

Abstract
Mast cells infiltrate the sites of inflammation associated with chronic atopic disease and during helminth and bacterial infection. This process requires receptor-mediated cell chemotaxis across a concentration gradient of their chemotactic ligands. In vivo, mast cells are likely to be exposed to several such agents, which can cooperate in a synergistic manner to regulate mast cell homing. Here, we report that chemotaxis of mouse bone-marrow-derived mast cells (BMMCs) in response to the chemoattractants stem-cell factor (SCF) and prostaglandin (PG)E2, is substantially enhanced following antigen-dependent ligation of the high-affinity receptor for IgE (FcεRI). These responses were associated with enhanced activation of phosphoinositide 3-kinase (PI3K), and downstream activation of the tyrosine protein kinase Btk, with subsequent enhanced phospholipase (PL)Cγ-mediated Ca2+ mobilization, Rac activation and F-actin rearrangement. Antigen-induced chemotaxis, and the ability of antigen to amplify responses mediated by SCF, adenosine and PGE2 were suppressed following inhibition of PI3K, and were impaired in BMMCs derived from Btk−/− mice. There were corresponding decreases in the PLCγ-mediated Ca2+ signal, Rac activation and F-actin rearrangement, which, as they are essential for BMMC chemotaxis, accounts for the impaired migration of Btk-deficient cells. Taken together, these data demonstrate that, by regulating signaling pathways that control F-actin rearrangement, Btk is crucial for the ability of antigen to amplify mast-cell chemotactic responses.
Keywords: Mast cells, FcεRI, Btk, Rac activation, Actin rearrangement, Chemotaxis, SCF, PGE2
Introduction
Mast cells are important players in host defence mechanisms associated with innate and adaptive immune responses (Galli et al., 2005; Marshall, 2004; Mekori, 2004). However, in afflicted individuals, inflammatory mediators released from activated mast cells can also induce atopic allergic inflammatory responses (Brown et al., 2008; Metcalfe et al., 1997). Mast cells infiltrate the sites of inflammation associated with chronic atopic disease, and helminth and bacterial infection (Echtenacher et al., 1996; Madden et al., 1991). These processes rely on the ability of mast cells to migrate to target tissues in response to the appropriate chemotactic stimuli. Mast cells express surface receptors for a number of endogenous ligands that are known to be potent mast-cell chemoattractants under experimental conditions. Of these, the most widely investigated is stem cell factor (SCF), the natural ligand for the growth-factor receptor KIT (Meininger et al., 1992; Nilsson et al., 1994). Others include several agonists of G-protein-coupled receptors (GPCRs), such as prostaglandin E2 (PGE2), which positively regulates mast-cell responses through the EP3 receptor (Weller et al., 2007). There is also evidence to suggest that IgE-sensitized mast cells migrate toward antigen, potentially through the release of chemoattractants from mast cells themselves (Ishizuka et al., 2001; Kitaura et al., 2005). This evokes a potential contributory role for the high-affinity receptor for IgE (FcεRI) in mast-cell homing, in addition to its role in release of the mast-cell mediator. As demonstrated in several cell types, chemotaxis is a Ca2+-dependent process that requires cytoskeletal rearrangement attributable to initial polymerization and then depolymerization of actin filaments (Nishida et al., 2005; Shimizu et al., 2009). In mast cells, sensitization with IgE alone enhances cortical F-actin-ring formation, as a consequence of F-actin polymerization, and subsequent IgE-FcεRI crosslinking with antigen induces disassembly of this structure (Allen et al., 2009; Nishida et al., 2005). The events that regulate SCF-mediated mast-cell chemotaxis might in part be regulated by phosphoinositide 3-kinase (PI3K)-mediated pathways (Kim et al., 2008; Samayawardhena et al., 2007)
Inflamed tissues in allergic individuals accumulate inflammatory mast-cell-activating agents including PGE2 (Harris et al., 2002; Vancheri et al., 2004) and adenosine (Spicuzza et al., 2006). In addition, chronically inflamed tissues undergo fibrotic changes that might lead to increased production of SCF by fibroblasts (Hogaboam et al., 1998). Therefore, it is likely that mast cells within these tissues are exposed to co-activating factors, which might thus contribute to tissue targeting of mast cells in a pathological environment through synergistic interactions with antigen. We therefore investigated whether such synergy can occur and, if so, the molecular mechanisms regulating these responses in mast cells derived from the bone marrow (BMMCs) of wild-type and gene-deficient mice. Signaling events induced by antigen were found to synergistically interact with those elicited by SCF and the GPCR agonists, adenosine and PGE2, to dramatically enhance chemotaxis in mouse BMMCs. Such synergy was associated with enhanced increases in Ca2+ mobilization and Rac-dependent cytoskeletal reorganization through actin polymerization and depolymerization. We finally demonstrate that, by regulating such synergistic signaling events, Btk is an integral component of the integrated signal-transduction pathway that permits antigen to amplify the chemotactic responses of mast cells.
Results
Mast-cell migration toward SCF and GPCR agonists is synergistically enhanced by antigen
To investigate potential synergistic interactions between the signaling events elicited through KIT and GPCRs in the context of FcεRI ligation, we first examined the ability of antigen to enhance the chemotactic response elicited by SCF and GPCR agonists in BMMCs. As expected, optimal concentrations of SCF (10 ng/ml) and, to a much lesser extent antigen (10 ng/ml), produced significant chemotaxis towards each stimulant over the course of 4 hours (Fig. 1A). However, the chemotactic response was enhanced approximately threefold when SCF and antigen were added simultaneously, and this was apparent at all time points examined (Fig. 1B). GPCR agonists were examined in a similar manner, and, on the basis of their efficacy in enhancing antigen-mediated degranulation in BMMCs (Kuehn et al., 2008a; Kuehn and Gilfillan, 2007; Zhong et al., 2003), PGE2 and adenosine were selected as examples for study. From Fig. 1C, it can be seen that the chemotactic response induced by an optimal concentration of PGE2 alone (100 nM) was more rapid than that produced by SCF (compare Fig. 1C with 1A). In combination with antigen, the response to PGE2 was substantially enhanced, and comparable with that observed for the combination of antigen and SCF. However, as the response of PGE2 alone was more robust than that produced by SCF alone, the degree of enhancement of the SCF response by antigen was of greater relative magnitude, at least at the 1 hour time point. There are no reports of the effect of adenosine on mast-cell chemotaxis but, unlike SCF and PGE2, we found that adenosine by itself had little or no ability to induce chemotaxis (Fig. 1D). However, the combination of adenosine and antigen challenge induced a chemotactic response approximately fourfold greater than that induced by antigen alone. A similar pattern has been noted for mast-cell degranulation where adenosine potentiates antigen-induced degranulation without inducing degranulation by itself (Ali et al., 1990). Nevertheless, the chemotactic response to the adenosine-antigen combination was lower than that observed with the combinations of SCF or PGE2 with antigen.
Fig. 1.

FcεRI aggregation synergistically enhances chemotactic responses elicited by KIT and GPCRs. (A-D) BMMCs were sensitized overnight with IgE in cytokine-free medium. In these, and other, studies described, 3×105 cells were added to the upper chambers. The chemotaxis assay was performed as described in the Materials and Methods. (E) BMMCs were preincubated with pertussis toxin (1 μg/ml) for 4 hours then washed and used for the assay. The indicated agonists [used at optimal concentrations (see supplementary material Fig. S1), antigen, Ag (10 ng/ml), SCF (10 ng/ml), adenosine (1 μM), PGE2 (100 nM)] were added in the lower chamber, and the migrated BMMCs were counted after 4 hours. Results are means ± s.e. of three separate experiments. *P<0.05 for comparison with same stimulation in control group by Student's t-test.
To determine whether or not the ability of antigen to enhance GPCR-agonist-induced chemotaxis was mediated through Gαi-dependent signaling, we examined the ability of the Gαi inhibitor, pertussis toxin, to block this response. As predicted, the chemotactic responses to SCF, antigen or the combination of the two, were unaffected by pertussis toxin pre-treatment (Fig. 1E, left panel). By contrast, such treatment completely blocked the chemotactic response to PGE2 as well as the synergistic enhancement of response to antigen caused by PGE2 and adenosine (Fig. 1E, middle and right panels). These results demonstrate that PGE2 and adenosine mediate their effects on chemotaxis exclusively through Gαi and that the responses to antigen or SCF are not due to the release of agonists of Gαi-coupled receptors, such as adenosine, which can then act in an autocrine or paracrine manner to induce chemotaxis.
Synergistic migration of BMMCs occurs by a direct chemotaxis response
To exclude the possibility that mast-cell products, other than those acting through Gαi, contribute to the responses described above, supernatants from BMMCs that had been previously stimulated with the various stimulants, or combinations thereof, were applied to the lower wells of the chemotactic chambers. Sensitized (with IgE) or non-sensitized BMMCs were then placed in the upper chambers. No differences were observed in the migration of sensitized cells towards the chambers containing the supernatants from previously stimulated cells (Fig. 2A) compared with cells that were stimulated in the conventional manner (compare data to Fig. 1). Virtually no migration of non-sensitized BMMCs occurred when the lower chambers contained supernatants from cultures stimulated with antigen, even though normal migration was still apparent when the lower chambers contained supernatants from cells stimulated with SCF or PGE2 (Fig. 2A). Furthermore, the synergy between antigen and the other stimulants was substantially decreased for non-sensitized cells. To further establish that the observed responses were not dependent on the generation and release of a chemotactic protein following antigen challenge, we examined whether such chemotaxis was prevented by incubation of the cells with the RNA-synthesis inhibitor, actinomycin D. As shown in Fig. 2B, actinomycin D did not inhibit BMMC migration induced by antigen, SCF or PGE2 alone, or in combination. Taken together, these data demonstrate that, in sensitized BMMCs, the synergistic chemotactic responses elicited by antigen in combination with SCF, PGE2 and adenosine are likely to be, in large part direct, rather than due to the release of a secondary chemotactic agent.
Fig. 2.

Synergistic cell migration is primarily dependent on chemotaxis. (A) IgE-sensitized BMMCs were washed three times with HEPES buffer containing 0.5% BSA, and then cells were stimulated with indicated agonists [antigen, Ag (10 ng/ml), SCF (10 ng/ml), adenosine (1 μM), PGE2 (100 nM)]. After 3 hours, cell-free supernatants (Sup) from antigen and/or other agonist-stimulated BMMCs were applied to the lower chamber. Sensitized or unsensitized BMMCs were placed in the upper chambers. After incubation for 4 hours, BMMCs migrating to the lower chambers were collected and counted. (B) Sensitized BMMCs were preincubated with or without actinomycin D (5 μg/ml) for 30 minutes, and cell migration was measured. (C) To test whether the cell migration was chemotaxis or chemokinesis, indicated agonists were placed in the lower chamber or both upper and lower chambers. Sensitized BMMCs were placed in upper chambers. After 4 hours, BMMCs migrating to the lower chambers were collected and counted. Results are means ± s.e. of three separate experiments. *P<0.05 for comparison with the same stimulation in control group by Student's t-test.
To verify that the responses were due to directed chemotaxis rather than chemokinesis (random migration), we examined the migration of sensitized BMMCs, with or without a concentration gradient of stimuli across the membranes. Without a gradient, the migration of BMMCs towards antigen, SCF and PGE2 was largely abrogated, as was the synergy between antigen and other stimulants (Fig. 2C). These data demonstrate that the migration of sensitized BMMCs towards the various stimulants was primarily due to chemotaxis and not chemokinesis. As the data show that SCF and GPCR-mediated responses were similarly potentiated by antigen, the detailed mechanistic studies were investigated with PGE2. Similar responses were, however, observed when cells were challenged with SCF or adenosine in the absence of antigen (data not shown).
Synergy in chemotaxis is associated with synergistic F-actin cytoskeletal rearrangement and an enhanced Ca2+ signal
We next explored whether the synergy in chemotactic responses was accompanied by a similar enhancement of actin polymerization and depolymerization, as monitored by FITC-labeled phalloidin, which interacts with F-actin but not with G-actin (monomeric, globular G-actin). As shown in Fig. 3A, F-actin staining with FITC-phalloidin was reduced by antigen at early time points (<2 minutes) and then restored after 10 minutes, indicating antigen-induced actin depolymerization and re-polymerization. By contrast, FITC-phalloidin staining indicated that PGE2 increased actin polymerization; however, when added to the cells concurrently with antigen, it accentuated F-actin disassembly. To establish a link between these observations and BMMC chemotaxis, we used cytochalasin B, an inhibitor of actin polymerization (MacLean-Fletcher and Pollard, 1980). Cytochalasin B had no effect on degranulation and chemokine generation (data not shown) and little effect on antigen-induced actin depolymerization (Fig. 3B). By contrast, cytochalasin B significantly blocked actin polymerization whether cells were stimulated with antigen, PGE2 or a combination of the two (Fig. 3B). Cell chemotaxis was also blocked (Fig. 3C).
Fig. 3.

The role of cytoskeletal reorganization and Ca2+ in mast-cell migration. (A,B) Sensitized BMMCs were preincubated with or without cytochalasin B (10 μM) for 20 minutes. After preincubation with cytochalasin B, cells were stimulated with indicated agonists [Ag (10 ng/ml), PGE2 (100 nM)] for the indicated time, fixed, then permeabilized. Actin rearrangement was measured by using FITC-labeled phalloidin staining of the cells, followed by flow cytometry. Data are mean values of fluorescence intensities of phalloidin staining. (C) Sensitized BMMCs were preincubated with or without Cytochalasin B (10 μM) for 20-30 minutes, and cell migration was measured. (D) Sensitized BMMCs were preincubated with U73122 (1 μM), 2APB (50 μM), or EDTA (5 mM) in upper chambers and placed in 600 μl HEPES buffer containing 0.5% BSA and indicated inhibitors for 30 minutes, and then upper chambers were placed in the lower chambers containing the indicated agonists. Results are means ± s.e. of three separate experiments. *P<0.05 for comparison with the same stimulation in control group by Student's t-test.
Our previous observation that PGE2 enhances antigen-induced PLCγ1-dependent Ins(1,4,5)P3 production and the associated Ca2+ signal in BMMCs (Kuehn et al., 2008a), suggests that the Ca2+ signal could be one candidate for propagation of synergistic signals. The absolute requirement for a PLC- or Ins(1,4,5)P3-mediated Ca2+ signal was verified by use of U73122 (a PLC inhibitor), 2APB [an Ins(1,4,5)P3-receptor antagonist and inhibitor of store-operated Ca2+ entry] and EDTA (a Ca2+ chelator). As can be seen from Fig. 3D, all three agents largely or completely suppressed BMMC chemotaxis. These inhibitors also significantly suppressed actin polymerization by antigen and/or PGE2 (data not shown). Taken together, these data establish that the synergistic enhancement of PGE2-mediated mast-cell chemotaxis by antigen is dependent upon PLCγ-dependent Ca2+ mobilization and enhanced actin-filament reorganization.
PI3K is required for migration mediated by antigen, GPCR agonist and SCF
We next investigated the signal transduction events underlying the synergy in Ca2+ flux and F-actin reorganization. We hypothesized that PI3K might be a common upstream molecule that regulates these processes. Our previous studies have shown that 100 nM wortmannin effectively blocks AKT phosphorylation induced by antigen (Kim et al., 2008), SCF (Kim et al., 2008) and PGE2 (Kuehn et al., 2008a), which acts as a surrogate marker for PI3K activation. As shown in Fig. 4A, wortmannin completely abrogates the migration of BMMCs in response to antigen and/or PGE2. Similar inhibition was observed when wortmannin-treated cells were challenged with antigen in the presence and absence of SCF and adenosine (data not shown). These data demonstrate that PI3K is indeed essential for chemotactic responses to all stimulants regardless of whether they were added individually or in combination with antigen.
Fig. 4.
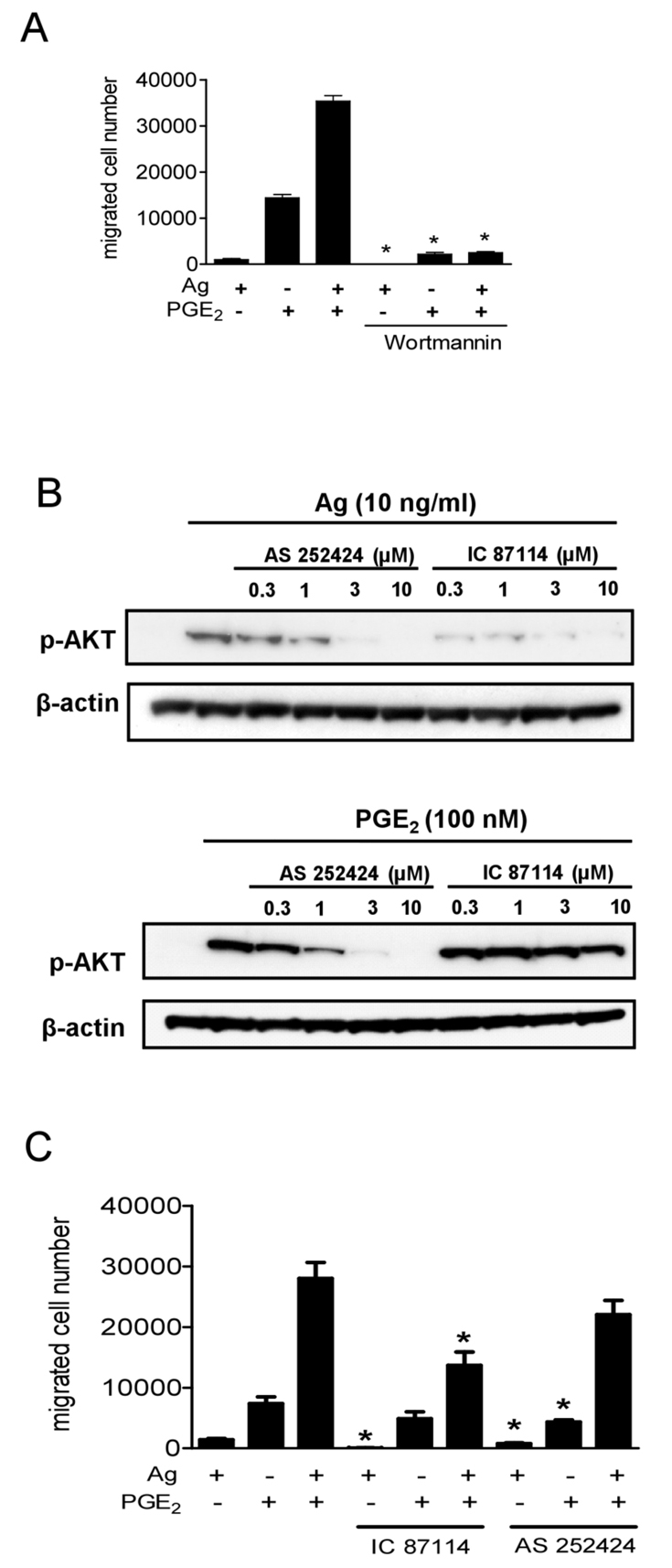
The role of PI3K in synergistic chemotactic responses. (A) Sensitized BMMCs were preincubated with or without wortmannin (100 nM), in the upper chamber and placed in 600 μl HEPES buffer containing 0.5% BSA and wortmannin for 30 minutes. The upper chambers were then replaced in the lower chambers containing the indicated agonists. (B) Sensitized BMMCs were preincubated with indicated inhibitors for 20 minutes and then stimulated with the indicated agonists for 5 minutes. Following electrophoresis and membrane transfer, proteins were probed using anti-phospho-AKT (Ser473-P). To normalize protein loading, membranes were stripped and probed for β-actin, or alternatively identically loaded samples were probed for β-actin. The data shown are from three separate experiments, each repeated at least three times, with identical results, on separate cell preparations. (C) Sensitized BMMCs were preincubated with or without AS 252424 (3 μM) or IC 87114 (3 μM) in the upper chambers and placed in 600 μl HEPES buffer containing 0.5% BSA and indicated inhibitors for 30 minutes, and then upper chambers were replaced in the lower chamber containing the indicated agonists. Results in A and C are means ± s.e. of three separate experiments. *P<0.05 for comparison with the same stimulation in control group by Student's t-test.
We next defined the role of the specific subfamilies of PI3K in the chemotactic responses. The PI3K family has been divided into three subfamilies (I, II and III) on the basis of their structural characteristics, activation mechanisms, and substrate specificity (Vanhaesebroeck et al., 2005). Type IA and type IB family members, PI3Kδ and PI3Kγ, are linked to receptors that signal through tyrosine kinases and G-proteins, respectively. Recently, isoform-specific inhibitors have been described, namely, IC 87114 and AS 252424, which selectively block PI3Kδ and PI3Kγ respectively (Ali et al., 2008). The effects of these compounds were thus examined to gain insight on the particular PI3K isoform involved in chemotaxis.
We initially determined the optimal concentrations of IC 87114 and AS 252424 on antigen- and PGE2-mediated AKT phosphorylation (Fig. 4B). As predicted, the PI3Kδ inhibitor IC87114 inhibited antigen-induced AKT phosphorylation without markedly affecting the phosphorylation of AKT in response to PGE2 (Fig. 4B). Since near-maximal inhibition was observed at 3 μM, this concentration was selected for subsequent studies. The PI3Kγ inhibitor AS 252424 inhibited both antigen- and PGE2-induced AKT phosphorylation at concentrations of 3 μM (Fig. 4B). These latter observations are consistent with previous observations that antigen-mediated responses might be partly regulated by PI3Kγ-dependent pathways (Ali et al., 2008). Nevertheless, a concentration of 3 μM AS 25224 was selected for subsequent studies
IC 87114 significantly attenuated the chemotactic response elicited by antigen with minimal or no inhibition of the response to PGE2 but substantial inhibition of the response to the combination of stimulants (Fig. 4C). By contrast, AS 252424 only partially reduced the chemotactic responses to antigen and PGE2, and failed to significantly diminish the response to both in combination. The synergy in chemotactic responses to antigen in combination with SCF or adenosine was similarly sensitive to the inhibitory effects of IC 87114 (data not shown). These results suggest that, although PI3Kγ might have a role in the responses mediated by GPCRs, PI3Kδ is the major isoform regulating the responses to antigen alone, or in combination with other stimulants.
Antigen-mediated and synergistic chemotactic responses require Btk
Btk is one of several downstream regulators of PI3K-dependent responses in hematopoietic cells, including B-cells (de Gorter et al., 2007) and mast cells (Kuehn et al., 2008b). We used BMMCs from Btk−/− mice to investigate whether Btk regulates chemotactic responses downstream of PI3K. Lack of Btk in these cells was confirmed by western blot analysis (Fig. 5A). Toluidine Blue staining demonstrated that Btk−/− BMMC morphology was similar to that of WT BMMCs (Fig. 5B). Btk−/− BMMCs are also known to express KIT and FcεRI to the same extent as WT BMMCs (Hata et al., 1998).
Fig. 5.

The role of Btk in synergistic chemotactic responses. (A) To confirm knock out of Btk, cell lysates were prepared from WT, Btk−/− BMMCs and proteins were probed using an anti-Btk antibody. (B) 4-week-old BMMCs from WT and Btk−/− mice were stained with Toluidine Blue and images were obtained with an original magnification of 100×. (C) Sensitized WT and Btk−/− BMMCs were washed and placed in the upper chambers. Indicated agonists [Ag (10 ng/ml), SCF (10 ng/ml), PGE2 (100 nM), adenosine (1 μM)] were added to the lower chamber. Migrated cells were counted after incubation for 4 hours. (D) Sensitized WT and Btk−/− BMMCs were stimulated with indicated agonists [antigen, Ag (10 ng/ml), PGE2 (100 nM)] for 30 seconds or 2 minutes, and then membrane fractions were prepared to analyse the activation status. Following electrophoresis and membrane transfer, proteins were probed using the following antibodies: anti-phospho-Btk (Tyr551-P), anti-Btk, anti-phosphorylated AKT (Ser473-P). To normalize protein loading, membranes were probed for KIT. (E) Data were generated by scanning the blots in three independent experiments, and then normalizing to the response at 2 minutes obtained with Ag in WT BMMCs. Results are means ± s.e. of three separate experiments. *P<0.05 for comparison with the same stimulation in WT BMMCs by Student's t-test.
The chemotactic response to antigen was virtually absent in the Btk−/− BMMCs and yet these cells responded fully to SCF and PGE2 (Fig. 5C). As in normal BMMCs (Fig. 1), adenosine was an ineffective chemotactic stimulant for Btk-deficient cells (Fig. 5C) Nevertheless, the synergistic chemotactic responses induced upon interactions of antigen with SCF, PGE2 and adenosine, were largely abrogated in the Btk−/− BMMCs. These results suggest that antigen-induced migration and the synergistic responses elicited by antigen are mediated by Btk downstream of PI3K, but the chemotactic responses induced by SCF or PGE2 alone are regulated by a PI3K-regulated, but Btk-independent, pathway.
Activation of Btk requires coordinated translocation to the cell membrane and tyrosine phosphorylation at positions Y223 and Y551. We thus next examined the ability of PGE2 to enhance the antigen-mediated phosphorylation of Btk in the membrane fractions of BMMCs derived from WT and Btk−/− mice, using an antibody that recognized Btk phosphorylated at Y551. To verify PI3K activation in these experiments, we also determined AKT phosphorylation in the membrane fractions.
Consistent with previous results (Kuehn et al., 2008a), and the ability of wortmannin to block BMMC chemotaxis (Fig. 4A), antigen-mediated phosphorylation of AKT was enhanced by PGE2 and this enhancement was most prominent at the 2 minute time point (Fig. 5D,E). This response was still observed in the Btk−/− BMMCs, which verified that activation of PI3K was upstream of Btk. Unlike antigen, which induced Btk phosphorylation in the membrane fraction of WT BMMCs, PGE2 had minimal effect on Btk phosphorylation in the absence of antigen at both time points examined (30 seconds and 120 seconds) (Fig. 5D,E). This was consistent with the finding that PGE2-mediated chemotaxis is not dependent on Btk as demonstrated in Btk−/− BMMCs, Nevertheless, in combination with antigen, PGE2 produced a marked synergistic enhancement of membrane-associated Btk phosphorylation (Fig. 5D,E). This was associated with a net increase in Btk protein level within the membrane fraction. Hence the increase in membrane-associated phosphorylated Btk could be attributed, at least in part, to increased Btk translocation rather than increased phosphorylation. These responses were, as predicted, absent in the Btk−/− BMMCs.
Collectively, these data demonstrate that, by enhancing antigen-mediated membrane translocation of Btk, PGE2 and antigen cooperate to increase the amount of phosphorylated Btk associated with the cell membrane, and that the activation of Btk in this manner is crucial for the synergistic intereaction between PGE2 and antigen for the enhancement of mast-cell chemotaxis.
The role of Btk in the synergistic enhancement of the Ca2+ signal and cytoskeletal reorganization in response to PGE2 and antigen
We next investigated the impact of Btk deficiency on activation of PLC, Ca2+ mobilization and actin-related cytoskeletal reorganization, because of the importance of these processes in chemotaxis. FcεRI-mediated Ca2+ mobilization requires activation of PLCγ through its translocation to the cell membrane and phosphorylation, with resulting generation of Ins(1,4,5)P3. In contrast to the amplification of PGE2-induced phosphorylation of membrane-associated PLCγ1 (Fig. 6A,B), production of Ins(1,4,5)P3 (Fig. 6C) and Ca2+ mobilization observed in response to antigen in WT BMMCs (Fig. 6D), these responses were no longer apparent in the Btk−/− BMMCs. These data provide support for the conclusion that Btk regulates the enhanced chemotactic response, at least in part, via enhanced PLCγ-dependent enhanced Ca2+ mobilization. The observed lack of reduction in the Ca2+ signal induced by PGE2 alone provides an explanation as to why the effects of PGE2 alone on chemotaxis were not reduced in the Btk−/− BMMCs.
Fig. 6.

The role of Btk in PGE2-enhanced, antigen-mediated Ca2+ signaling. (A) Sensitized WT and Btk−/− BMMCs were stimulated with the indicated agonists [antigen, Ag (10 ng/ml), PGE2 (100 nM)] for 30 seconds or 2 minutes, and then membrane fractions were prepared. Following electrophoresis and membrane transfer, proteins were probed using anti-phospho-PLCγ (Tyr783-P). To normalize protein loading, membranes were probed for KIT. (B) The data were generated by scanning the blots from three independent experiments, and then normalizing to the response at 2 minutes obtained with Ag in WT BMMCs. The data are presented as the means + s.e. of three separate experiments. (C) WT and Btk−/− BMMCs were stimulated with antigen (Ag, 10 ng/ml), PGE2 (100 nM) or Ag and PGE2. After 30 seconds, the samples were processed and the Ins(1,4,5)P3 levels determined. The data are presented as means + s.e. of four separate experiments conducted in duplicate. (D) WT Btk−/− BMMCs were loaded with Fura-2 AM, and then changes in intracellular Ca2+ levels were determined after challenge with Ag (10 ng/ml) or PGE2 (100 nM) or Ag and PGE2. The Ca2+ data are from three representative experiments conducted on separate cell preparations. Results are means ± s.e. of three separate experiments. *P<0.05 for comparison with same stimulation in WT by Student's t-test.
Rac activation is a crucial step in actin polymerization and depolymerization associated with cytoskeletal re-organization (Ridley, 2006). To investigate whether Rac activation leading to F-actin rearrangement is also dependent on Btk, a PAK-1-PBD binding assay was used to assess the Rac-GTP interaction as an index of Rac activation. Rac activation was increased in response to both PGE2 and antigen when added separately (Fig. 7A); however, when they were added together, this activation was markedly enhanced (Fig. 7A). In the Btk−/− BMMCs, antigen-induced Rac activation and the potentiation of this response by PGE2 were markedly attenuated (Fig. 7B). In addition, F-actin rearrangement in response to antigen was much diminished in the absence or presence of PGE2 (Fig. 7C-E). However, the increased polymerization induced by PGE2 alone was unchanged. Wortmannin treatment also significantly reduced F-actin rearrangement in response to antigen, both in the absence and presence of PGE2 (data not shown). These results demonstrated that PI3K-Btk regulates antigen-mediated actin rearrangement, which is required for cell migration.
Fig. 7.

The role of Btk in PGE2-enhanced, antigen-mediated cytoskeletal reorganization. (A,B) Rac activation was determined using a PAK-1 PBD assay kit. BMMCs were stimulated with indicated agonists [antigen, Ag (10 ng/ml) or PGE2 (100 nM)] for 2 minutes and lysates were mixed with GST-PBD bound to glutathione-agarose, and incubated for 4 hours at 4°C. For a positive control, cell lysates were incubated with 100 μM GTPγS. Precipitates were washed and suspended in sample buffer. Proteins were separated and blotted with an anti-Rac1 antibody. To establish equal Rac content in the reactions, cell lysates from each sample were probed by immunoblot analysis prior to reaction with GST-PBD. The western blots represent data from four separate experiments. (C-E) Cells were stimulated with indicated agonist Ag (10 ng/ml), PGE2 (100 nM) for indicated time (C) or 2 minutes (D,E), and then fixed and permeabilized. Actin polymerization was measured using FITC-labeled phalloidin staining of the cells, followed by flow cytometry (C,D) and imaging (E). Data are from four representative experiments conducted on separate cell preparations.
Taken together, these data demonstrate that, by regulating the required Ca2+ signaling and F-actin reorganization, Btk is crucial for FcεRI-induced mast-cell migration and its ability to enhance migration mediated by PGE2.
Discussion
In this study, we report for the first time that the signal-transduction pathways elicited by FcεRI aggregation integrate with those induced by SCF and the GPCR agonists, adenosine and PGE2, to amplify mast-cell chemotaxis and that the PI3K-Btk axis is central to this amplification. In addition, we show that Btk regulates both FcεRI-mediated Rac activation and actin polymerization, which are essential events for the chemotactic response.
Although it is widely recognized that antigen-induced aggregation of FcεRI initiates degranulation, as well as production of eicosanoids and cytokines, whether it also leads to mast-cell chemotaxis is less clear. It has been reported that both IgE and IgE-antigen induce mast-cell migration (Ishizuka et al., 2001; Jung et al., 2009; Kitaura et al., 2005), whereas another report noted that these responses might merely reflect chemokinesis or the autocrine-paracrine action of chemotactic factors that are released from the activated mast cells (Kitaura et al., 2005; Taub et al., 1995). In the current study, we opted not to use fibronectin-coated membranes as used previously (Kitaura et al., 2005) to avoid the potential activation of integrin receptors that might augment chemotactic responses. Although the mast-cell chemotactic responses to antigen were much lower than that to SCF or PGE2 (Fig. 1), our observations support the conclusion that FcεRI aggregation directly stimulates chemotaxis rather than chemokinesis (Fig. 2). This conclusion was based on the minimal chemotactic response of non-activated cells to supernatants taken from stimulated mast-cell cultures or of non-sensitized cells to antigen or supernatants (Fig. 2). The insensitivity of antigen-mediated chemotaxis to pertussis toxin (Fig. 1), the PI3Kγ inhibitor AS 252424 (Fig. 4), and the RNA-synthesis inhibitor actinomycin D (Fig. 2), also argues against an indirect mechanism through release of GPCR ligands from mast cells. Regardless of the physiological significance of the modest chemotactic response to antigen, our data clearly demonstrate that antigen has a marked capacity to enhance or potentiate the action of PGE2, SCF and adenosine (Fig. 1). Therefore, in addition to their ability to potentiate antigen-mediated activation of mast cells (Ali et al., 1990; Hundley et al., 2004; Kuehn et al., 2008a; Zhong et al., 2003) already present in tissues, KIT and GPCR ligands might also enable expansion of mast-cell populations at sites of antigen infiltration. We note, however, that the effects of SCF noted here were a consequence of short-term exposure to SCF. Long-term exposure to SCF might downregulate FcεRI-mediated chemotaxis (Sawada et al., 2005).
The enhancement of antigen-induced chemotaxis by KIT and GPCR ligands appears to be dependent on PI3K and Btk. This is in contrast to PGE2-mediated enhancement of antigen-mediated degranulation, which is not dependent on either PI3K (Kuehn et al., 2008a) or Btk (H.S.K. and A.M.G., unpublished observations). The participation of PI3K in the chemotactic responses was illustrated by the inhibition of chemotaxis by wortmannin, whether induced by antigen or PGE2 alone, or in combination (Fig. 4A). The studies with the isoform-specific PI3K inhibitors AS 252424 (PI3Kγ) and IC 87114 (PI3Kδ), further indicated that the PI3Kδ isoform was solely responsible for the synergistic chemotactic responses produced by antigen. However, a significant portion of the synergistic response to antigen and PGE2 was refractory to either inhibitor alone (Fig. 4C) or in combination (data not shown). The fact that this response was completely blocked by wortmannin suggests that additional PI3K isoforms also contribute to the ability of the antigen to enhance PGE2- mediated chemotaxis.
Chemotaxis was also severely impaired in Btk-deficient BMMCs (Fig. 5). Our previous studies provided evidence that Btk activation is PI3K dependent (Kuehn et al., 2008b) and that both are crucial for degranulation, as well as production of cytokines and reactive oxygen species with respect to antigen and SCF (Hata et al., 1998; Iwaki et al., 2005; Kuehn et al., 2008b). In assessing whether this was true for chemotaxis, we observed that the Btk-dependent synergy in the chemotactic response to PGE2 plus antigen was indeed accompanied by a similar synergistic increase in the amount of membrane-associated phosphorylated Btk (Fig. 5D,E). This was, however, probably due to an increase in total membrane-associated Btk rather than enhanced phosphorylation. However, the mechanism for this increase is unclear. Although it might be argued that this reflects an enhanced PI3K signal, leading to binding of the PH domain of Btk to the PtdIns(3,4,5)P3 formed at the cell membrane, the lack of Btk translocation observed with PGE2 alone despite robust AKT phosphorylation would suggest that additional signals are required to stabilize this interaction.
The lack of antigen-regulated chemotactic responses in Btk−/− BMMCs might be directly attributed to the impaired Ca2+ signal and F-actin rearrangement in response to antigen in the presence or absence of PGE2 (Figs 6,7). In several cell types, increased cytosolic Ca2+ levels and cytoskeletal reorganization associated with actin polymerization and depolymerization are crucial events for cell migration (Allen et al., 2009; Jung et al., 2009; Shimizu et al., 2009). The antigen-and PGE2-induced Ca2+ signal in mast cells is initially dependent on the activation of PLCγ and PLCβ, respectively, with resulting generation of Ins(1,4,5)P3, which liberates Ca2+ from intracellular stores (Kuehn et al., 2008a; Ma and Beaven, 2009). PLCγ activation, Ins(1,4,5)P3 generation and the resulting Ca2+ signal were substantially enhanced in BMMCs when stimulated concurrently with antigen and PGE2 (Kuehn et al., 2008a) (Fig. 6), as was Rac activation leading to F-actin rearrangement (Fig. 7). The requirement for these signals was also demonstrated by the fact that inhibitors of PLCγ- and Ins(1,4,5)P3-dependent Ca2+ mobilization (Fig. 3D) and prevention of cytoskeletal reorganization with cytochalasin B (Fig. 3B,C) ablated the chemotactic responses to antigen and PGE2 individually, or in combination. In addition, all these events were impaired in Btk−/− BMMCs (Figs 6,7). Irrespective of the PI3K isoform used by the individual receptors, these latter observations provide a direct insight as to how the PI3K-Btk axis might regulate antigen-mediated chemotaxis and its ability to enhance the responses elicited by GPCR agonists. We have previously demonstrated that the ability of SCF to enhance the antigen-dependent PLCγ1-mediated Ca2+ signal required for degranulation is also regulated by Btk (Iwaki et al., 2005). Thus, the synergistic enhancement of chemotaxis produced by SCF and antigen is likely to occur by a similar mechanism as that required by PGE2 in conjunction with antigen.
As outlined in Fig. 8, our data as a whole demonstrate that simultaneous challenge of mast cells with antigen, specific GPCR ligands and SCF result in activation of receptor-proximal signaling pathways leading to integrated activation of PI3Ks with a resulting synergistic activation of membrane-associated Btk (Fig. 5D). This in turn induces an enhancement in the Ca2+ signal and Rac-dependent F-actin rearrangement, which in combination lead to the observed synergy in mast-cell chemotaxis. We thus conclude that the signal-transduction events elicited by FcεRI act co-ordinately with those initiated by GPCRs and the SCF receptor KIT to amplify mast-cell chemotaxis and that the PI3K-Btk axis is required for these responses. This model could therefore provide a paradigm for how signal-transduction cascades initiated by several classes of receptors might integrate to promote migration of mast cells, or indeed other inflammatory cells, into inflammatory tissues or infected sites in disease states in a physiological setting.
Fig. 8.

Proposed integrated signaling pathway responsible for the synergistic chemotactic responses elicited by antigen in the presence of SCF or GPCR agonists. For clarity, the pathways by which GPCRs and KIT regulate chemotaxis in the absence of FcεRI aggregation have not been included. Dotted lines indicate an amplification pathway and does not imply direct regulation. Concurrent ligation of KIT or GPCRs with FcεRI aggregation leads to enhanced activation of PI3Ks, which would result in elevated production of phosphoinositide-3,4,5-trisphosphate [PtdIns(3,4,5)P3 or PIP3] from phosphoinositide-4,5-bisphosphate [PtdIns(4,5)P2 or PIP2]. This would allow the synergistic translocation and activation of Btk (Fig. 5D). This in turn induces an enhancement in the Ca2+ signal and Rac-dependent F-actin rearrangement, which in combination lead to the observed synergy in mast-cell chemotaxis.
Materials and Methods
Cell isolation and sensitization
Mouse bone-marrow-derived mast cells (BMMCs) were obtained by flushing bone marrow cells from the femurs of C57BL/6 mice (The Jackson Laboratory), then culturing the cells for 4-6 weeks in RPMI 1640 containing IL-3 (30 ng/ml) (Peprotech) as described (Kuehn et al., 2008a). The Btk−/− mice, kindly provided by Anne B. Satterthwaite (University of Texas Southwestern Medical Center, Dallas, TX), and the wild-type (WT) mice used in this study have been described (Iwaki et al., 2005). Mice were backcrossed with C57BL/6 (The Jackson Laboratory) over six generations and age-matched 8- to 10-week-old littermate mice were used as controls. The genotype of these mice was confirmed by RT-PCR of tail biopsies (data not shown).
BMMCs were sensitized overnight with an optimal concentration (100 ng/ml) of mouse IgE anti-DNP (clone, SPE-7) (Sigma) in cytokine-free medium. The cells were then rinsed three times with HEPES buffer (10 mM HEPES, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 0.4 mM Na2HPO4•7H2O, 5.6 mM glucose, 1.8 mM CaCl2•2H2O, and 1.3 mM MgSO4•7H2O) containing 0.04% BSA (Sigma) to remove excess IgE. Cells were then resuspended in this buffer at the appropriate cell density for a specific assay.
Chemotaxis assay
Chemotaxis assays were performed using Transwell® permeable support with 5.0 μM pore polycarbonate membranes on 6.5 mm inserts (Costar) placed within 24-well polystyrene plates. Sensitized BMMCs from WT or Btk−/− (3×105 cells/100 μl in HEPES buffer containing 0.5% BSA) were placed in the upper Transwell support chamber. The upper chambers were then preincubated within each well of the plate (functionally, the lower chamber) containing 600 μl HEPES buffer containing 0.5% BSA for 30 minutes. The upper chambers were then placed in the lower chambers containing the agonists, DNP-human serum albumin [DNP-HSA (antigen, Ag)] (Sigma), SCF, PGE2 and adenosine, as indicated. After incubation for 4 hours at 37°C, cells migrating to the lower chambers were collected and counted by microscopy. For the inhibitor studies, BMMCs were preincubated with pertussis toxin (1 μg/ml) (Sigma) for 4 hours, washed, then chemotaxis assessed as above. In other studies, cells were pre-incubated with or without wortmannin (100 nM), AS 252424 (3 μM), IC 87114 (3 μM), U73122 (1 μM), 2APB (50 μM), EDTA (5 mM), actinomycin D (5 μg/ml) or cytochalasin B (10 μM) in the upper chambers and placed in 600 μl HEPES buffer containing 0.5% BSA and indicated inhibitors for 30 minutes. The upper chambers were replaced in the lower chamber containing indicated agonists. Wortmannin, U73122, 2APB, cytochalasin B were purchased from EMD Biosciences. EDTA, actinomycin D were purchased from Sigma. AS 252424 and IC 87114 were Serono (Geneva, Switzerland).
Fractionation of cells and immunoblotting
To prepare membrane fractions, BMMCs (2×106 cells/sample) were sensitized and washed as above, then stimulated with antigen (10 ng/ml) and/or PGE2 (100 nM) at 37°C for the indicated times. For experiments in which the effects of inhibitors were examined, BMMCs were preincubated with the indicated inhibitors for 20 min before the addition of the indicated agonists. The reactions were terminated by washing with ice-cold PBS followed by the addition of 200 μl ice-cold lysis buffer [50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 2 mM DTT, 1 mM sodium orthovanadate, 50 mM sodium pyrophosphate, 50 mM sodium fluoride, protease inhibitor cocktail (Roche) and Sigma phosphatase inhibitor cocktails 1 and 2 (Sigma)]. The cells were lysed by sonication. Sonicates were then centrifuged at 700 g for 10 minutes to remove intact cells and nuclei. The recovered supernatants were further centrifuged at 20,000 g for 30 minutes to recover the membrane fractions from the resulting pellet. Proteins in the pellet fractions were solubilized with lysis buffer containing 1% Triton X-100, 1% NP40, and 0.1% SDS for 30 minutes on ice followed by centrifugation at 15,000 g for 15 minutes. The recovered supernatants were saved as membrane fractions. Proteins from the membrane fractions were separated by electrophoresis on 4-12% NuPAGE Bis-Tris gels (Invitrogen) and the proteins were probed for immunoreactive proteins utilizing the following antibodies (Abs): anti-phospho-AKT (Ser473-P) and anti-KIT (Cell Signaling); anti-phospho-PLCγ1 (Tyr783-P) (Biosource) and anti-phospho-Btk (Tyr551-P) and anti-Btk (BD Biosciences). Protein loading of the membrane fractions was normalized by stripping and probing for KIT; or alternative by probing identically loaded samples.
For immunoblot analyses, BMMC lysates were prepared as described (Tkaczyk et al., 2002) and proteins separated by electrophoresis on 4-12% NuPAGE BisTris gels (Invitrogen). Following membrane transfer, proteins were probed using the following antibodies: anti-β-actin mAb (clone AC-15) (Sigma); anti-phospho-AKT (Ser473-P) (Cell Signaling). To normalize protein loading, membranes were stripped and probed for β-actin, or alternatively identically loaded samples were probed for β-actin. To quantify changes in protein phosphorylation, the ECL films were scanned using a Quantity One scanner (Bio-Rad).
Rac activation assay
Affinity precipitation with GST-PBD (p21 Binding Domain) was performed using PAK-1 PBD (Rac effector protein, p21 activated kinase-1) assay kit (Upstate) according to the manufacturer's instructions, BMMC lysates were mixed with 10 μl GST-PBD bound to glutathione-agarose, and incubated for 4 hours at 4°C. For a positive control, cell lysates were incubated for 15 minutes at 30°C with 100 μM GTPγS in the presence of 1 mM EDTA. The reaction was stopped by the addition of 60 mM MgCl2. GTPγS-loaded lysates were incubated with GST-PBD for 30 minutes at 4°C. Finally, precipitates were washed three times with MLB and suspended in Laemmli sample buffer. Proteins were separated on 4-12% NuPAGE BisTris gels, transferred onto nitrocellulose membrane, and blotted with anti-Rac1 antibody. To establish equal Rac content in the reactions, cell lysates from each sample were probed by immunoblot analysis before reaction with GST-PBD.
Intracellular Ca2+ determination
Ca2+ flux was measured in sensitized and activated WT or Btk−/− BMMCs following loading of the cells with Fura-2 AM ester (Molecular Probes) as described (Tkaczyk et al., 2003). Cells were loaded with Fura-2 AM (2 μM) for 30 minutes at 37°C, rinsed, and resuspended in HEPES buffer containing 0.04% BSA and sulfinpyrazone (0.3 mM) (Sigma), and then placed in a 96-well black culture plate (2×104 cells/well) (CulturPlat-96 F, PerkinElmer Life Sciences). Fluorescence was measured at two excitation wavelengths (340 and 380 nm) and an emission wavelength of 510 nm. The ratio of the fluorescence readings was calculated following subtraction of the fluorescence of the cells that had not been loaded with Fura-2 AM.
Ins(1,4,5)P3 assay
Sensitized WT or Btk−/− BMMCs (2×106) were stimulated with antigen (10 ng/ml) and/or PGE2 (100 nM) in the same buffer (400 μl). After 30 seconds, the reaction was stopped by adding 80 μl ice-cold 100% trichloroacetic acid. Ins(1,4,5)P3 was then extracted from the trichloroacetic acid precipitates using 1,1,2-trichlorofluorethane-trioctylamine. Cellular Ins(1,4,5)P3 content was determined using a commercially available kit (Amersham Biosciences) according to the manufacturer's instructions. The results are expressed as picomoles Ins(1,4,5)P3 per 2×106 cells.
Measurement of F-actin (polymeric, filamentous actin) content by flow cytometry
Sensitized BMMCs (1×106 cells/sample) were washed, then challenged with DNP-HSA (Ag, 10 ng/ml) and/or PGE2 (100 nM) for 2 minutes as indicated. Next, cells were fixed at RT by the addition of 1 ml of 4% paraformaldehyde for 15 minutes. The cells were then permeabilized with 0.1% Saponin-PBS for 5 minutes and stained with 2 μg/ml FITC-labelled phalloidin (Sigma) in 1% BSA, 0.1% Saponin-PBS for 1 hour in the dark at room temperature. After washing three times with PBS, cellular F-actin content was determined using FACScan flow cytometer by gating on 10,000 living cells.
Fluorescence microscopy
For imaging, sensitized BMMCs (50,000/sample) were stimulated with Ag (10 ng/ml) and/or PGE2 (100 nM) for 2 minutes. After stimulation, cells were immediately attached to glass slides using a cytospin centrifuge (450 r.p.m., 3 minutes, RT). Cells were then fixed and permeabilized and stained with FITC-labeled phalloidin as described above. After washing the slides carefully with PBS, images were obtained using fluorescence microscopy.
Toluidine Blue staining
Four-week-old WT or Btk−/− BMMCs (50,000 cells/sample) were stained with Toluidine Blue as described (Kirshenbaum and Metcalfe, 2006). Images were obtained using confocal microscopy (100×).
Statistical analysis
Data are represented as the mean ± s.e. The statistical analyses were performed by unpaired Student's t-test. Differences were considered significant when P<0.05. The n values represent experiments from several preparations.
Supplementary Material
Acknowledgments
This work was supported by the Division of Intramural Research of NIAID and NHLBI within the National Institutes of Health. We thank Anne B. Satterthwaite, University of Texas Southwestern Medical Center, for kindly providing Btk−/− mice. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/15/2576/DC1
References
- Ali H., Cunha-Melo J. R., Saul W. F., Beaven M. A. (1990). Activation of phospholipase C via adenosine receptors provides synergistic signals for secretion in antigen-stimulated RBL-2H3 cells. Evidence for a novel adenosine receptor. J. Biol. Chem. 265, 745-753 [PubMed] [Google Scholar]
- Ali K., Camps M., Pearce W. P., Ji H., Ruckle T., Kuehn N., Pasquali C., Chabert C., Rommel C., Vanhaesebroeck B. (2008). Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J. Immunol. 180, 2538-2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. D., Jaffer Z. M., Park S. J., Burgin S., Hofmann C., Sells M. A., Chen S., Derr-Yellin E., Michels E. G., McDaniel A., et al. (2009). p21-activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 113, 2695-2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. M., Wilson T. M., Metcalfe D. D. (2008). The mast cell and allergic diseases: role in pathogenesis and implications for therapy. Clin. Exp. Allergy 38, 4-18 [DOI] [PubMed] [Google Scholar]
- de Gorter D. J., Beuling E. A., Kersseboom R., Middendorp S., van Gils J. M., Hendriks R. W., Pals S. T., Spaargaren M. (2007). Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 26, 93-104 [DOI] [PubMed] [Google Scholar]
- Echtenacher B., Mannel D. N., Hultner L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381, 75-77 [DOI] [PubMed] [Google Scholar]
- Galli S. J., Nakae S., Tsai M. (2005). Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135-142 [DOI] [PubMed] [Google Scholar]
- Harris S. G., Padilla J., Koumas L., Ray D., Phipps R. P. (2002). Prostaglandins as modulators of immunity. Trends Immunol. 23, 144-150 [DOI] [PubMed] [Google Scholar]
- Hata D., Kawakami Y., Inagaki N., Lantz C. S., Kitamura T., Khan W. N., Maeda-Yamamoto M., Miura T., Han W., Hartman S. E., et al. (1998). Involvement of Bruton's tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187, 1235-1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogaboam C., Kunkel S. L., Strieter R. M., Taub D. D., Lincoln P., Standiford T. J., Lukacs N. W. (1998). Novel role of transmembrane SCF for mast cell activation and eotaxin production in mast cell-fibroblast interactions. J. Immunol. 160, 6166-6171 [PubMed] [Google Scholar]
- Hundley T. R., Gilfillan A. M., Tkaczyk C., Andrade M. V., Metcalfe D. D., Beaven M. A. (2004). Kit and FcepsilonRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood 104, 2410-2417 [DOI] [PubMed] [Google Scholar]
- Ishizuka T., Okajima F., Ishiwara M., Iizuka K., Ichimonji I., Kawata T., Tsukagoshi H., Dobashi K., Nakazawa T., Mori M. (2001). Sensitized mast cells migrate toward the antigen: a response regulated by p38 mitogen-activated protein kinase and Rho-associated coiled-coil-forming protein kinase. J. Immunol. 167, 2298-2304 [DOI] [PubMed] [Google Scholar]
- Iwaki S., Tkaczyk C., Satterthwaite A. B., Halcomb K., Beaven M. A., Metcalfe D. D., Gilfillan A. M. (2005). Btk plays a crucial role in the amplification of Fc epsilonRI-mediated mast cell activation by kit. J. Biol. Chem. 280, 40261-40270 [DOI] [PubMed] [Google Scholar]
- Jung I. D., Lee H. S., Lee H. Y., Choi O. H. (2009). FcεRI-mediated mast cell migration: signaling pathways and dependence on cytosolic free Ca2+ concentration. Cell Signal 21, 1698-1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. S., Kuehn H. S., Metcalfe D. D., Gilfillan A. M. (2008). Activation and function of the mTORC1 pathway in mast cells. J. Immunol. 180, 4586-4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshenbaum A. S., Metcalfe D. D. (2006). Growth of human mast cells from bone marrow and peripheral blood-derived CD34+ pluripotent progenitor cells. Methods Mol. Biol. 315, 105-112 [DOI] [PubMed] [Google Scholar]
- Kitaura J., Kinoshita T., Matsumoto M., Chung S., Kawakami Y., Leitges M., Wu D., Lowell C. A., Kawakami T. (2005). IgE- and IgE+Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood 105, 3222-3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn H. S., Gilfillan A. M. (2007). G protein-coupled receptors and the modification of FcvarepsilonRI-mediated mast cell activation. Immunol. Lett. 113, 59-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn H. S., Beaven M. A., Ma H. T., Kim M. S., Metcalfe D. D., Gilfillan A. M. (2008a). Synergistic activation of phospholipases Cγ and Cβ: a novel mechanism for PI3K-independent enhancement of FcεRI-induced mast cell mediator release. Cell. Signal. 20, 625-636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn H. S., Swindle E. J., Kim M. S., Beaven M. A., Metcalfe D. D., Gilfillan A. M. (2008b). The phosphoinositide 3-kinase-dependent activation of Btk is required for optimal eicosanoid production and generation of reactive oxygen species in antigen-stimulated mast cells. J. Immunol. 181, 7706-7712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H. T., Beaven M. A. (2009). Regulation of Ca2+ signaling with particular focus on mast cells. Crit. Rev. Immunol. 29, 155-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean-Fletcher S., Pollard T. D. (1980). Mechanism of action of cytochalasin B on actin. Cell 20, 329-341 [DOI] [PubMed] [Google Scholar]
- Madden K. B., Urban J. F., Jr, Ziltener H. J., Schrader J. W., Finkelman F. D., Katona I. M. (1991). Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J. Immunol. 147, 1387-1391 [PubMed] [Google Scholar]
- Marshall J. S. (2004). Mast-cell responses to pathogens. Nat. Rev. Immunol. 4, 787-799 [DOI] [PubMed] [Google Scholar]
- Meininger C. J., Yano H., Rottapel R., Bernstein A., Zsebo K. M., Zetter B. R. (1992). The c-kit receptor ligand functions as a mast cell chemoattractant. Blood 79, 958-963 [PubMed] [Google Scholar]
- Mekori Y. A. (2004). The mastocyte: the “other” inflammatory cell in immunopathogenesis. J. Allergy Clin. Immunol. 114, 52-57 [DOI] [PubMed] [Google Scholar]
- Metcalfe D. D., Baram D., Mekori Y. A. (1997). Mast cells. Physiol. Rev. 77, 1033-1079 [DOI] [PubMed] [Google Scholar]
- Nilsson G., Butterfield J. H., Nilsson K., Siegbahn A. (1994). Stem cell factor is a chemotactic factor for human mast cells. J. Immunol. 153, 3717-3723 [PubMed] [Google Scholar]
- Nishida K., Yamasaki S., Ito Y., Kabu K., Hattori K., Tezuka T., Nishizumi H., Kitamura D., Goitsuka R., Geha R. S., et al. (2005). FcεRI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J. Cell Biol. 170, 115-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J. (2006). Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell. Biol. 16, 522-529 [DOI] [PubMed] [Google Scholar]
- Samayawardhena L. A., Kapur R., Craig A. W. (2007). Involvement of Fyn kinase in Kit and integrin-mediated Rac activation, cytoskeletal reorganization, and chemotaxis of mast cells. Blood 109, 3679-3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada J., Shimizu S., Tamatani T., Kanegasaki S., Saito H., Tanaka A., Kambe N., Nakahata T., Matsuda H. (2005). Stem cell factor has a suppressive activity to IgE-mediated chemotaxis of mast cells. J. Immunol. 174, 3626-3632 [DOI] [PubMed] [Google Scholar]
- Shimizu T., Owsianik G., Freichel M., Flockerzi V., Nilius B., Vennekens R. (2009). TRPM4 regulates migration of mast cells in mice. Cell Calcium 45, 226-232 [DOI] [PubMed] [Google Scholar]
- Spicuzza L., Di Maria G., Polosa R. (2006). Adenosine in the airways: implications and applications. Eur. J. Pharmacol. 533, 77-88 [DOI] [PubMed] [Google Scholar]
- Taub D., Dastych J., Inamura N., Upton J., Kelvin D., Metcalfe D., Oppenheim J. (1995). Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. J. Immunol. 154, 2393-2402 [PubMed] [Google Scholar]
- Tkaczyk C., Metcalfe D. D., Gilfillan A. M. (2002). Determination of protein phosphorylation in Fc epsilon RI-activated human mast cells by immunoblot analysis requires protein extraction under denaturing conditions. J. Immunol. Methods 268, 239-243 [DOI] [PubMed] [Google Scholar]
- Tkaczyk C., Beaven M. A., Brachman S. M., Metcalfe D. D., Gilfillan A. M. (2003). The phospholipase Cγ1-dependent pathway of FcεRI-mediated mast cell activation is regulated independently of phosphatidylinositol 3-kinase. J. Biol. Chem. 278, 48474-48484 [DOI] [PubMed] [Google Scholar]
- Vancheri C., Mastruzzo C., Sortino M. A., Crimi N. (2004). The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 25, 40-46 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Ali K., Bilancio A., Geering B., Foukas L. C. (2005). Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 30, 194-204 [DOI] [PubMed] [Google Scholar]
- Weller C. L., Collington S. J., Hartnell A., Conroy D. M., Kaise T., Barker J. E., Wilson M. S., Taylor G. W., Jose P. J., Williams T. J. (2007). Chemotactic action of prostaglandin E2 on mouse mast cells acting via the PGE2 receptor 3. Proc. Natl. Acad. Sci. USA 104, 11712-11717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H., Shlykov S. G., Molina J. G., Sanborn B. M., Jacobson M. A., Tilley S. L., Blackburn M. R. (2003). Activation of murine lung mast cells by the adenosine A3 receptor. J. Immunol. 171, 338-345 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.