 75–99% ee under phase transfer catalysis using chiral cinchonidinium catalysts. They later disclosed a similar, albeit less stereoselective, alkylation of specifically phenethyl 2-naphthylacetate.6 While promising, these examples have only been demonstrated for a 2-naphthyl substituent and require superstoichiometric amounts of base and alkylating agents. In this report we describe the first catalytic asymmetric allylic alkylation (AAA) of a broad range of 2-acylimidazole-derived enol carbonates, the products of which are easily converted to a variety of ketone and carboxylic acid derivatives.
The transformation of 2-acylimidazoles to carboxylic acid derivatives was first reported by Ohta and coworkers.7 They found that while 2-acylimidazoles did not undergo acyl transfer reactions, alkylation of the imidazole nitrogen generated an activated leaving group that allowed substitution to occur (eq. 1). Based on this finding, we envisioned expanding the scope of our AAA reaction of enol carbonates to include 2-acylimidazoles as the first efficient and general example of catalytic asymmetric alkylations of ester enolate equivalents.
 |
(1) |
Our initial studies focused on the decarboxylative allylic alkylation reaction of allyl enol carbonates 1 catalyzed by Pd2(dba)3·CHCl3 (2) and L1 (Table 1, entries 1–4). While the reaction proceeded in a number of solvents, conducting it in dioxane generated alkylated product 3a with the highest yield and ee (entry 1). Varying the N-substituent on the imidazole portion of the enol carbonate from methyl (1a) to phenyl (1b) led to higher yields and higher ee of the product 3b (entry 5). Further varying R to 2-methylphenyl (1c) or 1-naphthyl (1d) gave similar yields but led to a small decrease in the ee of the products 3c–d (entries 6–7). In addition, varying the ligand from L1 to L2–L4 gave product 3b in good yields, but with lower enantioselectivity.
Table 1.
Selected optimization studies
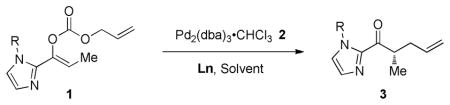 | |||||||
|---|---|---|---|---|---|---|---|
| entrya | solvent | Nu | R | Ln | 3 | Yield (%)b | ee (%)c |
| 1 | Dioxane | 1a | Me | L1 | 3a | 89 | 87 |
| 2 | CH2Cl2 | 1a | Me | L1 | 3a | 10 | n.d. |
| 3 | Toluene | 1a | Me | L1 | 3a | 83 | 76 |
| 4 | THF | 1a | Me | L1 | 3a | 65 | 80 |
| 5 | Dioxane | 1b | Ph | L1 | 3b | 96 | 92 |
| 6 | Dioxane | 1c | 2-Tol | L1 | 3c | 95 | 89 |
| 7 | Dioxane | 1d | 1-naph | L1 | 3d | 99 | 87 |
| 8 | Dioxane | 1b | Ph | L2 | 3b | 99 | 76 |
| 9 | Dioxane | 1b | Ph | L3 | 3b | 93 | 76 |
| 10 | Dioxane | 1b | Ph | L4 | 3b | 99 | 58 |
Reactions performed using 0.2 mmol substrate, 2.5 mol % 2, and 6 mol % Ln in 1 mL of solvent at ambient temperature for 16 hr.
Isolated yields.
Determined by chiral HPLC analysis. n.d = not determined.
The reaction scope under our optimized conditions (Table 1, entry 5) is summarized in Table 2. A variety of acyclic and cyclic allylic carbonates8 (4a–d) participate in the reaction to generate 2-acylimidazole products 5a–d in high yield and high ee (entries 2–5). Aryl and alkyl substitution on the enol carbonate are also tolerated (entries 6–9). The mild reaction conditions also permit the presence of alkynes (entry 6) and heteroaromatic substituents (entry 9). A variety of O-protected α-hydroxyketones can also be generated in good yield and ee using L2. To demonstrate the scalability of this process, reaction of enol carbonate 1b was performed on 2 mmol scale using just 1 mol % 2 and 2.5 mol % L1 to generate product 3b in 97% yield and 94% ee (entry 13).9
Table 2.
Reactions of different imidazole enol carbonates
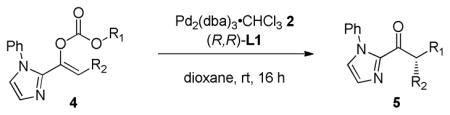 | |||||||
|---|---|---|---|---|---|---|---|
| entrya | 4 | R1 | R2 | 5 | Yield (%)b | drc | ee (%)d |
| 1 | 1b | Me | 3b | 96 | 92 | ||
| 2 | 4a | Me | 5a | 95 | 92 | ||
| 3 | 4b | Me | 5b | 69 | >95:5 | >99 | |
| 4 | 4c | Me | 5c | 91 | >95:5 | 97 | |
| 5e,f | 4d | Me | 5d | 80j | 46 | ||
| 6 | 4e | 5e | 91 | 93 | |||
| 7 | 4f | 5f | 75 | 94 | |||
| 8 | 4g | Ph | 5g | 99 | >95:5 | 86 | |
| 9 | 4h | 5h | 98 | >95:5 | 97 | ||
| 10g,h | 4i | OMOM | 5i | 85 | 71 | ||
| 11g | 4j | OBn | 5j | 89 | 73 | ||
| 12g | 4k | OTBDMS | 5k | 54 | 10:1 | >99 | |
| 13i | 1b | Me | 3b | 97 | 94 | ||
Unless otherwise noted, reactions were performed using 0.2 mmol substrate, 2.5 mol % 2, and 6 mol % L1 in 1 mL of solvent at ambient temperature for 16 hr.
Isolated yields.
Determined by 1H analysis of the crude reaction mixture.
Determined by chiral HPLC analysis.
Reaction run for 48hrs.
L4 was employed.
L2 was employed.
Reaction time was 30 min.
Performed on 2 mmol scale with 1 mol % 2 and 2.5 mol % L1 for 18 hr.
10:1 mixture trans:cis diene.
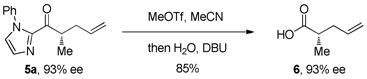
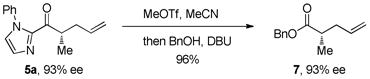
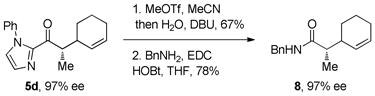
Our attention next turned to conversion of the 2-acylimidazole products to the corresponding carboxylic acid or ketone derivatives. While acyl transfer reactions of 2-acylimidazoles are well precedented,10 we were unsure whether the transformations could be performed without racemizing the α-keto stereocenter. We were delighted to find that hydrolysis of the imidazole under standard conditions (treatment with MeOTf in MeCN followed by addition of H2O and DBU) gave carboxylic acid 6 in 85% yield with complete retention of stereochemistry (eq 2). In a similar fashion, 2-acylimidazole 5a was converted to the benzyl ester derivative in 96% yield (eq 3). During our derivatization studies we found that direct conversion of the 2-acylimidazoles to amides or thioesters gave racemic products; converting the 2-acylimidazole first to the acid and then to the amide prevents racemization (eq 4). Conversion of imidazole ketone 5d to the corresponding alkyl ketone by treatment of the methylated imidazole with n-hexylmagnesium bromide also proceeded smoothly to give ketone 9 in good yield (eq 5). Synthesizing α-chiral aliphatic ketones using this methodology circumvents the otherwise difficult alkylation of unsymmetrical ketones where regioselective enolization is challenging.
 |
(2) |
 |
(3) |
 |
(4) |
 |
(5) |
The synthetic utility of this new methodology is demonstrated by the concise synthesis of cetiedil (Scheme 1). Cetiedil is currently used clinically in racemic form for the treatment of vascular disease.11 While enantiomerically pure cetiedil is generally obtained by fractional crystallization,12 one asymmetric synthesis has been reported.13 Ours begins with acylation of 1-phenylimidazole with Weinreb amide 10 to give 2-acylimidazole 11. Formation of enol carbonate 4i by O-acylation of the sodium enolate, followed by decarboxylative allylic alkylation of 4i generates 2-acylimidazole 5i in 98% yield and 97% ee. Hydrogenation of the double bond in 5i gives ketone 12 in 90% yield and 90% ee.14 Treatment of 12 with methyl triflate then amino alcohol 13 produces cetiedil in 89% yield and 90% ee.
Scheme 1.

Synthesis of cetiedil
In summary, we report the synthesis of highly enantioenriched 2-acylimidazoles by palladium-catalyzed decarboxylative asymmetric allylic alkylation of 2-imidazolo-substituted enol carbonates. The absolute configuration of the products of alkylation were established by comparison of carboxylic acid 615 and our synthetic cetiedil13 to known materials. In contrast to previously reported methods, the catalytic asymmetric alkylation methodology presented herein is quite general in scope for both the allylic electrophile and enolate nucleophile. The enantioenriched 2-acylimidazole products can easily be converted to the corresponding carboxylic acid, ester, amide, and ketone derivatives with complete retention of the enantiopurity.
Supplementary Material
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health, General Medical Sciences Grant GM33049, for their generous support of our programs. K. L. thanks the Deutscher Akademischer Austausch Dienst (DAAD) for a fellowship. D. J. M. thanks the NIH for a postdoctoral fellowship (F32 GM093467-01).
Footnotes
Supporting information available: Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Evans DA. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1983. pp. 83–110. Chapter 1.Meyers AI. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1983. pp. 213–274. Chapter 3.
- 2.For recent reviews, see: Prabhat; Qin H. Tetrahedron. 2000;56:917–947.Vicario JL, Badía D, Carrillo L, Reyes E, Etxebarria J. Curr Org Chem. 2005;9:219–235.
- 3.For selected examples of catalytic asymmetric alkylations of ketones and derivatives, see: Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J Am Chem Soc. 1994;116:8829–8830.Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. Tetrahedron. 2000;56:179–185.Doyle AG, Jacobsen EN. J Am Chem Soc. 2004;127:62–63. doi: 10.1021/ja043601p.Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x.Andrus MB, Hicken EJ, Stephens JC, Bedke DK. J Org Chem. 2006;71:8651–8654. doi: 10.1021/jo061395t.Doyle AG, Jacobsen EN. Angew Chem, Int Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901.Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948.For an example of an intramolecular alkylation of aldehydes see: Vignola N, List B. J Am Chem Soc. 2003;126:450–451. doi: 10.1021/ja0392566.
- 4.For reviews on the alkylation of glycine derivatives by chiral phase-transfer catalysis, see: Maruoka K, Ooi T. Chem Rev. 2003;103:3013–3028. doi: 10.1021/cr020020e.Jew S, Park H. Chem Commun. 2009;46:7090–7103. doi: 10.1039/b914028j.
- 5.Andrus MB, Christiansen MA, Hicken EJ, Gainer MJ, Bedke DK, Harper KC, Mikkelson SR, Dodson DS, Harris DT. Org Lett. 2007;9:4865–4868. doi: 10.1021/ol702197r. [DOI] [PubMed] [Google Scholar]
- 6.Andrus MB, Harper KC, Christiansen MA, Binkley MA. Tetrahedron Lett. 2009;50:4541–4544. [Google Scholar]
- 7.Ohta S, Hayakawa S, Nishimura K, Okamoto M. Chem Pharm Bull. 1987;35:1058–1069. and references therein. [Google Scholar]
- 8.Isomerically pure (Z)-enol carbonates (>95:5 Z:E as determined by 1H NMR analysis) were generally synthesized as seen in Scheme 1.
- 9.Lower catalyst loadings were not explored.
- 10.For selected examples of the utility of 2-acylimidazoles in their conversion to other groups such as acids, esters, ketones, etc see: Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu R. J Am Chem Soc. 2007;129:10029–10041. doi: 10.1021/ja072976i.Evans DA, Song HJ, Fandrick KR. Org Lett. 2006;8:3351–3354. doi: 10.1021/ol061223i.Davies DH, Haire NA, Hall J, Smith EH. Tetrahedron. 1992;48:7839–7856.Bakhtiar C, Smith EH. J Chem Soc, Perkin Trans. 1994;1:239–243.
- 11.Schmidt WF, III, Asakura T, Schwartz E. J Clin Invest. 1982;69:589–594. doi: 10.1172/JCI110485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roxburgh CJ, Ganellin CR, Shiner MAR, Benton DCH, Dunn PM, Ayalew Y, Jenkinson DH. J Pharm Parmacol. 1996;48:851–857. doi: 10.1111/j.2042-7158.1996.tb03986.x. [DOI] [PubMed] [Google Scholar]
- 13.Davies HML, Walji AM, Townsend RJ. Tetrahedron Lett. 2002;43:4981–4983. [Google Scholar]
- 14.The drop in the enantioselectivity upon hydrogenation of 5i is reflective on the conversion of both diastereomers in 5i to 12.
- 15.Riley RG, Silverstein RM. Tetrahedron. 1974;30:1171–1174. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.