

Abstract
The transcription factor hypoxia-inducible factor-1 (HIF-1) regulates the expression of more than 70 genes involved in cellular adaptation and survival under hypoxic stress. Activation of HIF-1 is associated with numerous physiological and pathological processes that include tumorigenesis, vascular remodeling, inflammation, and hypoxia/ischemia-related tissue damage. Clinical studies suggested that HIF-1 activation correlates directly with advanced disease stages and treatment resistance among cancer patients. Preclinical studies support the inhibition of HIF-1 as a major molecular target for antitumor drug discovery. Considerable effort is underway, in government laboratories, industry and academia, to identify therapeutically useful small molecule HIF-1 inhibitors. Natural products (low molecular weight organic compounds produced by plants, microbes, and animals) continue to play a major role in modern antitumor drug discovery. Most of the compounds discovered to inhibit HIF-1 are natural products or synthetic compounds with structures that are based on natural product leads. Natural products have also served a vital role as molecular probes to elucidate the pathways that regulate HIF-1 activity. Natural products and natural product-derived compounds that inhibit HIF-1 are summarized in light of their biological source, chemical class, ancd effect on HIF-1 and HIF-mediated gene regulation. When known, the mechanism(s) of action of HIF-1 inhibitors are described. Many of the substances found to inhibit HIF-1 are non-druggable compounds that are too cytotoxic to serve as drug leads. The application of high-throughput screening methods, complementary molecular-targeted assays, and structurally diverse chemical libraries hold promise for the discovery of therapeutically useful HIF-1 inhibitors.
Keywords: HIF-1, Natural Product, Tumor Hypoxia, Molecular-Targeted Drug Discovery, Small Molecule HIF-1 Inhibitor, Hypoxia Selective
Introduction
In the human body, oxygen is delivered along a concentration gradient from the site of uptake in the lung capillaries to the site of consumption, where most of the oxygen is consumed by mitochondria to generate ATP [1]. Interruption of blood flow, reduction in oxygen tension, decreased oxygen carrying capacity, and failure to transport oxygen from the microvasculature to cells can all lead to an insufficient oxygen supply (hypoxia) to meet the metabolic requirements of specific tissues. In solid tumors, rapid tumor growth outstrips the capability of existing blood vessels to supply oxygen and hypoxic regions commonly occur in solid tumors [2–5]. Unlike normal cells from the same tissue, tumor cells are often chronically hypoxic. Hypoxia triggers tumor angiogenesis and the newly formed tumor blood vessels often fail to mature. As a result, certain tumor regions are constantly under hypoxic stress due to sluggish and irregular blood flow. The extent of tumor hypoxia correlates positively with advanced stages and poor prognosis [2–5]. Hypoxic tumor cells are more resistant than normoxic tumor cells to radiation treatment and chemotherapy and these hypoxic cells are considered an important contributor to disease relapse. Currently, the general strategies to overcome tumor hypoxia are: 1) increasing tumor oxygenation by means such as breathing carbogen (95% O2, 5% CO2); 2) developing chemical sensitizers to increase the sensitivity of hypoxic cells to radiation; 3) developing hypoxic cytotoxins that selectively kill hypoxic cells; and 4) developing hypoxia-selective gene therapy [2,3,5,6]. These approaches target the direct effect of hypoxia - lack of cellular oxygen. However, hypoxia also exerts indirect effects on tumor cells by inducing the expression of genes that promote hypoxic adaptation and survival. For example, hypoxia provides a physiological pressure and selects for cells with diminished apoptotic potential in oncogenically transformed cells [7].
The transcription factor that plays a critical role in hypoxia-induced gene expression is hypoxia-inducible factor-1 (HIF-1), a heterodimer of the basic helix-loop-helix (bHLH) PER-ARNT-SIM (PAS) proteins HIF-1α and HIF-1β /aryl hydrocarbon receptor nuclear translocator (ARNT) [8–10]. The HIF-1α subunit is degraded rapidly under normoxic conditions and stabilized under hypoxic conditions, while HIF-1β is constitutively expressed. In general, the availability and activity of HIF-1α protein determines the bioactivity of HIF-1. Numerous post-translational modifications are involved in regulating the stability and activity of HIF-1α protein [11–15]. In particular, oxygen-dependent prolyl hydroxylation destabilizes HIF-1α protein by promoting the tumor suppressor von Hippel-Lindau protein (pVHL)-mediated proteasomal degradation, while oxygen-dependent asparaginyl hydroxylation inactivates HIF-1α protein by preventing the interaction between HIF-1α protein and a co-activator CBP/p300 [CREB (cAMP-response element-binding protein)-binding protein/E1A-binding protein, 300 kD] [16–18]. In addition to hypoxia, other physiological and pathological factors such as cytokines, growth factors, hormones, activated oncogenes, inactivated tumor suppressors, etc., can also activate HIF-1 [11,12,19–25]. Over seventy genes are known to be regulated by HIF-1, most of these genes are regulated in a cell type-specific manner [11,19]. The list of HIF-1 target genes is still growing rapidly. Clinical studies have indicated that tumor hypoxia-associated HIF-1α overexpression correlates positively with advanced disease stages and poor prognosis among cancer patients [26–33]. Preclinical studies in numerous animal models have demonstrated that HIF-1 inhibition suppresses angiogenesis, retards tumor growth, and enhances treatment outcomes when used in combination with chemotherapeutic agents or radiation [34–43]. However, one study indicated that HIF-1α deficiency inhibits angiogenesis while promoting embryonic stem cell-derived tumor growth [44]. One recent study that used genetically engineered transformed murine astrocytes as an in vivo mouse model for astrocytoma suggested that the outcome of HIF-1 inhibition is dependent on the tumor microenvironment: HIF-1α deficiency retards tumor growth in the poorly vascularized subcutaneous region, while HIF-1α deficiency enhances tumor growth in the highly vascularized brain parenchyma [45]. A number of recent reviews provide an extensive overview of HIF-1 as a molecular target for cancer therapy [19–25].
A growing number of HIF-1 inhibitors of natural and synthetic origin have recently been identified. Small molecule synthetic HIF-1 inhibitors (not based on natural product or natural product-like structures) have been described in several recent reviews [19–24]. The focus of this review is natural product-based HIF-1 inhibitors and their therapeutic potential for cancer. In general, the term "natural product" refers to low molecular weight secondary metabolites produced by animals, plants, and microbes for chemical defense and growth advantage. Natural products have been a major source of new drugs for centuries and the chemical diversity offered by natural products has not been matched by any other approach [46]. Statistics show that over 60% of the approved anticancer agents are of natural origin (natural products or synthetic compounds based on natural product models). In this review, the natural product-derived HIF-1 inhibitors are grouped by the mechanisms employed to achieve HIF-1 inhibition.
Inhibitors of HIF-1α Protein Synthesis
The availability and activity of HIF-1α protein plays an important role in HIF-1 activation. Many of the known HIF-1 inhibitors function by decreasing HIF-1α protein. The level of HIF-1α protein is controlled by an intricate balance between production and degradation. Decreased HIF-1α synthesis or increased degradation can each block the accumulation of HIF-1α protein, while the increased synthesis or decreased degradation can each induce HIF-1α protein [Fig.(1)]. The compounds that decrease HIF-1α protein synthesis may function through the following mechanisms: inhibit transcription, degrade HIF-1α mRNA, and decrease translation.
Figure 1.

The Streptomyces parvullus (formerly S. antibioticus) metabolite actinomycin D (1, dactinomycin) is an inhibitor of transcription that was shown to abolish hypoxia-induced HIF-1 binding activity in Hep3B human heptoma cells [47]. A later study revealed that actinomycin D (1) blocked the induction of HIF-1α protein by angiotensin II (Ang II) in rat vascular smooth muscle cells but not the induction by hypoxia [48]. The HIF-1 inhibitory effects exerted by 1 may be dependent on the cell type, stimulus, and drug concentration.
The compound GL331 (2) is a cytotoxic, semisynthetic podophyllotoxin-derived topoisomerase II inhibitor with IC50 values in the range of 0.5 to 2 μM against a panel of tumor cell lines [49]. In CL1-5 human lung adenocarcinoma cells, GL331 (10 μM) inhibited HIF-1 activation by decreasing HIF-1α mRNA level, presumably through transcriptional inhibition [50]. This inhibitory effect is unlikely to be HIF-1specific. At comparable concentrations, 2 is cytotoxic and inhibited expression of genes such as cyclin D1 in CL1-5 cells [51]. GL331 (2) was ineffective against gastric cancer in a clinical study [52].
Acting through yet to be determined mechanisms, a number of natural products have been shown to decrease HIF-1α mRNA levels. The Indian traditional medicine known as picroliv (a purified iridoid glycoside fraction from the roots of Picrorhiza kurrooa) reduced HIF-1α and vascular endothelial growth factor (VEGF) mRNA levels in vitro [53]. The major compounds found in picroliv are the iridoid glycosides picroside-I and kutkoside [54]. The precise chemical constituents responsible for the reported HIF-1 inhibitory activity have not been defined. When CF-1 mice were fed on a diet enriched with soy-derived sphingolipids (0.025 and 0.1%), 1,2-dimethylhydrazine-induced colonic cell tumorigenesis was suppressed [55]. At the concentrations tested, these plant 4,8-sphingadiene glucosylceramide-type sphingolipids (i.e. 3) decreased HIF-1α mRNA levels in the intestinal mucosal cells by more than 50% [55]. The effect of plant sphingolipids on HIF-1 activation and target gene expression is not known.
The antifungal antibiotic cycloheximide (4, isolated from Streptomyces griseus) inhibits general eukaryotic protein synthesis and blocked hypoxia-induced HIF-1α protein accumulation and HIF-1 activation [8]. Compounds such as cycloheximide (4) and actinomycin D (1) are known to non-selectively inhibit gene expression, it is likely that the use of these types of natural products for their antitumor properties will always be accompanied by a considerable level of treatment-associated toxicity.
In an effort to identify small molecule HIF-1 inhibitors, a U251 human glioma cell-based high-throughput screening (HTS) assay was used to examine compounds from the National Cancer Institute (NCI) Diversity Set (~2,000 pure compounds representing the maximal three-dimensional chemical diversity) for HIF-1 inhibitory activity [56]. The compounds found to be active include three camptothecin analogues [topotecan (5), camptothecin 20-ester (S) (6), and 9-glycineamido-20(S)-camptothecin HCl (7)] and a quinocarmycin analogue DX-52-1 (8)[56]. The best characterized compound topotecan (5), inhibited both hypoxia (1% O2)- and iron chelator (desferoxime; DFO)-induced HIF-1 activation at submicromolar concentrations. Topotecan (5) inhibits HIF-1 by decreasing HIF-1α protein translation in a topoisomerase I-dependent, oxygen-independent manner [56,57]. The indenoisoquinoline MJ-III-65 [58], a synthetic non-camptothecin Topo I inhibitor exhibited similar HIF-1 inhibitory activity as that observed for topotecan [57]. The HIF-1 inhibitory activity of topotecan is reversible and schedule-dependent in vitro (U251 cells), and requires a daily (not intermittent) administration schedule in vivo (U251 tumor xenograft model) [43]. Topotecan (5) is a DNA topoisomerase I inhibitor and has been used clinically as an antineoplastic agent. The therapeutic potential of topotecan (5) as a HIF-1 inhibitor has been recently reviewed [59].
One group of compounds reported to inhibit HIF-1α protein synthesis is microtubule disrupting agents (MDA). In human prostate PC-3 and breast MDA-MB-231 carcinoma cells, the natural estradiol metabolite 2-methoxyestradiol (2ME2, 9) inhibited both basal and hypoxia-induced HIF-1α protein expression [60]. Mechanistic studies indicated that 2ME2 (9) inhibits HIF-1 by suppressing HIF-1α protein synthesis. Other MDAs that include the Taxus brevifolia diterpenoid taxol (paclitaxol, 10) and the Catharanthus roseus (formerly Vinca rosea) alkaloid vincristine (11) also inhibited hypoxia-induced HIF-1α protein accumulation and HIF-1 activation [60]. A separate study reported that MDAs vinblastine, colchicine, and the synthetic nocodazole actually induce HIF-1α protein and activate HIF-1 in several cell lines [61]. Microtubule disruption is required for both the inhibition and the induction of HIF-1α protein [60,61]. The theory of how 2ME2 (9) inhibits HIF-1 has been challenged by a recent study that suggested 9 promotes HIF-1α protein degradation under hypoxia via inhibition of the mitochondrial electron transport chain (complex I) [62]. In addition, 2ME2 (9) did not inhibit the induction of HIF-1α protein by either N-mercaptopropionylglycine (2-oxoglutarate analogue) or DFO (iron chelator) in HEK 293 cells [62]. Although 2ME2 (9) is currently undergoing clinical evaluation for the treatment of cancer, the concentrations that inhibit HIF-1 and mitochondrial function are far above those required for the antitumor effects [60,62–64]. Whether the inhibition of HIF-1 contributes to the in vivo antitumor activity of 2ME2 (9) remains unclear.
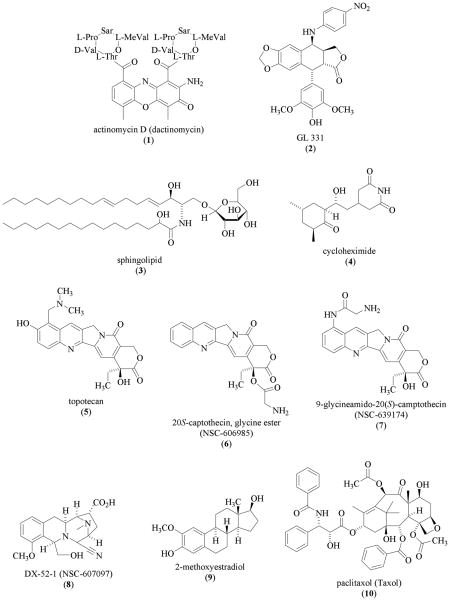
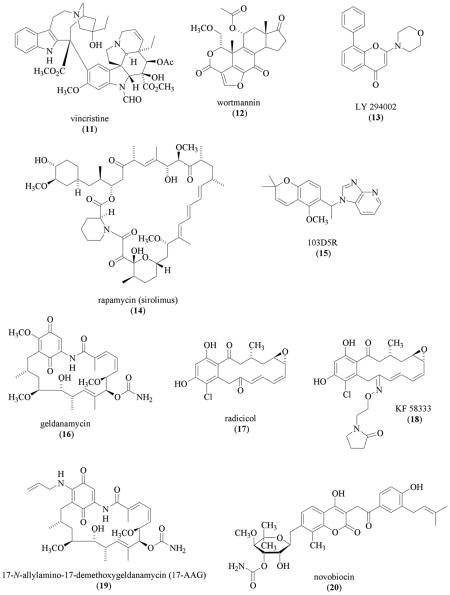
Studies from scores of research groups have contributed to the understanding of how the phosphoinositol 3-kinase/protein kinase B (PI3K/AKT) signaling pathway regulates HIF-1α protein accumulation. These findings were summarized in a number of recent reviews [19–24,65,66]. The PI3K inhibitors wortmannin (12, isolated from Penicillium fumiculosum) and LY294002 (13, flavonoid-like synthetic compound) inhibited HIF-1 activation by both hypoxia and non-hypoxic stimuli via inactivation of the downstream targets that regulate the translation of HIF-1α mRNA. Two recent studies suggest that the PI3K/AKT pathway is not involved in mediating the hypoxic induction of HIF-1α protein [67,68]. The regulatory effects exerted by the PI3K/AKT pathway on HIF-1 may depend on the specific cell type and the type of stimulus. One of the protein kinases that is regulated by AKT is the mammalian target of rapamycin (mTOR). The mTOR inhibitor rapamycin (14, isolated from Streptomyces hygroscopicus) was shown to inhibit HIF-1α protein accumulation by at least two mechanisms: inhibition of HIF-1α protein synthesis [69] and promotion of HIF-1α protein degradation [70].
A human glioma LN229 cell-based reporter assay was used to examine a combinatorial library of 10,000 "natural product-like" synthetic compounds that were based on a 2,2-dimethylbenzopyran scaffold [71]. The most active compound 103D5R (15) weakly inhibited HIF-1 activation by hypoxia (IC50 35 μM) and more potently suppressed LN229 cell proliferation (IC50 26 μM). The compound 103D5R (15) inhibited both hypoxia- and cobalt chloride-induced HIF-1 activation in a panel of cell lines by decreasing HIF-1α protein synthesis.
Natural Products that Promote HIF-1α Protein Degradation
Under normoxic conditions, HIF-1α protein is post-translationally modified and is rapidly degraded by the proteasome. Either a reduction in oxygen tension or the removal of enzyme cofactors can result in the stabilization of HIF-1α protein by inhibiting the enzymes that modify HIF-1α prior to proteasome degradation. Some HIF-1 inhibitors promote the degradation of HIF-1α protein in the presence of stimuli. For the purpose of this review, these types of HIF-1 inhibitors are grouped in the following text by the specific molecular target affected and the mechanism of action.
Since the first report of the interaction between HIF-1α protein and the molecular chaperone heat shock protein 90 (hsp90) [72], a number of studies have investigated the significance of this interaction using pharmacological agents that inhibit hsp90 [73–77]. Recent studies suggested that hsp90 binds to the HIF-1α PAS domains and stabilizes HIF-1α protein, and the HIF-1β /ARNT subunit competes with hsp90 for the same binding sites to stabilize HIF-1α protein [78,79]. The Streptomyces hygroscopicus metabolite geldanamycin (GA, 16) inhibits hsp90 by binding to the amino-terminal ATP/ADP binding pocket [80–82]. Geldanamycin (16) inhibited HIF-1 activation by promoting pVHL-independent proteasomal degradation of HIF-1α protein under both normoxic and hypoxic conditions [73,75]. The GA-induced, oxygen-independent, proteasome-mediated HIF-1α protein degradation is enhanced in the absence of serum in human prostate carcinoma PC-3 and LNCaP cells [74]. Geldanamycin (16) also inhibited heat induced nuclear accumulation of a transcriptionally inactive form of HIF-1α protein [76]. In contrast to 16, radicicol (17), another hsp90 inhibitor that also binds to the amino-terminal ATP/ADP binding pocket [83], inhibits HIF-1 in Hep3B cells by a different mechanism - blocking the binding of HIF-1 to the hypoxia-response elements (HREs) in promoters of HIF-1 target genes [77]. Using human breast carcinoma KPL-1 and KPL-4 cells as tumor models, an oxime derivative of radicicol KF58333 (18) was shown to decrease HIF-1α protein level in vitro and retard tumor growth in vivo [84]. Other hsp90 inhibitors that include the 17-N-allylamino-17-demethoxygeldanamycin (17-AAG, 19) and the structurally unique antibiotic novobiocin (20, produced by Streptomyces niveus and S. spheroides) also inhibited HIF-1α protein accumulation [75,76]. Geldanamycin (16) is unsuitable for clinical use due to its hepatotoxicity [85]. However, the geldanamycin derivative 17-AAG (19) is undergoing clinical trial for the treatment of cancer [85]. In preclinical models, 17-AAG (19) retarded tumor growth and enhanced the therapeutic outcome of radiation treatment [85,86]. Encouraging results from drug tolerance studies were obtained in a recently completed Phase I clinical study that used 17-AAG (19) to treat adult patients with solid tumors [87].
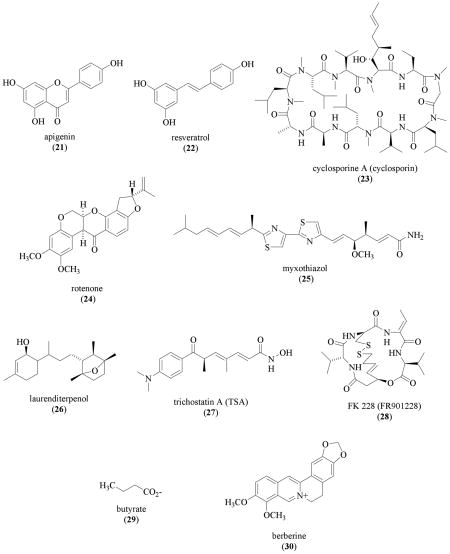
Apigenin (4',5,7-trihydroxyflavone, 21), a plant flavone found in many fruits and vegetables, has exhibited anti-tumor activity in numerous in vitro and in vivo models and is considered a chemopreventive agent for cancer [88–95]. Apigenin (100 μM, 21) inhibited both hypoxia (5% O2)-induced and transition metal (CoCl2)-induced HIF-1α protein in Hep3B cells [96]. This study suggested that 21 may destabilize HIF-1α protein by disrupting the interaction between HIF-1α protein and hsp90. A separate study demonstrated that 21 decreased HIF-1α mRNA levels and promoted the degradation of HIF-1α protein in human ovarian carcinoma A2780/CP70 and OVCAR-3 cells, independent of the oxygen status [97]. The HIF-1 inhibitory effect exerted by 21 is most likely mediated by multiple pathways. In addition to the PI3K/AKT/p70S6K1, hsp90, and HDM2 [human ortholog of Mdm2 (mouse double minute 2 homolog)]/p53 pathways discussed in these two related HIF-1 studies, apigenin (21) has been reported to affect other pathways/cellular targets that range from nuclear factor-κ B (NFκ B) to human epidermal growth factor receptor 2 (HER2/neu) at comparable or lower concentrations [88–95].
Resveratrol (trans-3,4,5'-trihydroxystilbene, 22) is a phytoalexin, plant metabolite produced in response to environmental stress such as fungal infection and injury. Resveratrol (22) has been found in various types of plants that range from grapes to peanuts [98–100]. The discovery that resveratrol inhibits a diverse set of cellular targets that are associated with the initiation, promotion, and progression of carcinogenesis [98] has sparked a tremendous amount of research on the potential of resveratrol as a cancer chemopreventive agent [99,100]. Resveratrol (22) was shown to inhibit basal, insulin-, and insulin-like growth factor 1 (IGF1)-induced expression of HIF-1α protein in A2780/CP70 and OVCAR-3 cells (IC50 values 20 to 30 μM) [101]. Resveratrol (22) inhibits HIF-1α protein expression by at least two mechanisms: inhibit HIF-1α protein synthesis and facilitate proteasome-mediated HIF-1α protein degradation [101]. In most of the experimental systems, 22 exerts its potential anti-tumor activities at the concentrations that range from 10 to 100 μM [99,100]. However, these concentrations (including the concentration required to inhibit HIF-1) are far beyond what is biologically available from food consumption [99]. It would be of considerable interest to investigate if any of the metabolites formed from resveratrol (22) can inhibit the activation of HIF-1 or its downstream targets at biologically relevant concentrations.
Prolyl hydroxylation is one of the earlier events during the destabilization and degradation of HIF-1α protein. Overexpression of the HIF prolyl hydroxylase 3 (HPH3, PHD1, EGLN2) decreases HIF-1α protein expression and inhibits tumor growth [102]. The fungal metabolite cyclosporine (cyclosporin A or CsA, 23, isolated from Tolypocladium inflatum Gams) is an immunosuppressive agent used to prevent the rejection of transplanted organs [103]. In rat glioma C6 cells, 23 (10 μM) inhibited hypoxia (2% O2)-induced HIF-1α protein accumulation and subsequent HIF-1 activation [104]. Cyclosporine (23) inhibits HIF-1 activation by preventing oxygen-dependent degradation domain (ODDD)-mediated HIF-1α protein stabilization. Biochemical studies suggest that 23 stimulates the prolyl hydroxylase(s) that modifies Pro-564 of the human HIF-1α protein. However, it remains unresolved regarding which prolyl hydroxylase(s) cyclosporine (23) activates or by what mechanism 23 activates the enzyme.
Hypoxia decreases the level of intracellular oxygen, inhibits the hydroxylases that modify HIF-1α protein, and leads to the stabilization HIF-1α protein and activation of HIF-1. The mitochondrion is the major cellular organelle that consumes oxygen. Mitochondrial inhibitors that include the plant derived natural product rotenone (24, isolated from Lonchocarpus spp.), diphenylene iodonium (DPI), myxothiazol (25, isolated from the myxobacteria Myxococcus fulvus/Stigmatella aurantiaca), and 4,4'-diisothiocyanatostilbene-2,2'-disulfonate (DIDS) inhibited hypoxia-induced HIF-1α protein accumulation, HIF-1 activity, and expression of HIF-1 target genes [105,106]. These compounds did not inhibit the induction of HIF-1α protein by stimuli such as DFO and CoCl2 [105,106]. The mitochondrial complex I inhibitor 1-methyl-4-phenylpiridinium also inhibited hypoxic induction of HIF-1α protein [107]. Other mitochondrial inhibitors that include sodium azide, antimycin A, and cyanide have been shown to inhibit hypoxia induced HIF-1α protein accumulation [108,109]. Under hypoxic conditions, mitochondrial inhibition increases the available intracellular oxygen level, promotes HIF-1α protein prolyl hydroxylation, and is followed by enhanced degradation of HIF-1α protein under hypoxic conditions [108].
While a growing number of natural products and natural product-derived compounds have been demonstrated to inhibit HIF-1, relatively few efforts have been directed at the discovery of novel HIF-1 inhibitors from natural product-rich extracts of plants, microbes, or marine organisms as the source of chemicals. Using a T47D human breast tumor cell-based reporter assay to monitor HIF-1 activity, our research group at the University of Mississippi evaluated the lipid-soluble extracts of several thousand plants and marine organisms for HIF-1 inhibitory activity [110,111]. Bioassay-guided fractionation of the lipid extract of a Jamaican sample of the red alga Laurencia intricata Lamouroux (Rhodomelaceae) yielded the structurally novel diterpene laurenditerpenol (26) [111]. Members of the genus Laurencia have been shown to be a rich source of several hundred unusual secondary metabolites. Laurenditerpenol (26) was determined to be a structurally novel bicyclic diterpene from the analysis of high-resolution spectroscopic and spectrometric data. The absolute configuration of position C-1 was determined by analysis of the proton nuclear magnetic resonance (1H-NMR) spectra of the modified Mosher ester derivatives that were prepared directly in NMR tubes. Laurenditerpenol is the first marine natural product found to inhibit hypoxia (1% O2)-induced HIF-1 activation. In T47D cells, 26 potently inhibited HIF-1 activation by hypoxia (IC50 0.4 μM) but not by the iron chelator 1,10-phenanthroline. Compound 26 inhibits HIF-1 by blocking the hypoxia-induced HIF-1α protein accumulation. Mitochondrial respiration studies revealed that laurenditerpenol (26) suppresses oxygen consumption at the concentrations that inhibit hypoxia-induced HIF-1 activation. Therefore, it is believed that laurenditerpenol is the first member of a structurally novel class of marine natural product-based mitochondrial inhibitors that block mitochondrial oxygen consumption and promote the degradation of HIF-1α protein under hypoxic conditions.
Following prolyl hydroxylation, the prolyl-hydroxylated HIF-1α protein is recognized by pVHL that is part of an E3 ubiquitin ligase complex. HIF-1α protein is then polyubiquitinized and degraded by the proteosome. Certain HIF-1 inhibitors are believed to function by increasing the expression of VHL under hypoxic conditions. Hypoxia increases the expression and activity of histone deacetylases (HDAC) that down-regulate tumor suppressors p53 and pVHL and up-regulate angiogenic factors such as VEGF through the induction of HIF-1α protein and activation of HIF-1 [112]. Histone deacetylases catalyze histone deacetylation and cause chromatin remodeling. The compound (R)-trichostatin A (TSA, 27), originally isolated from Streptomyces hygroscopicus as a fungistatic antibiotic, is a potent and specific HDAC inhibitor [113,114]. The compound TSA (27) was demonstrated to inhibit hypoxia-induced HIF-1α protein accumulation by inducing pVHL expression [112]. Another potent HDAC inhibitor FK228 dose-dependently inhibited hypoxia (1% O2)-induced HIF-1α protein accumulation and the downstream events in Lewis lung carcinoma tumor model in vitro and in vivo [115]. The compound FK228 (FR901228, 28) is a bicyclic depsipeptide produced by Chromabacterium violaceum that was originally isolated as an antitumor agent and later found to inhibit HDAC [116–119]. Butyrate (29), a weak inhibitor of HDAC, was also found to inhibit HIF-1 at millimolar concentrations [120–122]. One of the intended therapeutic applications of HIF-1 inhibitors is to augment the effects of radiation therapy. In preclinical models, HDAC inhibitors such as FK 228 (28) were shown to enhance radiation treatments [123,124]. A number of recent reviews have summarized the antitumor activity of HDAC inhibitors in preclinical models and their therapeutic potential for cancer [125–128]. Encouraging results have been reported from several phase I clinical trials that used FK228 (28) for cancer treatment [129–132].
Acetylation of HIF-1α protein at Lys-532 was reported to enhance the interaction between HIF-1α and pVHL [133]. The natural product berberine (30), a tautomeric alkaloid most commonly found in members of the Berberidaceae, was shown to inhibit hypoxia (2% O2)-induced HIF-1α protein accumulation by promoting the lysine acetylation of HIF-1α protein [134]. The concentration of 30 (7.5 μM) that inhibited the HIF-1 pathway is equal to the cytotoxic IC50 value in human gastric adenocarcinoma SC-M1 cells [134]. The inhibitory effect of berberine (30) is unlikely to be HIF-1 specific, since 30 also inhibited other transcription factors such as AP-1 at comparable concentrations [135].
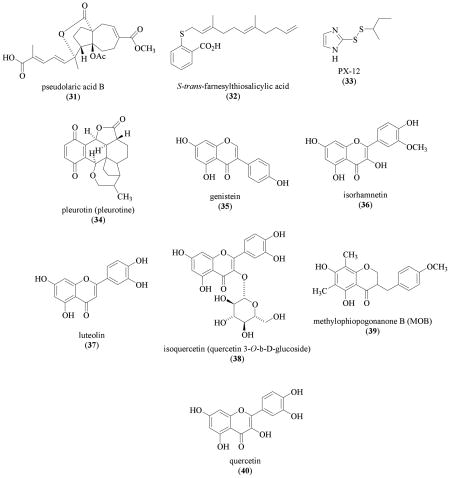
Pseudolaric acid B (31) is a cytotoxic diterpene isolated from Pseudolarix kaempferi Grord. (Pinaceae) (IC50 values that range from 0.02 to 1.23 μM in 39 tumor cell lines reported) [136]. In human breast carcinoma MDA-MB-468 and MDA-MB-435 cells, 31 (10 μM) inhibited hypoxia (1% O2)-induced HIF-1α protein by promoting proteasome-mediated degradation [137]. Pseudolaric acid B (31) suppressed the viability/proliferation of MDA-MB-468 cells at even lower concentrations (IC50 0.42 μM) [137]. Therefore, it is unlikely that the HIF-1 inhibition exerted by 31 is due to a specific effect on the pathways that regulate HIF-1. In addition to the reported cytotoxicity [136], 31 also inhibited VEGF-induced angiogenesis under normoxic conditions [137,138].
In glioblastoma multiforme cells, the Ras inhibitor trans-farnesylthiosalicylic acid (FTS, 32) (70 μM) inhibited hypoxia (1% O2) induced HIF-1α protein accumulation [139]. The observation that proteasome inhibitor MG132 blocked the FTS inhibition on HIF-1α protein suggests that 32 promotes HIF-1α protein degradation. The compound FTS (32) was shown to inhibit Ras downstream target the PI3K/AKT/mTOR pathway, but not the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) pathway. As previously discussed, the PI3K/AKT/mTOR pathway regulates the synthesis of HIF-1α protein. The mechanism by which trans-farnesylthiosalicylic acid (32) promotes HIF-1α protein degradation is not clear.
Compounds that Prevent HIF-1α Protein Accumulation
The following section of this review focuses on the compounds that inhibit HIF-1α protein accumulation with yet to be identified molecular mechanisms.
Early studies on HIF-1 implicated that cellular redox status regulates HIF-1 activity [140]. At the molecular level, redox proteins such as redox factor 1 (REF-1) and thioredoxin upregulate HIF-1 activity by enhancing the interaction between HIF-1α and transcriptional coactivators CBP/p300 and SRC-1 [141–143]. Overexpression of thioredoxin-1 in MCF-7 human breast tumor cells induced the accumulation of HIF-1α protein, HIF-1 activation, and expression of HIF-1 target genes without affecting the level of HIF-1α mRNA [144]. The compounds PX-12 (1-methylpropyl 2-imidazoyl disulfide, 33), an inhibitor of thioredoxin-1, and pleurotin (34), a toxic antibiotic obtained from the mushroom Pleurotus griseus that acts as an inhibitor of thioredoxin-1 reductase, each inhibited HIF-1α protein accumulation under both normoxic and hypoxic conditions [145]. In MCF-7 cells, PX-12 (33) and pleurotin (34) inhibited hypoxia-induced HIF-1α protein accumulation (IC50 values 7.2 and 7.6 μM, respectively); and suppressed cell proliferation/viability (IC50 values 1.9 and 0.9 μM, respectively). Similar HIF-1 inhibitory effects of 33 and 34 were also observed in HT-29 human colon carcinoma cells and pVHL-mutated RCC4 renal cell carcinoma cells. The compound 33 also inhibited the expression of HIF-1α protein in MCF-7 tumor xenografts. How 33 and the natural product pleurotin (34) decrease HIF-1α protein level is unclear. The compound PX-478 (S-2-amino-3-[4'-N,N-bis(2-chloroethyl)amino] phenyl propionic acid N-oxide dihydrochloride) was shown to decrease HIF-1α protein accumulation under both normoxic and hypoxic conditions in vitro, and in various tumor xenograft models [145]. The antitumor effects of PX-478 correlate directly with the level of tumor HIF-1α protein.
The plant isoflavone genistein (35), a broad-spectrum tyrosine kinase inhibitor, inhibits HIF-1 by blocking the induction of HIF-1α protein [146,147]. Subsequent studies have shown that several additional flavonoids and homoisoflavonoids are weak inhibitors of hypoxia-induced HIF-1 activation in a CHO (A4-4 clone) cell-based reporter assay [148]. Isorhamnetin (36), leuteolin (37), isoquercetin (38), and methylophiopogonanone B (39) inhibited HIF-1 activation at concentrations that range from 3 to 9 μg/mL (approximately 30 to 90 μM). At even higher concentrations (> 27 μg/mL), quercetin (40) also inhibited HIF-1 activation. The precise IC50 values were not determined. The compound MOB (39) was also shown to inhibited HIF-1 by blocking the induction of HIF-1α protein.
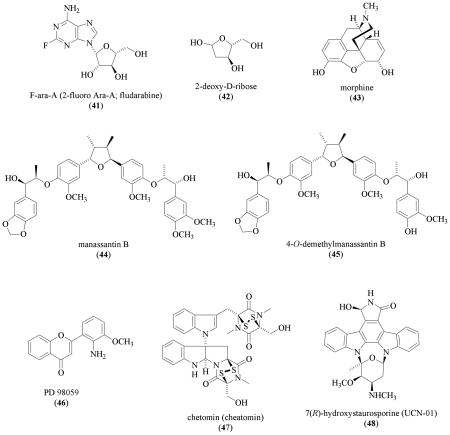
The antitumor nucleoside analog 9-β-D-arabinofuranosyl-2-fluoroadenine (F-ara-A, 41) interferes with DNA replication and inhibits VEGF expression in ovarian cancer cells (OVCAR-3 and A2780/CP70) [149]. The compound F-ara-A (41) significantly inhibited HIF-1α expression at concentrations that ranged from 2.5 to 10 μM [149]. The observation that AKT activation overcomes the inhibition by F-ara-A (41) suggests that 41 inhibits HIF-1 through the PI3K/AKT signaling pathway. Thymidine phosphorylase (TP; also known as platelet-derived endothelial growth factor or PD-ECGF) stimulates endothelial cell chemotaxis and is essential for angiogenesis [150]. Thymidine phosphorylase catalyzes the degradation of 2-deoxy-D-ribose-1-phosphate to 2-deoxy-D-ribose (42). This thymidine metabolite 42 (10 μM) was shown to produce a slight decrease in hypoxia-induced HIF-1α in HL-60 human leukemia cells [151]. The effect of 42 on either HIF-1 activation or HIF-1 target gene expression was not determined. In an unrelated study, the natural opium alkaloid morphine (43) was reported to inhibit HIF-1α protein expression in myocardial ischemia models in vitro and in vivo with a yet to be identified mechanism [152].
In order to discover novel, potent, and hypoxia-selective HIF-1 inhibitors, our research group initiated a natural product drug discovery effort that combines a traditional natural product-based chemical approach with molecular-targeted screening assays [110,111]. The lipid extract of the aquatic plant Saururus cernuus L. (Saururaceae) potently inhibited hypoxia-induced HIF-1 activation in the primary assay [110]. Saururus cernuus (also known as “lizards tail”) is a native aquatic plant found in the eastern United States that has a long history of medicinal use by both Native Americans and colonists, including the treatment of tumors [153,154]. Bioassay-guided chromatographic fractionation of the S. cernuus organic extract resulted in the isolation of manassantin B (44) and 4-O-demethylmanassantin B (45) [110]. From spectroscopic analysis of the Mosher MTPA ester derivatives of manassantin B, this study reported for the first time the absolute configurations of the chiral centers in each side chain of the representative manassantin-type dineolignans. Both 44 and 45 are among the most potent small molecule HIF-1 inhibitors discovered (IC50 values 3 and 30 nM, respectively). The manassantins selectively inhibited the activation of HIF-1 by hypoxia. Even at significantly higher concentrations (IC50 values >100 and 1000 nM, respectively), compounds 44 and 45 did not inhibit the activation of HIF-1 by the iron chelator 1,10-phenanthroline. Both compounds also inhibited the induction of the angiogenic factor VEGF by hypoxia. Further study revealed that manassantin B (44) selectively blocked the induction of HIF-1α protein, the oxygen-regulated HIF-1 subunit that determines HIF-1 activity.
Inhibitors of the Interaction between HIF-1α and Coactivators
The following section of this review discusses HIF-1 inhibitors that inhibit HIF-1 activation without decreasing the level of HIF-1α protein.
The observation that PD98059 (46) suppressed HIF-1 target gene expression suggested that the mitogen-activated protein kinase (MAPK) pathway regulates HIF-1 activity [155]. The synthetic compound PD98059 (46) is based on a flavone natural product-like structure that specifically inhibits MEK1 [156,157]. Activation of the MEK1/p42/p44 MAPK enhances HIF-1 activity and 46 inhibits HIF-1 activation [158,147]. The compound PD98059 (46) inhibits the activation of MAPK that enhances the interaction between the HIF-1α C-terminal transactivation domain (CTAD) and p300 without affecting HIF-1α protein level or the binding between HIF-1 and HRE [147,159,160].
After establishing that blocking the interaction between HIF-1α and p300/CBP proteins suppresses tumor growth in xenograft models, Kung and coworkers took a high-throughput screening (HTS) approach to discover small molecule disruptors of the HIF-1α/p300 interaction [38,161]. Using a time resolved fluorescence primary assay that monitors the interaction between the human HIF-1α CTAD domain (786–826) and the p300 CH1 domain (302–423) followed by secondary assays, the fungal natural product chetomin (47, isolated from Chetomium spp.) was identified as a disruptor of HIF-1α/p300 interaction from a library of >600,000 natural and synthetic compounds [161]. Chetomin (47), which was originally named "chaetomin," is an antibiotic isolated from Chaetomium cochliodes by Waksman and coworkers in 1944 [162,163]. Due to its complex nature, the chemical structure of 47 was not elucidated until decades later [164,165]. Chetomin (47) binds to the CH1 domain of p300 and disrupts the tertiary structure [161]. At submicromolar concentrations, 47 inhibited both hypoxia- and iron chelator-induced HIF-1 activation and its downstream targets in vitro as well as in vivo. Toxicity associated with the administration of chetomin (47) in animal models may limit its therapeutic potential for use in treating cancer.
Other HIF-1 Inhibitors
The protein kinase C (PKC) inhibitor 7(R)-hydroxystaurosporine (UCN-01, 48) was originally isolated from cultures of a Streptomyces sp. [166]. Compound 48 inhibits endothelial cell proliferation (human aortic endothelial cells; HAEC) at considerably lower concentrations (IC50 32 to 75 nM) than the concentrations required to suppress tumor cell lines (i.e. LNCaP cell IC50 1000 nM) [167]. Figg and coworkers demonstrated that 48 significantly decreases hypoxia-induced and desferoxamine-induced HIF-responsive promoter activity in human prostate tumor PC3M cells. The IC50 was not determined. Examination of the data reported would indicate that the IC50 value of 48 is between 40 and 200 nM. The effect of UCN-01 (48) on HIF-mediated gene expression was not determined.
Perspectives
Since oxygen is critical to our survival, it is not surprising to see that many toxins and other natural products inhibit HIF-1, a key regulator of oxygen homeostasis. Distinctly different signaling pathways regulate the availability and activity of HIF-1 in response to environmental, extracellular, and intracellular stimuli. While hypoxia appears to be a universal stimulus for HIF-1 activation, other stimuli act in a cell-type specific fashion. Correspondingly, certain HIF-1 inhibitors are more selective towards particular stimuli, while others act as more generalized inhibitors. Many drug discovery efforts are limited by bioassays that only monitor a narrow set of HIF-1 regulatory pathways. It may be necessary to incorporate both environmental and genetic factors into the in vitro model(s) when developing bioassays to screen chemical libraries for HIF-1 inhibitors. Since hypoxic tissues are intrinsically undervascularized, drug delivery will remain a critical factor that can hinder or contribute to the clinical success of HIF-1 inhibitors.
In general, the premise behind molecular-targeted drug discovery is the idea of identifying new therapeutically useful agents that selectively target a disease-specific molecular mechanism(s) or pathway(s). In terms of antitumor drug discovery, the goal of such efforts is to discover new chemotherapeutic agents that are directed only at tumor cells and would not cause general cytotoxicity-related side effects [168]. The discovery of new natural product-based inhibitors of HIF-1 holds, such promise. Therefore, it is surprising to see that so much effort in the discovery of HIF-1 inhibitors is currently spent characterizing small molecules that inhibit HIF-1 at cytotoxic concentrations [summarized in Table (1), reference 22]. It is possible that the ability of certain cytotoxic compounds to inhibit HIF-1 may provide some degree of selectivity to otherwise nonspecific cytotoxins. Many of the currently used chemotherapeutic drugs such as alkylating agents and antimitotics rely on relatively non-selective cytotoxic mechanisms. The fact that some of the compounds described as HIF-1 inhibitors inhibit HIF-1 at concentrations much higher than those required to inhibit proliferation and kill cells, does bring in the question as to the biological relevance of such studies. Efforts to discover therapeutically useful HIF-1 inhibitors, including natural product-based inhibitors, should include an early stage deselection criteria that exclude cytotoxic compounds. Molecular-targeted drug discovery programs that fail to consider the cytotoxic properties of the active compounds identified are destined to continue producing relatively nonselective cytotoxic antitumor agents. On the other hand, programs that combine well-designed bioassays and unique sources of chemical diversity, such as natural products, will have great potential to discover tumor-specific chemotherapeutic agents.
Acknowledgments
Support for this effort was provided by the NIH/NCI CA-98787-01, DOD PC040931, NOAA NURP/NIUST NA16RU1496, and USDA/Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009.
List of Abbreviations
- 17-AAG
17-N-allylamino-17-demethoxygeldanamycin
- 2ME2
2-methoxyestradiol
- AKT
Protein kinase B
- Ang II
Angiotensin II
- ARNT
Aryl hydrocarbon receptor nuclear translocator
- bHLH
Basic helix-loop-helix
- CBP
CREB (cAMP-response element-binding protein)-binding protein
- COX-2
Cyclooxygenase-2
- CsA
Cyclosporine (same as cyclosporin A)
- CTAD
C-terminal transactivation domain (same as TADC)
- DFO
Desferoxamine
- DIDS
4,4’-diisothiocyanatostilbene-2,2’-disulfonate
- DPI
Diphenylene iodonium
- ERK
Extracellular signal-regulated kinase (same as MAPK)
- FTS
S-trans-farnesylthiosalicylic acid [same as (E,E)-S- farnesylthiosalicylic acid]
- GA
Geldanamycin
- HAEC
Human aortic endothelial cells
- HER2/neu
Human epidermal growth factor receptor 2 (
- HDAC
Histone deacetylases
- Hdm2
Human ortholog of Mdm2 (Mouse double minute 2 homolog)
- HIF-1
Hypoxia-inducible factor-1
- HPH
HIF prolyl hydroxylase (same as PHD)
- HRE
Hypoxia response element
- hsp90
Heat shock protein (90 kD)
- HTS
High-throughput screening
- MAPK
Mitogen-activated protein kinase (same as ERK)
- MDA
Microtubule-disrupting agent
- MEK
Mitogen-activated protein kinase kinase (same as MAPKK; same as MAPK/ERK kinase)
- mTOR
Mammalian target of rapamycin
- NCI
U.S. National Cancer Institute
- NFκB
Nuclear factor-κB
- NMR
Nuclear magnetic resonance
- ODDD
Oxygen-dependent degradation domain
- PAS
PER-ARNT-SIM
- p300
E1A-binding protein, 300 kD
- PHD
Prolyl hydroxylase domain-containing protein (same as HPH)
- PI3K
Phosphoinositol 3-kinase
- PKC
Protein kinase C
- REF-1
Redox factor 1
- TP
Thymidine phosphorylase (same as platelet-derived endothelial growth factor or PD-ECGF)
- TSA
(R)-trichostatin A
- UCN-01
7(R)-hydroxystaurosporine
- VEGF
Vascular endothelial growth factor
- VHL
von Hippel-Lindau disease tumor suppressor
References
- 1.Moody EJ, Simon BA, Johns RA. In: The Pharmacological Basis of Therapeutics. Hardman JG, Limbird LE, Gilman AG, editors. McGraw-Hill Medical Publishing Division; New York: 2001. pp. 385–397. [Google Scholar]
- 2.Brown JM. Cancer Res. 1999;59(23):5863–5870. [PubMed] [Google Scholar]
- 3.Harris AL. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 4.Subarsky P, Hill RP. Clin Exp Metastasis. 2003;20(3):237–250. doi: 10.1023/a:1022939318102. [DOI] [PubMed] [Google Scholar]
- 5.Brown JM, Wilson WR. Nat Rev Cancer. 2004;4(6):437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 6.Sutphin PD, Chan DA, Giaccia AJ. Cell Cycle. 2004;3(2):160–163. [PubMed] [Google Scholar]
- 7.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL, Wang GL. Mol Cell Biol. 1992;12(12):5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang GL, Semenza GL. J Biol Chem. 1995;270(3):1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 10.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 12.Huang LE, Bunn HF. J Biol Chem. 2003;278(22):19575–19578. doi: 10.1074/jbc.R200030200. [DOI] [PubMed] [Google Scholar]
- 13.Poellinger L, Johnson RS. Curr Opin Genet Dev. 2004;14:81–85. doi: 10.1016/j.gde.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Mazure NM, Brahimi-Horn MC, Berta MA, Benizri E, Bilton RL, Dayan F, Ginouves A, Berra E, Pouyssegur J. Biochem Pharmacol. 2004;68(6):971–980. doi: 10.1016/j.bcp.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 15.Brahimi-Horn C, Mazure N, Pouyssegur J. Cell Signal. 2005;17(1):1–9. doi: 10.1016/j.cellsig.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 17.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim AV, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 18.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Science. 2002;295(5556):858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 19.Semenza GL. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 20.Giaccia A, Siim BG, Johnson RS. Nat Rev Drug Discov. 2003;2(10):803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- 21.Welsh SJ, Powis G. Curr Cancer Drug Targets. 2003;3(6):391–405. doi: 10.2174/1568009033481732. [DOI] [PubMed] [Google Scholar]
- 22.Powis G, Kirkpatrick L. Mol Cancer Ther. 2004;3(5):647–654. [PubMed] [Google Scholar]
- 23.Yeo EJ, Chun YS, Park JW. Biochem Pharmacol. 2004;68(6):1061–1069. doi: 10.1016/j.bcp.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 24.Paul SA, Simons JW, Mabjeesh NJ. J Cell Physiol. 2004;200(1):20–30. doi: 10.1002/jcp.10479. [DOI] [PubMed] [Google Scholar]
- 25.Hopfl G, Ogunshola O, Gassmann M. Am J Physiol Regul Integr Comp Physiol. 2004;286(4):R608–R623. doi: 10.1152/ajpregu.00538.2003. [DOI] [PubMed] [Google Scholar]
- 26.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Cancer Res. 1999;59(22):5830–5835. [PubMed] [Google Scholar]
- 27.Bos R, Zhong H, Hanrahan CF, Mommers EC, Semenza GL, Pinedo HM, Abeloff MD, Simons JW, van Diest PJ, van der Wall E. J Natl Cancer Inst. 2001;93(4):309–314. doi: 10.1093/jnci/93.4.309. [DOI] [PubMed] [Google Scholar]
- 28.Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, Semenza GL, van Diest PJ, van der Wall E. Cancer. 2003;97(6):1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- 29.Vleugel MM, Greijer AE, Shvarts A, van der Groep P, van Berkel M, Aarbodem Y, van Tinteren H, Harris AL, van Diest PJ, van der Wall E. J Clin Pathol. 2005;58(2):172–177. doi: 10.1136/jcp.2004.019885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi R, Tanaka S, Hiyama T, Ito M, Kitadai Y, Sumii M, Haruma K, Chayama K. Oncol Rep. 2003;10(4):797–802. [PubMed] [Google Scholar]
- 31.Sohda M, Ishikawa H, Masuda N, Kato H, Miyazaki T, Nakajima M, Fukuchi M, Manda R, Fukai Y, Sakurai H, Kuwano H. Int J Cancer. 2004;110(6):838–844. doi: 10.1002/ijc.20215. [DOI] [PubMed] [Google Scholar]
- 32.Theodoropoulos VE, Lazaris AC, Sofras F, Gerzelis I, Tsoukala V, Ghikonti I, Manikas K, Kastriotis I. Eur Urol. 2004;46(2):200–208. doi: 10.1016/j.eururo.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Zhong H, Semenza GL, Simons JW, De Marzo AM. Cancer Detect Prev. 2004;28(2):88–93. doi: 10.1016/j.cdp.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Jiang BH, Agani F, Passaniti A, Semenza GL. Cancer Res. 1997;57(23):5328–5335. [PubMed] [Google Scholar]
- 35.Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ. Proc Natl Acad Sci U S A. 1997;94 (15):8104–8109. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan HE, Lo J, Johnson RS. EMBO J. 1998;17(11):3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Cancer Res. 2000;60(15):4010–4015. [PubMed] [Google Scholar]
- 38.Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM. Nat Med. 2000;6(12):1335–1340. doi: 10.1038/82146. [DOI] [PubMed] [Google Scholar]
- 39.Hopfl G, Wenger RH, Ziegler U, Stallmach T, Gardelle O, Achermann R, Wergin M, Kaser-Hotz B, Saunders HM, Williams KJ, Stratford IJ, Gassmann M, Desbaillets I. Cancer Res. 2002;62(10):2962–2970. [PubMed] [Google Scholar]
- 40.Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. Oncogene. 2003;22(21):3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- 41.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. Nat Med. 2004;10(6):594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 42.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Cancer Cell. 2004;5(5):429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 43.Rapisarda A, Zalek J, Hollingshead M, Braunschweig T, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, Hewitt SM, Shoemaker RH, Melillo G. Cancer Res. 2004;64(19):6845–6848. doi: 10.1158/0008-5472.CAN-04-2116. [DOI] [PubMed] [Google Scholar]
- 44.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Nature. 1998;394(6692):485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 45.Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G. Cancer Cell. 2003;4(2):133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 46.Newman DJ, Cragg GM, Snader KM. J Nat Prod. 2003;66(7):1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 47.Wang GL, Semenza GL. J Biol Chem. 1993;268(29):21513–21518. [PubMed] [Google Scholar]
- 48.Page EL, Robitaille GA, Pouyssegur J, Richard DE. J Biol Chem. 2002;277(50):48403–48409. doi: 10.1074/jbc.M209114200. [DOI] [PubMed] [Google Scholar]
- 49.Huang TS, Shu CH, Shih YL, Huang HC, Su YC, Chao Y, Yang WK, Whang-Peng J. Cancer Lett. 1996;110(1–2):77–85. doi: 10.1016/s0304-3835(96)04464-3. [DOI] [PubMed] [Google Scholar]
- 50.Chang H, Shyu KG, Lee CC, Tsai SC, Wang BW, Lee YH, Lin S. Biochem Biophys Res Commun. 2003;302(1):95–100. doi: 10.1016/s0006-291x(03)00111-6. [DOI] [PubMed] [Google Scholar]
- 51.Lin S, Huang HC, Chen LL, Lee CC, Huang TS. Mol Pharmacol. 2001;60 (4):768–775. [PubMed] [Google Scholar]
- 52.Liu JM, Chen LT, Chao Y, Li AF, Wu CW, Liu TS, Shiah HS, Chang JY, Chen JD, Wu HW, Lin WC, Lan C, Whang-Peng J. Cancer Chemother Pharmacol. 2002;49 (5):425–428. doi: 10.1007/s00280-002-0429-3. [DOI] [PubMed] [Google Scholar]
- 53.Gaddipati JP, Madhavan S, Sidhu GS, Singh AK, Seth P, Maheshwari RK. Mol Cell Biochem. 1999;194(1–2):271–281. doi: 10.1023/a:1006982028460. [DOI] [PubMed] [Google Scholar]
- 54.Chander R, Kapoor NK, Dhawan BN. Biochem Pharmacol. 1992;44(1):180–183. doi: 10.1016/0006-2952(92)90054-m. [DOI] [PubMed] [Google Scholar]
- 55.Symolon H, Schmelz EM, Dillehay DL, Merrill AH., Jr J Nutr. 2004;134(5):1157–1161. doi: 10.1093/jn/134.5.1157. [DOI] [PubMed] [Google Scholar]
- 56.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G. Cancer Res. 2002;62(15):4316–4324. [PubMed] [Google Scholar]
- 57.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Cancer Res. 2004;64(4):1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 58.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. J Med Chem. 2000;43(20):3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 59.Rapisarda A, Shoemaker RH, Melillo G. Cell Cycle. 2004;3(2):172–175. [PubMed] [Google Scholar]
- 60.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. Cancer Cell. 2003;3(4):363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 61.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. J Biol Chem. 2003;278(9):7445–7452. doi: 10.1074/jbc.M209804200. [DOI] [PubMed] [Google Scholar]
- 62.Hagen T, D'Amico G, Quintero M, Palacios–Callender M, Hollis V, Lam F, Moncada S. Biochem Biophys Res Commun. 2004;322(3):923–929. doi: 10.1016/j.bbrc.2004.07.204. [DOI] [PubMed] [Google Scholar]
- 63.Pribluda VS, Gubish ER, Jr, Lavallee TM, Treston A, Swartz GM, Green SJ. Cancer Metastasis Rev. 2000;19(1–2):173–179. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]
- 64.Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, van Waes C. Clin Cancer Res. 2004;10(24):8665–8673. doi: 10.1158/1078-0432.CCR-04-1393. [DOI] [PubMed] [Google Scholar]
- 65.Mazure NM, Brahimi-Horn MC, Pouyssegur J. Curr Pharm Des. 2003;9(7):531–541. doi: 10.2174/1381612033391469. [DOI] [PubMed] [Google Scholar]
- 66.Liu L, Simon MC. Cancer Biol Ther. 2004;3(6):492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 67.Arsham AM, Plas DR, Thompson CB, Simon MC. J Biol Chem. 2002;277(17):15162–15170. doi: 10.1074/jbc.M111162200. [DOI] [PubMed] [Google Scholar]
- 68.Alvarez-Tejado M, Alfranca A, Aragones J, Vara A, Landazuri MO, del Peso L. J Biol Chem. 2002;277(16):13508–13517. doi: 10.1074/jbc.M200017200. [DOI] [PubMed] [Google Scholar]
- 69.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. Mol Cell Biol. 2001;21(12):3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Mol Cell Biol. 2002;22(20):7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Cancer Res. 2005;65(2):605–612. [PubMed] [Google Scholar]
- 72.Gradin K, McGuire J, Wenger RH, Kvietikova I, Whitelaw ML, Toftgard R, Tora L, Gassmann M, Poellinger L. Mol Cell Biol. 1996;16(10):5221–5231. doi: 10.1128/mcb.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 74.Mabjeesh NJ, Post DE, Willard MT, Kaur B, Van Meir EG, Simons JW, Zhong H. Cancer Res. 2002;62(9):2478–2482. [PubMed] [Google Scholar]
- 75.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. J Biol Chem. 2002;277(33):29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 76.Katschinski DM, Le L, Heinrich D, Wagner KF, Hofer T, Schindler SG, Wenger RH. J Biol Chem. 2002;277(11):9262–9267. doi: 10.1074/jbc.M110377200. [DOI] [PubMed] [Google Scholar]
- 77.Hur E, Kim HH, Choi SM, Kim JH, Yim S, Kwon HJ, Choi Y, Kim DK, Lee MO, Park H. Mol Pharmacol. 2002;62(5):975–982. doi: 10.1124/mol.62.5.975. [DOI] [PubMed] [Google Scholar]
- 78.Isaacs JS, Jung YJ, Neckers L. J Biol Chem. 2004;279(16):16128–16135. doi: 10.1074/jbc.M313342200. [DOI] [PubMed] [Google Scholar]
- 79.Katschinski DM, Le L, Schindler SG, Thomas T, Voss AK, Wenger RH. Cell Physiol Biochem. 2004;14(4–6):351–360. doi: 10.1159/000080345. [DOI] [PubMed] [Google Scholar]
- 80.Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. J Biol Chem. 1997;272(38):23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 81.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Cell. 1997;89(2):239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 82.Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90(1):65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 83.Schulte TW, Akinaga S, Murakata T, Agatsuma T, Sugimoto S, Nakano H, Lee YS, Simen BB, Argon Y, Felts S, Toft DO, Neckers LM, Sharma SV. Mol Endocrinol. 1999;13(9):1435–1448. doi: 10.1210/mend.13.9.0339. [DOI] [PubMed] [Google Scholar]
- 84.Kurebayashi J, Otsuki T, Kurosumi M, Soga S, Akinaga S, Sonoo H. Jpn J Cancer Res. 2001;92(12):1342–1351. doi: 10.1111/j.1349-7006.2001.tb02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Neckers L. Trends Mol Med. 2002;8(4 Suppl):S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 86.Bisht KS, Bradbury CM, Mattson D, Kaushal A, Sowers A, Markovina S, Ortiz KL, Sieck LK, Isaacs JS, Brechbiel MW, Mitchell JB, Neckers LM, Gius D. Cancer Res. 2003;63(24):8984–8995. [PubMed] [Google Scholar]
- 87.Grem JL, Morrison G, Guo XD, Agnew E, Takimoto CH, Thomas R, Szabo E, Grochow L, Grollman F, Hamilton JM, Neckers L, Wilson RH. J Clin Oncol. 2005;23(9):1885–1893. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 88.Birt DF, Walker B, Tibbels MG, Bresnick E. Carcinogenesis. 1986;7(6):959–963. doi: 10.1093/carcin/7.6.959. [DOI] [PubMed] [Google Scholar]
- 89.Wei H, Tye L, Bresnick E, Birt DF. Cancer Res. 1990;50(3):499–502. [PubMed] [Google Scholar]
- 90.Caltagirone S, Rossi C, Poggi A, Ranelletti FO, Natali PG, Brunetti M, Aiello FB, Piantelli M. Int J Cancer. 2000;87(4):595–600. doi: 10.1002/1097-0215(20000815)87:4<595::aid-ijc21>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 91.Tatsuta A, Iishi H, Baba M, Yano H, Murata K, Mukai M, Akedo H. Clin Exp Metastasis. 2000;18(8):657–662. doi: 10.1023/a:1013133803806. [DOI] [PubMed] [Google Scholar]
- 92.Gupta S, Afaq F, Mukhtar H. Biochem Biophys Res Commun. 2001;287(4):914–920. doi: 10.1006/bbrc.2001.5672. [DOI] [PubMed] [Google Scholar]
- 93.Way TD, Kao MC, Lin JK. J Biol Chem. 2004;279(6):4479–4489. doi: 10.1074/jbc.M305529200. [DOI] [PubMed] [Google Scholar]
- 94.Shukla S, Gupta S. Clin Cancer Res. 2004;10(9):3169–3178. doi: 10.1158/1078-0432.ccr-03-0586. [DOI] [PubMed] [Google Scholar]
- 95.Czyz J, Madeja Z, Irmer U, Korohoda W, Hulser DF. Int J Cancer. 2005;114(1):12–18. doi: 10.1002/ijc.20620. [DOI] [PubMed] [Google Scholar]
- 96.Osada M, Imaoka S, Funae Y. FEBS Lett. 2004;575(1–3):59–63. doi: 10.1016/j.febslet.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 97.Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. FASEB J. 2005;19 (3):342–353. doi: 10.1096/fj.04-2175com. [DOI] [PubMed] [Google Scholar]
- 98.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275(5297):218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 99.Gescher AJ, Steward WP. Cancer Epidemiol Biomarkers Prev. 2003;12(10):953–957. [PubMed] [Google Scholar]
- 100.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Anticancer Res. 2004;24(5A):2783–2840. [PubMed] [Google Scholar]
- 101.Cao Z, Fang J, Xia C, Shi X, Jiang BH. Clin Cancer Res. 2004;10(15):5253–5263. doi: 10.1158/1078-0432.CCR-03-0588. [DOI] [PubMed] [Google Scholar]
- 102.Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V. Cancer Res. 2003;63(24):8777–8783. [PubMed] [Google Scholar]
- 103.Anonymous. In: AHFS Drug Information 2004. McEvoy GK, editor. American Society of Health–System Pharmacists, Inc./American Hospital Formulary Service; Bethesda: 2004. pp. 3625–3638. [Google Scholar]
- 104.D'Angelo G, Duplan E, Vigne P, Frelin C. J Biol Chem. 2003;278(17):15406–15411. doi: 10.1074/jbc.M211293200. [DOI] [PubMed] [Google Scholar]
- 105.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Proc Natl Acad Sci USA. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. J Biol Chem. 2000;275(33):25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 107.Agani FH, Pichiule P, Chavez CJ, LaManna JC. Comp Biochem Physiol Part A. 2002;132(1):107–109. doi: 10.1016/s1095-6433(01)00535-9. [DOI] [PubMed] [Google Scholar]
- 108.Hagen T, Taylor CT, Lam F, Moncada S. Science. 2003;302(5652):1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 109.Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S. Biochem J. 2003;376(Pt 2):537–544. doi: 10.1042/BJ20031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hodges TW, Hossain CF, Kim YP, Zhou YD, Nagle DG. J Nat Prod. 2004;67(5):767–771. doi: 10.1021/np030514m. [DOI] [PubMed] [Google Scholar]
- 111.Mohammed KA, Hossain CF, Zhang L, Bruick RK, Zhou YD, Nagle DG. J Nat Prod. 2004;67(12):2002–2007. doi: 10.1021/np049753f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Nat Med. 2001;7(4):437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 113.Tsuji N, Kobayashi M, Nagashima K, Wakisaka Y, Koizumi K. J Antibiot (Tokyo) 1976;29(1):1–6. doi: 10.7164/antibiotics.29.1. [DOI] [PubMed] [Google Scholar]
- 114.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265(28):17174–17179. [PubMed] [Google Scholar]
- 115.Lee YM, Kim SH, Kim HS, Son MJ, Nakajima H, Kwon JH, Kim KW. Biochem Biophys Res Commun. 2003;300(1):241–246. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 116.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. J Antibiot (Tokyo) 1994;47(3):301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 117.Shigematsu N, Ueda H, Takase S, Tanaka H, Yamamoto K, Tada T. J Antibiot (Tokyo) 1994;47(3):311–314. doi: 10.7164/antibiotics.47.311. [DOI] [PubMed] [Google Scholar]
- 118.Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K. J Antibiot (Tokyo) 1994;47(3):315–323. doi: 10.7164/antibiotics.47.315. [DOI] [PubMed] [Google Scholar]
- 119.Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. Exp Cell Res. 1998;241(1):126–133. doi: 10.1006/excr.1998.4027. [DOI] [PubMed] [Google Scholar]
- 120.Pellizzaro C, Coradini D, Daidone MG. Carcinogenesis. 2002;23(5):735–740. doi: 10.1093/carcin/23.5.735. [DOI] [PubMed] [Google Scholar]
- 121.Zgouras D, Wachtershauser A, Frings D, Stein J. Biochem Biophys Res Commun. 2003;300(4):832–838. doi: 10.1016/s0006-291x(02)02916-9. [DOI] [PubMed] [Google Scholar]
- 122.Miki K, Unno N, Nagata T, Uchijima M, Konno H, Koide Y, Nakamura S. Shock. 2004;22(5):446–452. doi: 10.1097/01.shk.0000140664.80530.bd. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Y, Adach M, Zhao X, Kawamura R, Imai K. Int J Cancer. 2004;110(2):301–308. doi: 10.1002/ijc.20117. [DOI] [PubMed] [Google Scholar]
- 124.Zhang Y, Jung M, Dritschilo A, Jung M. Radiat Res. 2004;161(6):667–674. doi: 10.1667/rr3192. [DOI] [PubMed] [Google Scholar]
- 125.Monneret C. Eur J Med Chem. 2005;40(1):1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 126.Piekarz R, Bates S. Curr Pharm Des. 2004;10(19):2289–2298. doi: 10.2174/1381612043383980. [DOI] [PubMed] [Google Scholar]
- 127.Yoshida M, Matsuyama A, Komatsu Y, Nishino N. Curr Med Chem. 2003;10 (22):2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]
- 128.Zhu WG, Otterson GA. Curr Med Chem Anti-Cancer Agents. 2003;3(3):187–19. doi: 10.2174/1568011033482440. [DOI] [PubMed] [Google Scholar]
- 129.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, Chan KK, Grever MR. Blood. 2005;105(3):959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 130.Marshall JL, Rizvi N, Kauh J, Dahut W, Figuera M, Kang MH, Figg WD, Wainer I, Chaissang C, Li MZ, Hawkins MJ. J Exp Ther Oncol. 2002;2(6):325–332. doi: 10.1046/j.1359-4117.2002.01039.x. [DOI] [PubMed] [Google Scholar]
- 131.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Clin Cancer Res. 2002;8(3):718–728. [PubMed] [Google Scholar]
- 132.Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Blood. 2001;98(9):2865–2868. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]
- 133.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Cell. 2002;111(5):709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 134.Lin S, Tsai SC, Lee CC, Wang BW, Liou JY, Shyu KG. Mol Pharmacol. 2004;66(3):612–619. [PubMed] [Google Scholar]
- 135.Fukuda K, Hibiya Y, Mutoh M, Koshiji M, Akao S, Fujiwara H. Planta Med. 1999;65(4):381–383. doi: 10.1055/s-2006-960795. [DOI] [PubMed] [Google Scholar]
- 136.Pan DJ, Li ZL, Hu CQ, Chen K, Chang JJ, Lee KH. Planta Med. 1990;56 (4):383–385. doi: 10.1055/s-2006-960989. [DOI] [PubMed] [Google Scholar]
- 137.Li MH, Miao ZH, Tan WF, Yue JM, Zhang C, Lin LP, Zhang XW, Ding J. Clin Cancer Res. 2004;10(24):8266–8274. doi: 10.1158/1078-0432.CCR-04-0951. [DOI] [PubMed] [Google Scholar]
- 138.Tan WF, Zhang XW, Li MH, Yue JM, Chen Y, Lin LP, Ding J. Eur J Pharmacol. 2004;499(3):219–228. doi: 10.1016/j.ejphar.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 139.Blum R, Jacob–Hirsch J, Amariglio N, Rechavi G, Kloog Y. Cancer Res. 2005;65(3):999–1006. [PubMed] [Google Scholar]
- 140.Wang GL, Jiang BH, Semenza GL. Biochem Biophys Res Commun. 1995;212(2):550–556. doi: 10.1006/bbrc.1995.2005. [DOI] [PubMed] [Google Scholar]
- 141.Huang LE, Arany Z, Livingston DM, Bunn HF. J Biol Chem. 1996;271(50):32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 142.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y. EMBO J. 1999;18(7):1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carrero P, Okamoto K, Coumailleau P, O'Brien S, Tanaka H, Poellinger L. Mol Cell Biol. 2000;20(1):402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Welsh SJ, Bellamy WT, Briehl MM, Powis G. Cancer Res. 2002;62(17):5089–5095. [PubMed] [Google Scholar]
- 145.Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. Mol Cancer Ther. 2003;2(3):235–243. [PubMed] [Google Scholar]
- 146.Wang GL, Jiang BH, Semenza GL. Biochem Biophys Res Commun. 1995;216(2):669–675. doi: 10.1006/bbrc.1995.2674. [DOI] [PubMed] [Google Scholar]
- 147.Hur E, Chang KY, Lee E, Lee SK, Park H. Mol Pharmacol. 2001;59(5):1216–1224. doi: 10.1124/mol.59.5.1216. [DOI] [PubMed] [Google Scholar]
- 148.Hasebe Y, Egawa K, Yamazaki Y, Kunimoto S, Hirai Y, Ida Y, Nose K. Biol Pharm Bull. 2003;26(10):1379–1383. doi: 10.1248/bpb.26.1379. [DOI] [PubMed] [Google Scholar]
- 149.Fang J, Cao Z, Chen YC, Reed E, Jiang BH. Mol Pharmacol. 2004;66(1):178–186. doi: 10.1124/mol.66.1.178. [DOI] [PubMed] [Google Scholar]
- 150.Ciccolini J, Evrard A, Cuq P. Curr Med Chem Anti-Canc Agents. 2004;4(2):71–81. doi: 10.2174/1568011043482089. [DOI] [PubMed] [Google Scholar]
- 151.Ikeda R, Furukawa T, Kitazono M, Ishitsuka K, Okumura H, Tani A, Sumizawa T, Haraguchi M, Komatsu M, Uchimiya H, Ren XQ, Motoya T, Yamada K, Akiyama S. Biochem Biophys Res Commun. 2002;291(4):806–812. doi: 10.1006/bbrc.2002.6432. [DOI] [PubMed] [Google Scholar]
- 152.Roy S, Balasubramanian S, Wang J, Chandrashekhar Y, Charboneau R, Barke R. Surgery. 2003;134(2):336–344. doi: 10.1067/msy.2003.247. [DOI] [PubMed] [Google Scholar]
- 153.Phares DL. Am J Pharm. 1867;39:468. [Google Scholar]
- 154.Hartwell JL. Lloydia. 1971;34(2):204–255. [PubMed] [Google Scholar]
- 155.Salceda S, Beck I, Srinivas V, Caro J. Kidney Int. 1997;51(2):556–559. doi: 10.1038/ki.1997.78. [DOI] [PubMed] [Google Scholar]
- 156.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. Proc Natl Acad Sci USA. 1995;92(17):7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. J Biol Chem. 1995;270(46):27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 158.Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. J Biol Chem. 1999;274(46):32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 159.Lee E, Yim S, Lee SK, Park H. Mol Cells. 2002;14(1):9–15. [PubMed] [Google Scholar]
- 160.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. J Biol Chem. 2003;278(16):14013–14019. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6(1):33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 162.Waksman SA, Bugie E. J Bacteriol. 1944;48:527–530. doi: 10.1128/jb.48.5.527-530.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Geiger WA, Conn JE, Waksman SA. J Bacteriol. 1944;48:531–536. doi: 10.1128/jb.48.5.531-536.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McInnes AG, Taylor A, Walter JA. J Am Chem Soc. 1976;98(21):6741. doi: 10.1021/ja00437a074. [DOI] [PubMed] [Google Scholar]
- 165.Fujimoto H, Sumino M, Okuyama E, Ishibashi M. J Nat Prod. 2004;67(1):98–102. doi: 10.1021/np0302201. [DOI] [PubMed] [Google Scholar]
- 166.Takahashi I, Kobayashi E, Asano K, Yoshida M, Nakano H. J Antibiot (Tokyo) 1987;40(12):1782–1784. doi: 10.7164/antibiotics.40.1782. [DOI] [PubMed] [Google Scholar]
- 167.Kruger EA, Blagosklonny MV, Dixon SC, Figg WD. Invasion Metastasis. 1998–99;18(4):209–218. doi: 10.1159/000024514. [DOI] [PubMed] [Google Scholar]
- 168.Nagle DG, Zhou YD, Mora FD, Mohammed KA, Kim YP. Curr Med Chem. 2004;11(13):1725–1756. doi: 10.2174/0929867043364991. [DOI] [PMC free article] [PubMed] [Google Scholar]