

Abstract
Amyloid-β plaques (Aβ) are a hallmark of Alzheimer's disease (AD), begin deposition decades before the incipient disease, and are thought to be associated with neuronal loss, brain atrophy and cognitive impairment. We examine associations between 11C-PiB-PET measurement of Aβ burden and brain volume changes in the preceding years in 57 non-demented individuals (age 64-86; M = 78.7). Participants were prospectively followed through the Baltimore Longitudinal Study of Aging, with up to 10 consecutive MRI scans (M = 8.1) and an 11C-PiB scan approximately 10 years after the initial MRI. Linear mixed effects models were used to determine whether mean cortical 11C-PiB distribution volume ratios, estimated by fitting a reference tissue model to the measured time activity curves, were associated with longitudinal regional brain volume changes of the whole brain, ventricular CSF, frontal, temporal, parietal, and occipital white and gray matter, the hippocampus, orbito-frontal cortex, and the precuneus. Despite significant longitudinal declines in the volumes of all investigated regions (p < 0.05), no associations were detected between current Aβ burden and regional brain volume decline trajectories in the preceding years, nor did the regional volume trajectories differ between those with highest and lowest Aβ burden. Consistent with a threshold model of disease, our findings suggest that Aβ load does not seem to affect brain volume changes in individuals without dementia.
Keywords: Alzheimer's Disease, BLSA, Volumetric MRI, Normal Aging: PET, 11C-PiB
Introduction
Alzheimer's disease (AD) is characterized by neuropathological changes (Alzheimer, 1906, 1907; Braak and Braak, 1991, 1998) in combination with clinicopathological features, and represents the culmination of a process that evolves over decades (Godbolt et al., 2004; Thal and Braak, 2005; Torack, 1979; Troncoso et al., 1998). The definitive diagnosis of AD still requires post-mortem confirmation of neuropathological hallmarks – amyloid-β plaques (Aβ) and neurofibrillary tangles.
The preeminent explanation of AD pathogenesis, the ‘amyloid cascade theory’, suggests that Aβ is at the root of neurodegeneration, subsequent brain atrophy, cognitive impairment and ultimately dementia (Hardy and Selkoe, 2002.). It has become evident, through prospective longitudinal studies with autopsy data, that neuropathological changes precede neurodegeneration, atrophy, and clinical symptoms by perhaps decades (Bennett et al., 2006; Price and Morris, 1999), with clinical impairments becoming detectable much later after substantial neurodegeneration has taken place (Morris and Price, 2001).
The advent of radiotracers for amyloid imaging (Mintun, 2005; Nordberg, 2008) presents an opportunity to investigate prospective changes in amyloid deposition in vivo. The best validated of these radiotracers to date is the PET ligand known as Pittsburgh Compound B (11C-PiB; {N-methyl-11C} 2-(4′-methylaminophenyl)-6-hydroxybenzothiazole). 11C-PiB detects almost a two-fold increase in tracer retention in AD cases compared to non-demented older individuals (Klunk et al., 2004), has a topographical distribution (Rowe et al., 2007) that parallels autopsy findings (Braak and Braak, 1991), shows promise in discriminating individuals with mild cognitive impairment (MCI) who progress versus those who remain stable, and provides some utility for differential diagnosis of dementia, particularly AD from frontotemporal (Ng et al., 2007).
Brain atrophy, presumably a direct result of neurodegeneration, is a fundamental, although not a specific, feature of AD and has been extensively characterized antemortem by magnetic resonance imaging (MRI; Kantarci and Jack, 2004). Recently we reported that over a 10-year interval those with mild cognitive impairment (MCI) show accelerated changes in the volumes of the whole brain, ventricular cerebrospinal fluid, temporal gray matter, and orbito-frontal and temporal association cortices, including the hippocampus, compared to older, non-demented individuals (Driscoll et al., 2009). Accelerated change associated with MCI was detected against a background of widespread age-related regional volume loss, suggesting that cognitive impairment is associated with a unique pattern of structural vulnerability.
In the present report, we investigate whether amyloid burden, measured by 11C-PiB, is associated with rates of brain atrophy in the preceding years. Based on our structural findings (Driscoll et al., 2009), we predict that the same regions that distinguish between non-demented and those with cognitive impairment, such as frontal and temporal association cortices for example, will exhibit steeper volume declines in association with higher 11C-PiB burden.
Methods
Participants
The sample consists of 57 non-demented, highly educated, community-dwelling individuals (M (age)=78.7±6.2; age range 64-86; 90% Caucasian), who were prospectively followed through the neuroimaging (NI) sub-study of the Baltimore Longitudinal Study of Aging (BLSA; Resnick et al., 2000). They were ascertained from the initial 61 BLSA-NI participants consecutively assessed with [11C]PiB from June 2005-March 2007, after excluding two participants with clinical stroke, one with a brain injury and one unable to tolerate MRI. The 57 individuals were representative of the entire BLSA-NI with respect to baseline age, sex, race, and education. All participants were in generally good health at enrollment, with exclusionary criteria encompassing CNS disease, severe cardiovascular disease, severe pulmonary disease, or metastatic cancer. Sample characteristics are presented in Table 1. All studies were approved by the local institutional review boards, and all participants gave written informed consent prior to each assessment.
Table 1. Sample Demographics and Clinical Characteristics.
| Characteristics | |
|---|---|
| N | 57 |
| Sex (M/F) | 33/24 |
| Race (white/AA) | 49/7 |
| Education (yrs) M (SD) | 16.8 (2.5) |
| Age @ PiB (yrs) M (SD) | 78.7 (6.2) |
| Age @ Baseline (yrs) M (SD) | 67.7 (6.1) |
| # of MRI scans M (SD) | 8.5 (0.9) |
| Follow-up Interval (yrs) M (SD) | 8.1 (0.8) |
| Last MRI to PiB Lag (yrs) M (SD) | 2.9 (0.8) |
| MMSE M (SD) | 28.9 (1.5) |
| CDR (# with 0.5) | 6 |
M = males; F = females; AA = African American; M = mean; SD = standard deviation
Participants received up to 10 annual MRI scans (M=8.07±0.83). 11C-PiB images were acquired approximately 11 years after the initial MRI. Although MRI scans were also obtained concurrent with 11C-PiB imaging, the volumetric analyses are restricted to the first 10 years of available MR data due to changes in hardware, with an average interval between last MR and PiB scan of 2.9 years. The MRIs obtained concurrently with PiB-PET were used only for region of interest (ROI) definition in quantifying regional PiB retention. Participants also received routine neuropsychological and neurological exams. Interval medical history, medication review, and a structured informant and subject interview are documented at each visit (Driscoll et al., 2006; Kawas et al., 2003).
Cognitive status was determined based on standardized consensus diagnostic procedures for the BLSA (Driscoll et al, 2006), whereby MCI diagnosis requires a progressive impairment in one or more cognitive domains without functional loss in activities of daily living (Petersen and Morris, 2005). Although all participants were non-demented, six had a score of 0.5 on the Clinical Dementia Rating (CDR) Scale (Morris, 1993). The CDR Scale, typically informant-based, was administered both in conjunction with the 11C-PiB imaging and previously if participants scored 3 or greater on the Blessed-Information-Memory-Concentration test (Blessed et al., 1968).
MR Imaging
MRI Acquisition
Scanning was performed on a GE Signa 1.5 Tesla scanner (Milwaukee, WI) using a high-resolution volumetric spoiled-grass (SPGR) axial series (TR = 35 ms, TE = 5 ms, FOV = 24 cm, flip angle = 45°, matrix = 256×256, NEX = 1, voxel dimensions 0.94 × 0.94 × 1.5mm).
MRI Image Analysis
Image processing procedures have been previously described in detail and validated (Davatzikos et al., 2001; Driscoll et al., 2009; Goldszal et al., 1998; Resnick et al., 2003;). Briefly, images are corrected for head tilt and rotation, and reformatted parallel to the anterior-posterior commissure plane. Extracranial tissue is removed using a semi-automated procedure followed by manual editing. Next, images are segmented into white matter (WM), gray matter (GM), and cerebrospinal fluid (CSF). The final step involves stereotaxic normalization and tissue quantitation for specific regions of interest with a template-based deformation approach. An ICBM standard MRI (Montreal Neurologic Institute) serves as the template and a hierarchical elastic matching algorithm is used for deformation and determination of volumes of interest (Shen and Davatzikos, 2002). All images are normalized individually to the same template. Voxel-based analysis utilizes the RAVENS approach (Goldszal et al., 1998), whereby local values of tissue density maps (for GM, WM, and CSF) reflect the amount of respective tissue in the vicinity of a voxel. Tissue densities are mathematical quantities measuring local tissue volumes and do not reflect any microstructural physical density of brain tissue.
Pet Imaging
PET Acquisition
Dynamic 11C-PiB PET images were acquired on a GE Advance scanner in 3-dimensional mode with 37 time frames (90 min) obtained at rest. PET scanning immediately followed an intravenous bolus injection of a mean (SD) 14.5 (0.9) mCi [11C] PiB with a mean (SD) specific activity of 5.7 (3.1; range 0.98 - 14.62) Ci/μmol. Participants were fitted with a thermoplastic mask to minimize motion during scanning. Two-dimensional transmission scans were used for attenuation correction of the emission scans. Dynamic images were reconstructed using filtered backprojection with a ramp filter (image size, 128 × 128; pixel size, 2 × 2 mm; slice thickness, 4.25 mm) with a spatial resolution of about 4.50-mm full width at half maximum at the center of the field of view.
MRI-based ROI Definition for PiB-PET
MRI scans acquired concurrently or closest in time to the 11C-PiB PET scan were used for region of interest (ROI) definition. MR scans used for volumetric analyses were exclusively obtained on the 1.5 T GE Signa scanner, while MRI scans for ROI definition used either the SPGR volumetric scans from the 1.5 T GE Signa or a comparable T1-weighted volumetric imaging protocol on a Philips 1.5 T scanner, which replaced the GE scanner in 2006. Volumetric scans were co-registered to the mean of the first 20-min dynamic PET images for each participant using the mutual information method in the Statistical Parametric Mapping software (SPM2; Wellcome Department of Cognitive Neurology). ROIs were drawn manually on the co-registered MR images (Price et al., 2005). Cerebellum served as a reference region. The parametric images of distribution volume ratio (DVR) and R1 (=K1/K1(reference tissue), the target to reference tissue ratio of tracer transport rate constant from vascular space to tissue) were generated by using a simplified reference tissue model and linear regression with spatial constraint algorithm (Zhou et al., 2003, 2007). The ROI DVRs were obtained by applying ROIs to the DVR images. The mean cortical DVR was calculated by averaging DVR values from orbitofrontal, prefrontal, superior frontal, parietal, lateral temporal, occipital, and anterior and posterior cingulate regions (Villemagne et al., 2008). For voxelwise analysis, parametric images of DVR were spatially normalized using SPM2 with an R1 template generated in our previous study (Zhou et al., 2007).
Statistical Analyses
We employed linear mixed models, which estimated cross-sectional and longitudinal effects simultaneously to investigate the association between amyloid burden, measured by mean cortical DVR, and longitudinal changes in select regional brain volumes. The models are fit using PROC MIXED in SAS v9.1 (SAS Institute Inc.; Cary, NC). Brain volume data were longitudinal, with annual measurements from 1994 through 2003. PiB was measured once, obtained between year 2005 and 2006.
We used regional brain volumes as dependent measures. Stability measures for volumes across follow-ups indicated that measurements generally were consistent over time. Only regions with stability measures above 0.85 were included in analyses (Driscoll et al., 2009). Independent variables include ICV, baseline age (age at which first brain measurements were taken), sex, interval (years since the first brain measurements were taken) and mean DVR score. Fixed effects include all the predictors listed above and sex*interval, baseline age*interval, PiB*interval. All continuous predictors, except interval, are centered around their means, and effect coding was used for sex. This kind of parameterization allows us to look at longitudinal volumetric changes after adjusting for all other covariates; and the effect of mean DVR on these longitudinal changes at the same time. Random effects include intercept and interval, which means we allow the intercepts and rates of change to vary among individuals.
To investigate the possibility that associations with regional volume changes would not be uniform across the range of cortical DVR values (i.e., relationships may be evident in individuals with high but not low 11C-PiB), we separated participants into three equal groups (high-, medium-, and low-amyloid-binding) based upon the tertiles of the 11C-PiB mean cortical DVR values. The distribution of 11C-PiB mean cortical DVR values as a function of age and the division into tertiles is presented in Figure 1. The mixed model described in the primary analysis was repeated with PiB (high vs. low) as a class rather than a continuous variable, omitting the tertile with intermediate mean cortical DVR values.
Figure 1. Scatterplot of mean cortical PiB DVR by age in the BLSA neuroimaging sample.

Participants were also divided into equal tertiles (N=19 each) for additional analyses involving high and low PiB-binding groups. Males are denoted in blue, females in red.
Results
Mixed-model results for global and regional volumes with cortical DVR score as a continuous variable are summarized in Table 2. In this sample, volume decline accompanied by ventricular CSF increase (p=0.001) was evident for all brain regions investigated (p≤ 0.001). Main effects of PiB (mean cortical DVR) on regional brain volumes were not significant (p>0.1). We also found no interactions between mean cortical PiB burden and interval, indicating no significant effects of mean cortical DVR on trajectories of age-related volume changes in the regions of interest (p≥ 0.3). There was, however, a non-significant trend observed for the precuneus (p=0.08) with greater rates of volume loss in association with higher mean cortical DVR. Select trajectories of volume change over time for vCSF, GM, the hippocampus and the precuneus in relation to PiB burden are shown in Figure 2. No significant differences in the trajectories of volume decline for any of the brain regions investigated were observed between the high- and low-amyloid-burden groups (p's≥ 0.2; data not shown) despite evident differences in 11C-PiB retention in the two groups (Figure 3).
Table 2. Annual rates of change (cm3) in relation to current PiB retention.
Longitudinal volume decline (interval effect), accompanied by ventricular CSF increase, was evident for all brain regions investigated. Main effects of PiB mean cortical DVR on regional brain volumes were not significant. No significant effects of mean cortical PiB on trajectories of age-related volume changes in the regions of interest were observed.
| Brain Volumes | Interval | PiB* | PiB × Interval |
|---|---|---|---|
| Whole Brain | -7.54 (0.3)* | -18.13 (22.5) | 0.29 (1.1) |
| vCSF | 1.13 (0.1)* | -5.62 (6.0) | -0.13 (0.29) |
| GM - Total | -2.64 (0.25)* | -8.77 (13.75) | -0.56 (1.01) |
| - Frontal | -0.86 (0.09)* | -1.27 (4.5) | 0.01 (0.36) |
| - Temporal | -0.33 (0.08)* | -2.94 (3.83) | -0.24 (0.33) |
| - Parietal | -0.72 (0.05) | -4.19 (3.33) | 0.02 (0.22) |
| - Occipital | -0.38 (0.05)* | 0.52 (2.86) | 0.08 (0.18) |
| WM - Total | -4.81 (0.24) | -2.17 (15.38) | 0.59 (0.99) |
| - Frontal | -2.07 (0.1)* | -1.26 (6.54) | 0.23 (0.41) |
| - Temporal | -1.73 (0.08)* | -0.11 (4.17) | 0.27 (0.31) |
| - Parietal | -0.39 (0.07)* | -3.93 (3.99) | 0.06 (0.27) |
| - Occipital | -0.21 (0.06)* | 0.89 (2.31) | -0.13 (0.23) |
| Hippocampus | -0.02 (0.01)* | -0.21 (0.28) | -0.01 (0.02) |
| OFC | -0.07 (0.02)* | -1.01 (1.01) | -0.02 (0.07) |
| Cingulate Gyrus | -0.2 (0.02)* | 0.03 (1.25) | -0.07 (0.09) |
| Precuneus | -0.04 (0.01)* | 0.34 (0.41) | -0.04 (0.02) |
Results are expressed as β-coefficients (SE).
PiB = mean cortical DVR; OFC = Orbito-frontal Cortex
PiB mean cortical DVR is an average of orbito-frontal, prefrontal, superior frontal, parietal, temporal, occipital, and anterior and posterior cingulated regions.
p < 0.05
Figure 2. Predicted longitudinal trajectories by tertiles of mean PiB DVR for select brain volume changes (cm3).
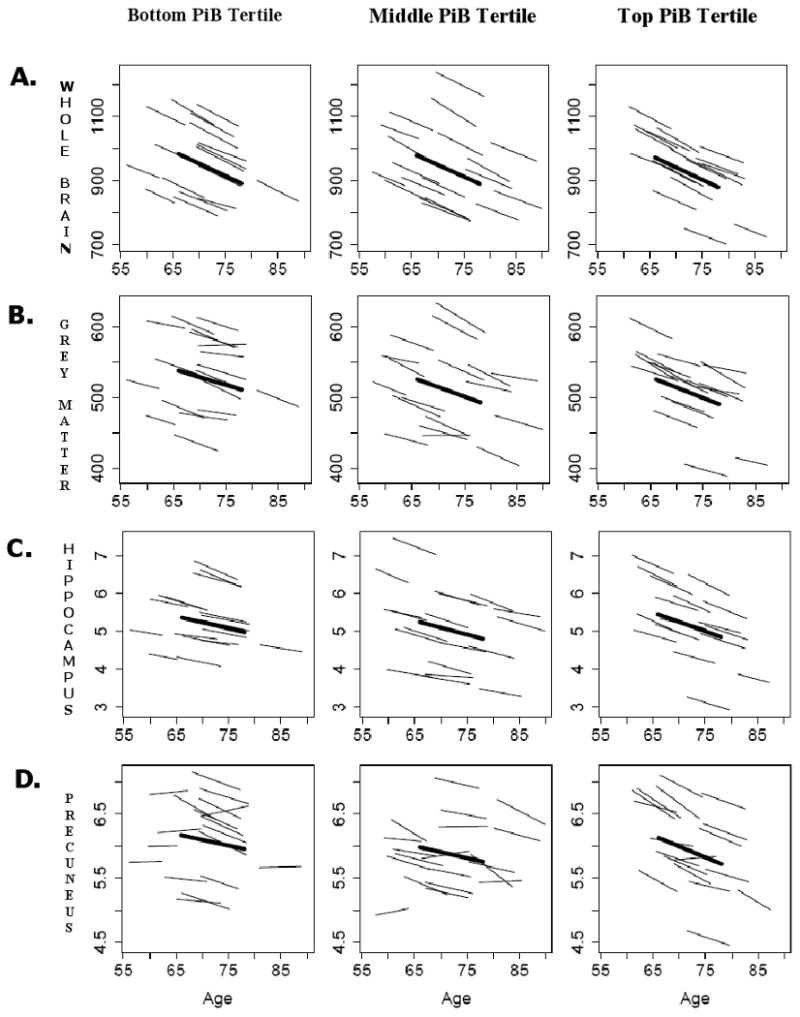
Age-related changes (y-axis) in (A) whole brain volume, (B) total grey matter, (C) the hippocampus, and (D) the precuneus were not related to mean cortical PiB retention. Thin lines represent individual volumes over time for a random sample of participants; bold lines represent mean trajectories for each tertile.
Figure 3. Group differences in cortical PiB DVR between High- and Low-amyloid retention groups; all non-demented.
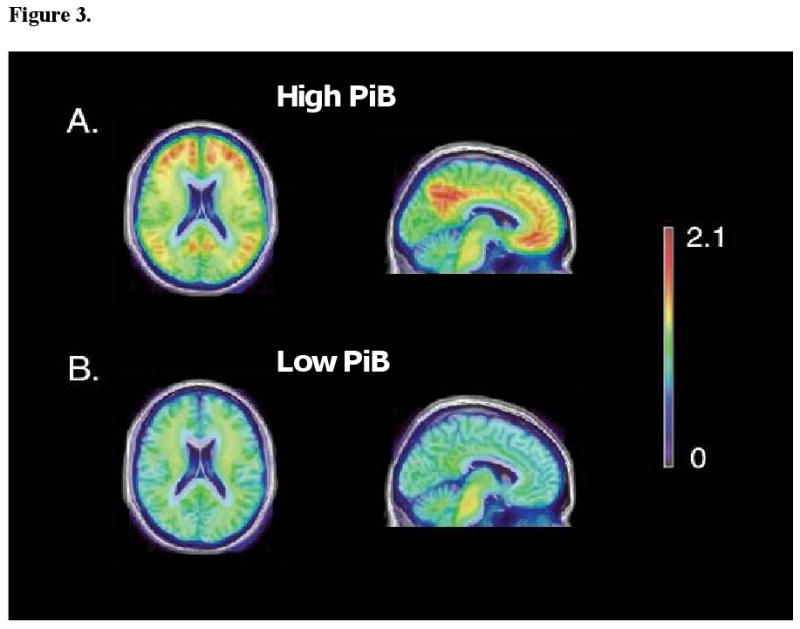
The mean of spatially normalized parametric PiB images (Zhou et al., 2007) for the 19 individuals in the upper tertile and 19 individuals in the lower tertile determined using SPM5 (Wellcome Department of Cognitive Neurology, London, England) were overlaid on an MRI template using Amide software (Loening and Gambhir, 2003). Being in the high-amyloid group or having a mean cortical DVR score in the upper tertile does not imply that an individual has an AD-like PiB scan. The scans of participants in the present study are easily distinguishable from those in other studies examining AD patients, despite the qualitative differences between the High- and Low-amyloid groups in this non-demented sample.
Discussion
The main goal of the present study was to examine the relationships between amyloid burden measured with 11C-PiB and trajectories of regional brain volume changes. Consistent with our findings of structural brain changes in the complete BLSA neuroimaging sample (Driscoll et al., 2009), we found significant age-related regional brain atrophy in this sub-sample of non-demented, community-dwelling older individuals who underwent PiB-PET imaging and were prospectively followed and extensively characterized through the BLSA. We found no significant associations, however, between cortical amyloid load and regional brain volume changes preceding the 11C-PiB measurement.
We considered the possibility that PiB associations with regional volume changes may not be uniform across the range of cortical DVR values and may not be evident in individuals with low 11C-PiB values. We compared and found no differences in trajectories of the investigated brain regions between high- and low-amyloid-retention groups, despite the qualitative group differences in mean PiB retention (Figure 3). It is important to note that the designation of high amyloid binding or having a mean cortical DVR score in the upper tertile in this study does not imply that an individual has an AD-like PiB-PET scan. Although many in the upper tertile would qualify as PiB positive controls, the scans of participants in the present study could be distinguished quantitatively and qualitatively from those of AD patients (Klunk et al., 2004; Ng et al., 2007).
There are several possible interpretations of the absence of differences in the rates of regional brain atrophy preceding the in vivo measurement of amyloid burden or between the groups with high and low PiB retention in this non-demented sample. One interpretation is that age-related structural brain volume changes, specifically declines in regional brain volumes, are independent of amyloid deposition. Another interpretation may be that there is a threshold beyond which AD neuropathology causes negative functional outcomes. We cannot rule out a threshold interpretation whereby a combined liability including neuropathology, vascular, and other risk factors move an individual toward a threshold beyond which cognitive impairment is evident.
Our observations are in agreement with the two existing reports on PiB and brain volume (Jack et al., 2008, 2009). Jack and colleagues (2008) originally reported little correspondence between PiB retention and brain volume loss. Moreover, PiB and MRI seemed to provide complementary information such that clinical diagnostic classification using both methods was superior to using either in isolation. In a more recent study, Jack and colleagues (2009) investigated MRI and PIB studies obtained at two time points, approximately one year apart, to gain insight into the sequence of pathologic events in AD. They reported a dissociation between the rate of amyloid deposition and the rate of neurodegeneration late in life over one year follow-up, with amyloid deposition proceeding at a constant slow rate irrespective of clinical status while neurodegeneration accelerates in association with clinical symptoms.
Seminal work by Fagan and colleagues (2006, 2009), however, reports that CSF Aβ42 does correlate with whole brain volume in non-demented individuals and is suggestive of a threshold hypothesis. CSF Aβ42 levels less than 500pg/ml were a good surrogate marker for the presence of amyloid in the brain; those with positive PiB binding had the lowest CSF Aβ42, while those with negative PIB binding had the CSF Aβ42 levels above 500pg/ml threshold. Furthermore, non-demented individuals with CSF Aβ42 levels below 500pg/ml had significantly smaller brain volumes compared to those with CSF Aβ42 levels above the threshold. The mean hippocampal volume was also smaller in non-demented individuals with low CSF Aβ42 compared with those with CSF Aβ42 values above 500pg/ml. The relationships do not hold up in those who have already succumbed to age-related impairment or dementia, presumably because by the time individuals become cognitively impaired cortical amyloid deposition may have already reached its maximum levels (Ingelsson et al., 2004; Price and Morris, 1999). Together, these observations suggest that alterations in CSF Aβ42 metabolism seem to be associated with structural change before the ability to detect any cognitive impairment. Although we do not have the CSF measurements in our sample, we would hypothesize that, given the lack of associations between PiB and brain volume changes over 10 years prior, the majority of our participants would have CSF Aβ42 values above 500pg/ml. Given the 3 year interval between last MRI assessment and PiB in our study, we are less likely to capture brain volume changes closer in temporal proximity to cognitive impairment. Our findings coupled with those of Fagan et al. (2009) also suggest that majority of cross-sectional studies or those with shorter follow-ups may not be as effective at screening out individuals who will eventually go on to become impaired. Conversely, our prospective design with a long follow-up over regular intervals may have afforded us an opportunity to better screen our sample.
Our study is not without limitations. Our sample is not population-based; most participants are highly educated and white, limiting the generalizability of our findings. However, the incidence of AD in the BLSA (Kawas et al., 2000) and the rates of brain changes (Resnick, et al., 2003) are comparable to other samples. Also, our PiB measurements in these analyses are cross-sectional and do not yet provide information on the trajectory of amyloid deposition. These potential limitations should not overshadow the unique aspects of our study, including a large number of individuals without dementia who are prospectively followed for many years, relatively short intervals between assessments, up to 10 serial MRI evaluations, and the extensive clinical, neuropsychological, and pathological characterizations of this sample. The relative homogeneity of the sample may also be seen as an asset because it diminishes possible confounding effects of race and education and the majority of our sample has good access to medical care and has remained relatively healthy over the follow-up interval. Long MRI follow-up allows us the advantage over other studies to detect even small differences in brain volume trajectories across different levels of PIB retention.
Our study also has several strengths. Neuroimaging participants were followed prospectively for an average of 8 years prior to initial [11C]PiB evaluation, and cognitive status was determined within the context of these longitudinal assessments. While we do not yet know which individuals will eventually develop cognitive impairment, our analyses represent a snapshot in time and the fact that most individuals have had stable cognition over ≥8 years increases the likelihood that the majority of participants will remain clinically normal through the next few years of follow-up.
Overall, our findings suggest that in non-demented, elderly individuals, amyloid accumulation does not affect the rate of brain atrophy beyond that already observed as a part of the normal aging process. A large number of repeat MRI assessments ensured sufficient power to detect even small changes in rates of regional brain tissue loss. Concurrent evaluations of longitudinal brain atrophy and amyloid deposition in relation to cognitive status are ongoing and are needed to more fully investigate the interplay between amyloid deposition and the structural integrity of the brain, as well as their functional and clinical significance. Both larger numbers of participants and follow-up visits per participant over longer intervals will be crucial in determining the sensitivity of in vivo PiB imaging to structural and functional changes in normal and pathological aging with greater confidence. In addition, the strength of amyloid imaging may lie in longitudinal studies that track the progression of amyloid deposition. Further development, refinement, and application of different physiologic and molecular probes, in concert with longitudinal and perhaps more focused and refined clinical observations, will bring us closer to understanding the pathophysiology of MCI and AD and may aid in determining which individuals at risk for AD are most likely to progress to disease and who will remain healthy.
Acknowledgments
We thank the staff of the PET facility at Johns Hopkins University and the neuroimaging staff of the BLSA project National Institute on Aging (NIA) for their assistance. Dr. Driscoll had full access to the data in the study and takes responsibility for the integrity of the data and the accuracy of the analysis. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (NIA), and N01-AG-3-2124 and K24 DA00412 (DFW). A portion of that support was through a R&D contract with MedStar Research Institute. Dr. Klunk and Mathis's contributions were supported by NIA grants P01-AG025204, R37 AG025516 and P50-AG005133.
Footnotes
Disclosure: GE Healthcare holds a license agreement with the University of Pittsburgh based on the PIB technology described in this manuscript. Drs. Klunk and Mathis are co-inventors of PIB and, as such, have a financial interest in this license agreement.
Disclosure Statement
Ira Driscoll - no actual or potential conflicts of interest.
Yun Zhou - no actual or potential conflicts of interest.
Yang An - no actual or potential conflicts of interest.
Jitka Sojkova - no actual or potential conflicts of interest.
Christos Davatzikos - no actual or potential conflicts of interest.
Michael A Kraut - no actual or potential conflicts of interest.
Weiguo Ye - no actual or potential conflicts of interest.
Luigi Ferrucci - no actual or potential conflicts of interest.
Chester A Mathis - GE Healthcare holds a license agreement with the University of Pittsburgh based on the PiB technology described in this manuscript. Dr. Mathis is a co-inventor of PiB and, as such, has a financial interest in this license agreement.
William E Klunk - GE Healthcare holds a license agreement with the University of Pittsburgh based on the PiB technology described in this manuscript. Dr. Klunk is a co-inventor of PiB and, as such, has a financial interest in this license agreement.
Dean F Wong - no actual or potential conflicts of interest.
Susan M Resnick – no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alzheimer A. Über einen eigenartigen schweren Krankheitsprozess der Hirnrinde. Neurologisches Centralblatt. 1906;25:1134. [Google Scholar]
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und psychisch-gerchtliche Medizin. 1907;64:146–148. [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer's disease. J Neural Transm Suppl. 1998;53:127–140. doi: 10.1007/978-3-7091-6467-9_11. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Genc A, Xu D, Resnick SM. Voxel-based morphometry using the RAVENS maps: methods and validation using simulated longitudinal atrophy. Neuroimage. 2001;14:1361–1369. doi: 10.1006/nimg.2001.0937. [DOI] [PubMed] [Google Scholar]
- Driscoll I, Resnick SM, Troncoso JC, An Y, O'Brien R, Zonderman AB. Impact of Alzheimer's pathology on cognitive trajectories in nondemented elderly. Ann Neurol. 2006;60:688–695. doi: 10.1002/ana.21031. [DOI] [PubMed] [Google Scholar]
- Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut MA, Resnick SM. Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology. 2009;72:1906–1913. doi: 10.1212/WNL.0b013e3181a82634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Godbolt AK, Cipolotti L, Watt H, Fox NC, Janssen JC, Rossor MN. The natural history of Alzheimer disease: a longitudinal presymptomatic and symptomatic study of a familial cohort. Arch Neurol. 2004;61:1743–1748. doi: 10.1001/archneur.61.11.1743. [DOI] [PubMed] [Google Scholar]
- Goldszal AF, Davatzikos C, Pham DL, Yan MX, Bryan RN, Resnick SM. An image-processing system for qualitative and quantitative volumetric analysis of brain images. J Comput Assist Tomogr. 1998;22:827–837. doi: 10.1097/00004728-199809000-00030. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. Review. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Alzheimer's Disease Neuroimaging Initiative. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Jack CR., Jr Quantitative magnetic resonance techniques as surrogate markers of Alzheimer's disease. NeuroRx. 2004;1:196–205. doi: 10.1602/neurorx.1.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas CH, Corrada MM, Brookmeyer R, Morrison A, Resnick SM, Zonderman AB, Arenberg D. Visual memory predicts Alzheimer's disease more than a decade before diagnosis. Neurology. 2003;60:1089–1093. doi: 10.1212/01.wnl.0000055813.36504.bf. [DOI] [PubMed] [Google Scholar]
- Kawas CH, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 2000;54:2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Mintun MA. Utilizing advanced imaging and surrogate markers across the spectrum of Alzheimer's disease. CNS Spectr. 2005;10:13–16. doi: 10.1017/s1092852900014188. [DOI] [PubMed] [Google Scholar]
- Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imag. 2003;2:131–137. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer's disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. Review. [DOI] [PubMed] [Google Scholar]
- Ng SY, Villemagne VL, Masters CL, Rowe CC. Evaluating atypical dementia syndromes using positron emission tomography with carbon 11 labeled Pittsburgh Compound B. Arch Neurol. 2007;64:1140–1144. doi: 10.1001/archneur.64.8.1140. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Amyloid plaque imaging in vivo: current achievement and future prospects. Eur J Nucl Med Mol Imaging. 2008;35:S46–50. doi: 10.1007/s00259-007-0700-2. Review. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Morris JC. Mild cognitive impairment as a clinical entity and treatment target. Arch Neurol. 2005;62:1160–1163. doi: 10.1001/archneur.62.7.1160. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Goldszal AF, Davatzikos C, Golski S, Kraut MA, Metter EJ, Bryan RN, Zonderman AB. One-year age changes in MRI brain volumes in older adults. Cereb Cortex. 2000;10:464–472. doi: 10.1093/cercor/10.5.464. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–3301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O'Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL. Villemagne. V.L., 2007. Imaging beta-amyloid burden in aging and dementia. Neurology. 68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Shen D, Davatzikos C. IEEE Trans Med Imaging. Vol. 21. 2002. HAMMER: hierarchical attribute matching mechanism for elastic registration; pp. 1421–1439. [DOI] [PubMed] [Google Scholar]
- Thal DR, Braak H. Post-mortem diagnosis of Alzheimer's disease. Pathologe. 2005;26:201–213. doi: 10.1007/s00292-004-0695-4. [DOI] [PubMed] [Google Scholar]
- Torack RM. Adult dementia: history, biopsy, pathology. Neurosurgery. 1979;4:434–442. doi: 10.1227/00006123-197905000-00011. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Cataldo AM, Nixon RA, Barnett JL, Lee MK, Checler F, Fowler DR, Smialek JE, Crain B, Martin LJ, Kawas CH. Neuropathology of preclinical and clinical late-onset Alzheimer's disease. Ann Neurol. 1998;43:673–676. doi: 10.1002/ana.410430519. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Darby D, Maruff P, Savage G, Ng S, Ackermann U, Cowie TF, Currie J, Chan SG, Jones G, Tochon-Danguy H, O'Keefe G, Masters CL, Rowe CC. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer's disease. Neuropsychologia. 2008;46:1688–1697. doi: 10.1016/j.neuropsychologia.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Endres CJ, Brasic JR, Huang SC, Wong DF. Linear regression with spatial constraint to generate parametric images of ligand-receptor dynamic PET studies with a simplified reference tissue model. Neuroimage. 2003;18:975–989. doi: 10.1016/s1053-8119(03)00017-x. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Resnick SM, Ye W, Fan H, Holt DP, Klunk WE, Mathis CA, Dannals R, Wong DF. Using a reference tissue model with spatial constraint to quantify [11C]Pittsburgh compound B PET for early diagnosis of Alzheimer's disease. Neuroimage. 2007;36:298–312. doi: 10.1016/j.neuroimage.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]