

Abstract
TTN-1, a predicted titin-like protein in C. elegans, is encoded by a single gene, and consists of multiple Ig and Fn3 domains, a protein kinase domain and several regions containing tandem short repeat sequences. We have characterized TTN-1’s sarcomere distribution, protein interaction with key myofibrillar proteins as well as the conformation malleability of representative motifs of five classes of short repeats. We report that two antibodies developed to portions of TTN-1 detect a ~2 MDa polypeptide on western blots. In addition, by immunofluorescence staining, both of these antibodies localize to the I-band and may extend into the outer edge of the A-band in the obliquely striated muscle of the nematode. Six different 300 residue segments of TTN-1 were shown to variously interact with actin and/or myosin in vitro. Conformations of synthetic peptides of representative copies of each of the five classes of repeats: 39-mer PEVT, 51-mer CEEEI, 42-mer AAPLE, 32-mer BLUE and 30-mer DispRep were investigated by circular dichroism at different temperatures, ionic strengths and solvent polarities. The PEVT, CEEEI, DispRep and AAPLE peptides displayed a combination of a polyproline II helix and an unordered structure in aqueous solution and converts in trifluoroethanol to α-helix (PEVT, DispRep) and β-turn (AAPLE) structures, respectively. The octads in BLUE motifs form unstable α-helix-unstructured coil in aqueous solution and negligible heptad-based coiled-coils, as predicted based on sequence analysis. The α-helical structure, as modeled by threading and molecular dynamics simulations, tends to form helical bundles and crosses based on its 8-4-2-2 hydrophobic helical patterns and charge arrays on its surface. Our finding indicates that APPLE, PEVT, DispRep regions are all intrinsically disordered and highly reminiscent of the conformational malleability and elasticity of vertebrate titin PEVK segments. The presence of long, modular and unstable α-helical oligomerization domains in the BLUE region of TTN-1 could oligomerize and bundle TTN-1 and stabilize oblique striation of the sarcomere.
Keywords: C. elegans titin, polyproline II helix, staggered helical bundle, Circular dichroism, force sensor, passive tension, intrinsically disordered proteins
Introduction
In vertebrate striated muscle, titin functions both in myofibril assembly and in providing passive tension for muscle. Single titin polypeptides are 3–4 MDa, are 1.2 μm long at rest, and span half of a sarcomere, with N-termini at the Z-disk and C-termini at the M-line (recently reviewed1). The complete sequence of one human titin isoform contains approximately 166 copies of Ig and 132 copies of Fn3 domains, a single protein kinase domain, and a “PEVK region.” The Ig and Fn3 domains are arranged into different patterns or super-repeats, in different regions of the sarcomere. The A-band portion of titin is tightly associated with the shaft of the thick filament, and specific regions of titin interact with myosin, thick filament accessory proteins and M-line proteins.2 Differential splicing of the titin gene results in multiple isoforms varying from 700–3700 kDa.3 Most of this variation is in the I-band portion created by varying numbers of tandem Ig domains and the length of the PEVK domain. Most of the passive tension of muscle arises from the reversible extension of the I-band portion of titin. Both poly-Ig and PEVK regions are considered distinct spring elements. For skeletal muscle titins, the poly-Ig region straightens at modest sarcomere stretch (without unfolding of Ig domains), and the PEVK region extends at higher physiological stretch. In cardiac titins, there is a third spring element formed by the “N2B unique sequence” which extends together with the PEVK region at higher physiological stretch.
In addition to titin’s structural and elastic functions, there is increasing evidence that titin is involved in several signaling pathways. At least three regions of titin form complexes with other proteins that are implicated in signaling. In titin’s Z-line region, repeats Z1-Z2 interacts with T-cap/telethonin,4 which itself interacts with a potassium channel subunit,5 myostatin (a muscle growth factor),6 and the muscle LIM protein (MLP).7 Z-line repeat Z4, and the 700 kDa alternative titin isoform “novex-3 titin” (located in the I-band near the Z-line) interact with obscurin, a ~700 kDa protein that is involved in regulating Rho-like GTPases. Titin’s M-line region interacts with the zinc RING finger protein MURF-1 that may have a role in regulating gene expression in the nucleus.3 Titin’s PEVK region contains abundant tandem repeats of SH3 binding motifs/sites and is thought to be a stress sensitive scaffolding adaptor for SH3 containing signaling proteins.8
Autosomal dominant mutations in human titin result in various forms of cardiomyopathy or muscular dystrophy: some cases of dilated cardiomyopathy, tibial muscular dystrophy; a late-onset, distal myopathy of skeletal muscle without cardiac involvement, or hereditary myopathy with early respiratory failure (HMERF).9 The involvement of titin in muscular dystrophies goes beyond mutations in titin itself; mutations in several proteins that interact with titin also cause other forms of muscular dystrophy. These include the muscle specific protease calpain-3, myotilin, and Tcap/telethionin.10
The striated muscle of the model genetic organism, C. elegans, contains three titin-like giant proteins: twitchin (754 kDa) located in the polar regions of the A-band11–13, UNC-89 (multiple isoforms up to 900 kDa) in the M-line14,15, and TTN-1 (previously called Ce titin).16 A single gene, ttn-1, was predicted to encode 3 titin-related polypeptides with masses of 2.2, 1.2 and 0.3 MDa, designated as TTN–1 (2.2MDa), TTN-1 (1.2MDa), TTN-1 (0.3MDa).16 As all work for the current study was based on the longest isoform TTN-1 (2.2 MDa), the remainder of this description will refer to this isoform of C. elegans titin as simply “TTN-1.” TTN-1 resembles twitchin and UNC-89 in that it contains multiple Ig (56 total) and Fn3 (11 total) domains, and a single protein kinase domain (Figure 1). In addition, TTN-1 contains 5 classes of short, 14–51 residue, repeat motifs arranged mostly as tandem copies: 39-residue repeats in the PEVT/K region, similar in amino acid composition to PPAK repeats of PEVK region of vertebrate titin; 51-residue “CEEEI” repeats which interrupt the PEVT/K repeats, similar to the E rich repeats that interrupt the PEVK region PPAK repeats; 14-residue repeats of the AAPLE region; 16-residue repeats that make up an approximately 1500 residue region predicted to form coiled-coil structure; and a 30 residue repeat present in fifteen dispersed copies that punctuates other predicted coiled-coil regions. The TTN-1 protein kinase domain has in vitro protein kinase activity towards a peptide derived from vertebrate myosin light chains.16 Single-molecule force spectroscopy experiments suggest that TTN-1 kinase may function as a force sensor.17 The kinase domain of TTN-1 is most similar to the kinase domains of twitchin (54% identical) and vertebrate MLCK (51–53% identical), and least similar to vertebrate titin kinase (39% identical). Thus, nematode TTN-1 can be viewed as a “hybrid” between invertebrate twitchin due to its homologous kinase domain, and vertebrate titin due to its multiple tandem repeat regions which are in several ways similar to PEVK, the main elastic region of titin.
Fig. 1. The domain organization, predicted disorder regions and sites for protein expression, antibody production and peptide synthesis in TTN-1.

Domain organization: Purple boxes represent Ig domains, green boxes represent Fn3 domains, yellow indicates the 2400 residue PVET/K region, the blue box represents the 1500 residue BLUE region predicted to form a coiled-coil, gray denotes the 250 residue region composed of AAPLE repeats, orange represents additional predicted coiled-coil sequences, and the thin blue boxes with projecting lines denote the 30 residue dispersed repeat (DispRep). Also, note the serine/threonine protein kinase domain near the C-terminus. Above the schematic is a bar showing the possible location of the TTN-1 domains in the scaromere. Expressed proteins and peptides: Black boxes denote the position and extent of ~300 residue regions that were expressed in E. coli (5/6, 1/2, 3/4, 9/10, 11/12 and 7/8), and peptides that were synthesized (AAPLE42, CEEEI, PEVT39, BLUE32, DispRep). Four polyclonal antibodies (EU145, EU102, 9/10 and EU143) were generated to the indicated regions. Immediately below the schematic are indicated whether or not (+ or −) a given region was able to bind to F-actin or myosin in vitro. The graph is the PONDR VLXT disorder probability profile for TTN-1.22 Disorder probability above 0.5 implies disorder.
In the present study, we have characterized TTN-1 further to reveal that indeed a 2.2 MDa protein can be detected on western blot and that this protein resides almost exclusively in I-bands. In addition, 6 different segments, each about 300 residues in length, were shown to variously bind actin and myosin in vitro. Synthetic peptides representing one or several copies of the repeating motifs were studied by CD and this revealed conformational motifs similar to those found in the intrinsically disordered PEVK region, one of the main elastic elements of vertebrate titin. In addition, a 1500 residue BLUE region was found to consist of nearly 100 tandem octad repeats that tend to form unstable and interconvertible α-helices and distinct helical bundles and could oligomerize TTN-1 in the I-bands of these obliquely striated muscles. These results extend the known types of sequence elements that are likely to be elastic, and provide insight into the role of TTN-1 in the elasticity, protein/protein interactions and myofilament off-set in the organization of the obliquely striated muscle of the nematode.
Results
A 2.2 MDa TTN-1
At a predicted 2.2 MDa, the longest isoform of TTN-1 lends itself well to providing several “well-spaced” epitopes for antibody production (Figure 1). These new antibodies were evaluated with western blots containing total proteins from C. elegans (Figure 2A). The N-terminal based antibody (EU145) reacted with four polypeptides 386, 117, 71 and 59 kDa. The EU102 antibody was generated to a ~350 residue sequence containing 2 Ig domains and a unique sequence just N-terminal to the PEVT region.16 As shown in Figure 2A, EU102 reacts cleanly with an approximately 460 kDa polypeptide. As the size of this protein is similar to the sequence-based size of kettin (472 kDa),18 another poly Ig domain protein of invertebrate muscle,19,20 we tested the idea that EU102 recognizes kettin. Lysates from wild type and from the kettin intragenic deletion mutant, ketn-1(ok1641), were run side by side and duplicate blots were reacted with EU102 and MH44, a monoclonal antibody previously shown to react with kettin.18 As shown in Figure 2B, both antibodies react with a ~460 kDa protein from wild type and a slightly truncated protein from the kettin mutant. Therefore, we conclude that although EU102 was generated to TTN-1 sequence, it actually recognizes kettin on immunoblots. The 9/10 antibody was made to a TTN-1 fragment derived from sequence just N-terminal of the kinase domain. As seen in Figure 2a, 9/10 reacts with a ~2 MDa polypeptide, near the T2 of rabbit skeletal muscle titin and above nebulin at 700 kDa (as detected by monoclonal antibodies RT11 and NB2) (Figure 2), as well as a smaller band near 87 kDa. The EU143 antibody was made to a fragment containing the C-terminal end of the longest isoform of TTN-1. This antibody recognizes a polypeptide of ~2 MDa (Figure 2A), the size expected from our sequence analysis for full-length TTN-1.
Fig. 2. Identification of a 2.2 MDa TTN-1 by immunoblot and site-specific antibodies.

(A) TTN-1 specific antibodies: Total proteins from a mixed-stage population of C. elegans (Ce), or rabbit myofibril proteins (Rb) were separated on a 2–8% gradient gel and transferred to PVDF membrane. Individual strips were incubated with either antibodies to vertebrate titin (3.2 and 2.2. MDa) or nebulin (700 kDa) or a mixture of antibodies to nematode myosin heavy chain (MHC, 200 kDa), paramyosin (90 kDa) or actin (43 kDa) to serve as size markers. Of the four antibodies produced to different regions of TTN-1(EU145, EU102, 9/10 and EU143), only 9/10 and EU143 detect a polypeptide from worm extracts running at approx. 2 MDa, the size predicted for TTN-1 from previous sequence analysis.16
(B) Kettin-specific antibody: Total proteins from either wild type (wt) or a strain carrying an intragenic deletion in the kettin gene (ok1641) were separated on a 5% gel, transferred to membrane and reacted with either EU102 or MH44, an monoclonal shown previously to recognize kettin.18 As shown, both antibodies recognize a protein of approx. 500 kDa, the size of kettin, from wild type and detect a slightly smaller protein in ok1641. Thus, although EU102 was generated to TTN-1 sequence, it clearly detects a different protein, kettin and displayed no reactivity with TTN-1 on immunoblot.
Several conclusions can be made: (1) Reaction to a ~2 MDa polypeptide on western blot using antibodies generated to two different regions, verifies the existence of a 2.2 MDa TTN-1 protein predicted from sequence analysis and molecular biology experiments. (2) Although the immunogen for EU102 was part of TTN-1, the resulting antibodies recognize only the related Ig domain protein called kettin. (3) The EU145 antibody, generated to the predicted N-terminus of TTN-1, does not recognize full-length predicted TTN-1, but instead, reacts to smaller proteins of either arising from the ttn-1 gene or perhaps other genes. Two of these proteins may represent splice isoforms that contain this N-terminus predicted on WormBase. The 71 kDa band may represent either the predicted W06H8.8a protein (724 aa) or the predicted W06H8.8b protein (703 aa). The 386 kDa band may represent the predicted protein W06H8.8c (3215 aa) (note: W06H8.8 is the cosmid-based name for TTN-1 on WormBase).
Immunolocalization of TTN-1 to I-bands
In contrast to vertebrate cross-striated skeletal and cardiac muscles in which myofibrils fill the entire cell, in C. elegans body wall muscle cells the myofilament lattice is obliquely striated and the lattice resides entirely in a narrow 1.5 to 2 μm zone adjacent to the muscle cell membrane (Fig. 3N).21 All of the dense bodies (the worm analogs of Z-discs) and M-lines attach to the muscle cell membrane. The same two antibodies, 9/10 and EU143 (developed to epitopes near the C-terminus), that detected ~2 MDa TTN-1 polypeptides on western blot, were used to localize TTN-1 in muscle by immunofluorescence microscopy (Fig. 3). As markers, we co-stained with antibodies to myosin heavy chain B (MHC B) and twitchin for A-bands, and α-actinin for dense bodies. As shown in Fig. 3, both EU143 and 9/10 antibodies broadly labeled the I-band, both between dense bodies, and outside of dense bodies. EU143 and 9/10 also stained the outer edges of the A-band (Fig. 3, panels A–C, G–I, and J–L), as shown by partial co-localization of part of EU143 and 9/10 with either MHC B or twitchin (this can been seen as the thin yellow stripes of co-localization in panels C, I and L). We conclude that the C-terminal TTN-1 epitopes reside throughout the I-band, except for dense bodies, and extend to the outer edge of the A-band. This localization is clearly different from the dense body localization of the predicted N-terminal 96 residues fused to GFP.16 This distribution is summarized in the schematic in Fig. 3M.
Fig. 3. Immunofluorescence localization of TTN-1 to the I-band region of the sarcomere.

Each panel of three (A–C, D–F etc.) confocal images shows results of co-staining adult body wall muscle with the indicated pairs of antibodies. (A–C) Anti-TTN-1 antibodies EU143 localize both between dense bodies and outside of dense bodies in the I-band. Both myosin heavy chain B (MHC B) and twitchin had been shown previously to localize to the A-band except for the middle of the A-band. Note that co-staining of antibodies generated to the C-terminus of TTN-1 (EU143) with either MHC B (panels A–C) or with twitchin (G–I) shows some overlap at the outer edge of the A-band (yellow in panels C and I). (J–L) Anti-TTN-1 antibodies 9/10 localize in the same region as the EU143 antibody. (M) Drawing of a portion of body wall muscle indicating the organization of the myofilament lattice in this obliquely striated muscle. The drawing is oriented such that the I-bands and A-bands have the same orientation as in the immunofluorescence images. Dense bodies are analogous to Z-disks in mammalian cross striated muscle. (N) Proposed model of TTN-1 orientation, maximal span and variable environment in the sarcomere. The TTN-1 molecules span from the dense bodies toward the A-band in an N to C orientation, with some reaching as far as the edge of the thick filaments. Such molecules are mis-registrated across the sarcomere (bottom three filaments), due to the shape of the dense body. The molecules are also offset significantly between adjacent sarcomeres due to the lateral offset of the sarcomeres in this obliquely striated muscle (top two filaments). Scale bar, 5 μm.
TTN-1 interacts with F-actin and myosin
The I-band location of TTN-1 in the sarcomere suggests the potential for interaction with thin filament proteins. Four bacterially expressed 6His tagged segments, which were used to generate various anti-TTN-1 antibodies (Fig. 1), were used in actin pelleting assays by titrating a fixed amount of F-actin with varying concentrations of each fragment (fragment 7/8 is shown in Fig. 4A). The binding curves of all four fragments demonstrate saturable binding with Kd’s in the low micromolar range. These data suggest that indeed multiple portions of TTN-1 sampled from throughout the polypeptide interact with F-actin. Additionally, synthetic peptides of each of the five classes of sequence repeats were tested for actin binding. DispRep pelleted with F-actin, whereas BLUE did not (data not shown). AAPLE and PEVT were unfortunately not detectable on the gel system that is designed for small peptides; perhaps they did not bind SDS well enough or they diffused out of the gels during the Coomassie staining procedure. The result that the DispRep interacts with actin shows that yet another region of TTN-1 interacts with actin.
Fig. 4. TTN-1 interacts with F-actin and myosin.
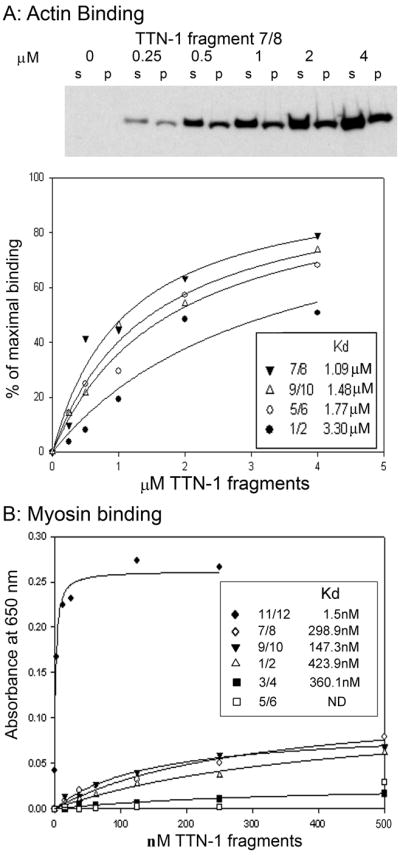
(A) Data demonstrating actin binding to some TTN-1 fragments. (Upper panel) Cosedimentation of TTN-1 fragment 7/8 with F-actin. Fragment 7/8 at 0, 0.25, 0.5, 1.0, 2.0 and 4.0 μM were incubated with 10 μM F-actin, and then pelleted by ultracentrifugation. Pellets (p) and supernatants (s) were resolved by SDS–PAGE (10% Tris-Glycine) and visualized by Coomassie Blue staining. (Lower panel) Plots of F-actin binding derived from cosedimentation experiments with fragments 7/8, 9/10, 5/6 and 1/2. (B) Data demonstrating myosin binding to some TTN-1 fragments. Total conventional myosin from nematodes was purified and adsorbed to the wells of a microtiter plate. The indicated 6His tagged TTN-1 fragments were added at 0 to 500 nM and an ELISA was conducted using anti-6His antibodies for detection.
Given the data suggesting that TTN-1 extends into the edge of the A-band (Fig. 3, panels A–C, G–I and J–L), the TTN-1 fragments were also tested for binding to purified nematode conventional myosin with a microtiter plate assay. As shown in Fig. 4C, TTN-1 fragment 11/12 showed strong binding to myosin, with an apparent Kd of 1.5 nM. The other fragments showed much weaker binding, with apparent Kd’s that are 100–300 fold higher. Since at least some labeling with antibodies generated to the C-terminus of TTN-1 occurred in the A-band, and the 11/12 region which showed myosin binding in vitro is near the C-terminus, this suggests that the C-terminal region of TTN-1 might indeed interact with myosin in situ.
Intrinsically disordered segments in the I-bands of TTN-1
The search for intrinsically disordered regions of TTN-1 was first done by subjecting the sequence to several predictive tools that calculate disorder propensity and the disorder probability profile from the PONDR VLXT predictor22 is shown in Fig. 1 (bottom panel). Three major regions of strikingly high disorderedness index (near 0.9) occur in the AAPLE, PEVT (including the CEEEI inserts), and BLUE segments. These regions of TTN-1 consist of proline-rich tandem short motifs similar in amino acid composition, but with no sequence homology to the tandem motifs that make up vertebrate titin PEVK.16 Given that CD and NMR studies showed that the 28 residue PEVK motif of vertebrate titin consists of random coil and PPII helix that interconvert with other secondary conformations,23,24 we sought physical evidence that might support similar structures in the TTN-1 repeating motifs. Circular dichroism (CD) studies on the above-described peptides from TTN-1 were conducted at different temperatures, ionic strengths and solvent polarity. These CD data, after deconvolution of spectra to basic components, suggest that the PEVT peptide (Supplemental Fig. S1G–L), the AAPLE peptide (Supplemental Fig. S1A–F) and additionally, the DispRep peptide (Supplemental Fig. S1M–R), CEEEI (Supplemental Fig. S2) even without a high disorder index, all consist of a combination of PPII and unordered coil in aqueous solution. In the presence of TFE, they convert in a distinct fashion, either to α-helix (PEVT, CEEEI), DispRep) or to β-turn (AAPLE) structures, respectively (Supplemental Figs. S1 and S2 and summarized in Fig. 9). This conformational malleability is prevalent in the titin PEVK segment24 and is a characteristic signature for intrinsically disordered proteins.
Fig. 9. Conformations of APPLE42, PEVT39, BLUE32 and DispRep peptides from distinct modules of TTN-1.

Conformations as determined from circular dichroism spectroscopy, including single α-helix (H, dark green), bundled α-helix (CC, pink), polyproline II helix (PPII, green), β-turn (βT, grey) and unordered structure (U, line) at various temperatures, ionic strengths and solvent polarity were characterized by circular dichroism. Peptides from PEVT, DispRep and AAPLE consist of a combination of PPII helix and unordered structure in aqueous solution. In trifluoroethanol (TFE) they are converted to single α-helix (for PEVT, DispRep) or β-turn (for AAPLE), respectively. Urea increases PPII contents of AAPLE, PEVT and DispRep peptides. NaCl decreases PPII contents. The BLUE32 peptide from the long 1500 residue region forms higher ordered helical structure. Its single α-helix content decreases with higher temperatures (converts to unordered) and increases with higher concentration of TFE, converted from an unordered structure and the unraveling of a coiled-coil helix. Urea denatures and NaCl destabilizes α-helical structure.
BLUE is an intrinsically disordered segment of an octad-based α-helix
The BLUE region of TTN-1 consists of 92 tandem repeats of a 16 residue consensus sequence with two octad repeats of 8 residues (Supplemental Fig. S3. The disorder probability profile of BLUE from the PONDR VLXT predictor22 (Fig. S4A) shows a very high disorder index, with a periodic and gentle fluctuation every 32 residues. Moreover, three major low disorder spikes were seen nearly every 400 residues at 7200, 7600, 8000. In contrast, the BLUE sequence has a very high α-helix propensity throughout this region (not shown). The analysis of coiled-coil propensity with the paircoil2 prediction (Supplemental Fig. S4B) showed that on average, the BLUE sequence is at best a marginal heptad repeat-based coiled-coil by the criteria used, since the index fluctuates around the cut off of 9.8 for the coil predictor. For a comparison with a well-characterized heptad coiled-coil protein (Supplemental Figs. S4C & D), most of the tropomyosin sequence has high disordered index, high helical propensity as well as high coiled-coil score well over the cutoff. The contrast between BLUE and tropomyosin suggests that BLUE might not be a typical two-stranded superhelix.
To investigate this dilemma further, circular dichroism measurements were carried out on a two module BLUE peptide (BLUE32) (Fig. 5). At low temperature (2 °C, Fig. 5A), the BLUE32 peptide in aqueous solution (10 mM K-phosphate, p H.7.0, 150 mM KCl) exhibited CD spectra with a characteristic α-helix pattern with double minima at 206 and 222 nm and a ratio of [θ]222 to [θ]208 of 0.93. At room temperature (25 °C, Fig. 5A), however, the peptide had lost most of its secondary structure. It became mostly unordered above 50 °C. The thermal melting curve also showed a destabilizing effect of high temperature with a transition temperature around 16 °C and the complete transition to random coil above 50 °C (Fig. 5F).
Fig. 5. Secondary structure interconversion, disorderness and coiled-coil of BLUE32.

(A–F). CD spectra of BLUE32 peptide (50 μM) in 10 mM K-phosphate, pH 7.0, 150 mM KCl: (A) at 2, 25, 50 and 75 °C; (B) in 0%, 20%, 40% and 80% (v/v) TFE. (C, D) Changes in the relative content (mole fractions) of conformational states of BLUE32 at different temperature (C) and in different concentrations of TFE (D) (●, ▲, ■ for component I, II and III, respectively). (E) Three conformational states I, II and III resulting from CCA deconvolution of all spectra at 2 (with and without 150 mM KCl), 25 (with and without 150 mM KCl), 50, 75 °C and in 0%, 20%, 40%, 80% (v/v) TFE. (F) Thermal titration of Blue 32 was monitored by continuous measurement of CD value at 222 nm from 2 to 80 °C; (G) in the presence of 0, 2, 5 M urea; and (H) the addition of 4 M NaCl.
A transition from random coil to α-helix was observed with increasing TFE concentration at room temperature (Fig. 5B). In 80% TFE, CD spectra revealed two minima at 207 and 222 nm with a ratio of [θ]222 to [θ]208 at 0.85, close to the ratio for non-interacting α-helices (0.83). Analysis of the spectra indicated that three components were sufficient to reconstruct the spectra within experimental error (Figs. 5C & 5D). Component I (Fig. 5E) had two strong negative maxima at 208 and 222 nm, which was assigned to the α-helical conformation. Component II had a ratio of [θ]222 to [θ]208 being close to 1.0, suggesting the presence of interacting helices such as coiled-coils, 3–10 or π-helices. Therefore, component II was tentatively assigned as helical bundles. Component III had a less intense negative band around 199 nm and a slight positive shoulder at 217 nm, which is a prominent feature of an unordered conformation. The temperature dependence of the α-helical signal at 222 nm (Fig. 5F) indicated that the helical content decreases significantly between 2 and 30 °C.
The relative contents of the three components at different temperatures indicated that the α-helical conformation decreases with higher temperatures. BLUE32 contained 88% helical bundles and no single α-helices and at 2 °C and no bundles and only residual (17%) α-helix at 75 °C. It is interesting to note that both α-helical and bundles increased upon titration of BLUE32 with up to 20% TFE and α-helical content further increased with addition of TFE up to 40% while bundles remained unchanged. However, at 80 % TFE, all structures were single α-helical. Ionic strength had less effect on BLUE32 in aqueous solution at 25 °C than at 2 °C. In the latter case, 400 mM KCl decreased the ellipticity of CD spectrum at 206 and 222 nm, indicating the reduction of bundles. In contrast to the PPII containing PEVT, DispRep and AAPLE peptides (Supplemental Figs. S1S, U, W), BLUE32 is denatured by a high concentration of urea (at 5 M, Fig. 5G) and of NaCl (at 4 M, Fig. 5H). These two results indicate that the α-helix and its aggregates of BLUE32 are dependent upon both hydrophobic and electrostatic interaction for their stability and very sensitive to temperature.
Modeling of BLUE octad repeats and helical bundles
To explore the structure of the helices and bundles formed by the unique and highly conserved octad repeats in BLUE (Supplemental Fig. S3), the helical net pattern of a portion of the BLUE sequence was analyzed (Fig. 6A, with yellow circles for hydrophobic residues, red circles for E and D, blue circles for K and green circles for Q and N. The half green, half blue circles are the locations of either N or K residues at position 13 in the BLUE module). While the consensus sequence in BLUE is 16 residues long, each repeat consists of two octads of eight residues, designated with a, b, c, d, e, f, g, h. These residues are indicated on the helical net in Fig. 6A. There are two striking features: two (i, i + 4) strips of alternating charged groups (colored cyan and magenta in Fig. 6A) and a continuous (i, i + 4) strip of hydrophobic residues that is only broken by a N or K residue at position 13 of the module (Fig. 6C). The final strip also has alternating charged groups, but also polar (G and N) and hydrophobic (A) groups. This pattern of hydrophobic residues on the BLUE α-helix has a pattern of 8-4-2-2 (Fig. 6A). The 8-4-2-2 pattern is also shown in Fig. 6D with only the location of the hydrophobic residues highlighted for clarity.
Fig. 6. Helical nature of BLUE-32 and comparison with tropomyosin.

(A) Helical net diagram of hydrophobic residues in BLUE peptide sequence, residues 7036–7088. Large yellow circles are hydrophobic residues with the 8-4-2-2 pattern. The split green/blue circles are the locations of the As or Lys residues between the two leucines separated by 8 residues in the 8-4-2-2 pattern. Small colored circles are charged (red and blue) and polar (green) residues. (B) Space filling model of the BLUE-16 repeat. Structure of BLUE16 was generated by ROSETTA and then subjected to 4 ns of MD in a water box at low ionic strength. Structure orientated to show the residues QAKD that are in the white strip on the left side of the inset helical net between the two structures. (C) Same BLUE-16 structure rotated 180 degrees to show the hydrophobic strip as in the helical net, residues LALNL. Sequence of peptide with 8-4-2-2 repeat pattern shown at top between structures. (D) The 8-4-2-2 pattern is shown with only the location of the hydrophobic residues highlighted for clarity. (E) Helical net typically found in globular proteins.25 (F) Helical net found in coiled-coil proteins such as tropomyosin. (G) Helical net for residues 61–113 of porcine tropomyosin that the BLUE-32 peptide threaded to.
For comparison, the hydrophobic pattern (4-4-4) is shown in Fig. 6E. This pattern of hydrophobic groups on α-helices has been described to be important in the formation of crossed helices in globular proteins with helical bundles.25 The pattern of hydrophobic residues for an ideal left-handed coiled-coil of parallel right-handed helices (7-7-7) is shown in Fig. 6F. The helical net for the well characterized coiled-coil protein, tropomyosin is shown in Fig. 6G. Although there are some oppositely charged groups at i, i + 4 positions in tropomyosin, there are no long range strips of alternating charge (a charge train) as found in BLUE. The hydrophobic net of BLUE32 has greater similarity to the (4-4-4) pattern for packing helices than the (7-7-7) pattern for the formation of coiled-coils.26
While the consensus sequence in BLUE is 16 residues long, the two halves of the sequence are highly similar and the overall sequence repeat in α-helical notation is a, b, c, d, e, f, g, h. These residues are indicated on the helical net in Figure 6A. With 16 residues in 4 turns, the average separation of the hydrophobic residues is 16/4 = 4, which is much larger than that found in the average α-helix of 3.6–3.64. The hydrophobic strip will be right handed like the helix and has been seen in other α-helices that form nearly parallel coiled-coils or right handed coiled-coils.27 To understand what helical structures BLUE may form, the first 110 residues of BLUE were used in an ab initio structural simulation using ROSETTA++.28 From over 30,000 decoys, the best scoring structure of the top cluster was used for further analysis. The region of this structure which contained the BLUE32 peptide was a straight alpha-helix and these 16 residues were place in a water box and subjected to 4 ns of MD simulation with NAMD.29 The results are shown in Fig. 6B–C. The relaxed BLUE32 structure retained its α-helical conformation and some of the alternating charged groups have formed salt-bridges. Figure 6B is oriented to show the QAKD (i, i + 4) strip and was rotated 180 degrees to show the hydrophobic LALNL (i, i + 4) strip in Fig 6C. These structures indicate further that salt bridges are important for the stability of the BLUE helix. Further analysis of the parallel and antiparallel coiled-coils with the helical net approach and examination of the hydrophobic residues at a/d and polar and charge/polar residues at e/g positions has been ambiguous, probably due to the major departure of an octad repeat from a heptad repeat, a hallmark of helical packing into superhelical assembly in fibrous proteins.
Structural-based Modeling of BLUE octad helical bundles
To explore further the conformation of the BLUE region, the sequence was analyzed structurally by threading onto proteins with known 3D structures with the Protein Homology/analogY Recognition Engine (PHYRE). The threading, as expected, gave a list of helical proteins (single α-helices, coiled-coils, helical bundles, etc.), and the first 280 residues hits moesin, tropomyosin, colicin 1a, myosin, α-actinin with the lowest E-values and greatest estimated precision.30 The alignment and threaded structures based on tropomyosin (as parallel coiled-coil) and α-actinin (antiparallel helical bundles), were used as a starting point for further molecular dynamics simulations.
The structure of the first 250 residues of BLUE threaded onto tropomyosin is shown in Fig. 7A. This structure was generated with the threading program NEST31 which optimizes the sidechain conformations for the threaded model and makes as small as possible changes to the backbone to accommodate the different residues. The backbone of the structure is very similar to that of the starting tropomyosin structure and the coiled-coil conformation was well maintained. After minimization and 15 ns of MD simulation (Fig. 7B), there have been large changes in the structure. The N-terminal end has opened up and several turns of the α-helix have been lost at several locations. The most striking change in the structure is that the helices have straightened out at several locations as indicated by the asterisks. The alternating charges on two sides of the helices are likely to resist the bending of the helices to form the supercoiling of the coiled-coils. The BLUE32 region of this model is enlarged in Fig. 7C. Before the MD simulation, the helices are close together as expected for a coiled-coil with hydrophobic interactions along the core. However, there are only two interacting hydrophobic residues, L7029 at the start of the BLUE32 peptide and L7053, 24 residues down the helix. After minimization and 15 ns of MD simulation (Fig. 7C), the coiled-coil structure has altered significantly. The helices have separated and a cluster of salt-bridges has formed between them. For clarity, the salt bridges on the (i, i + 4) strips are not shown as all of the residues are in salt bridges at some time. The two interacting leucine residues at the i and i + 24 positions are maintained during the simulation.
Fig. 7. BLUE helices in parallel configuration.

(A) Model of the first 250 residues of BLUE threaded onto tropomyosin before MD. The BLUE32 peptide is highlighted to show residue type. (B) The structure after 15 ns of MD. Asterisks indicate regions of the α-helices that transition from the curved form as found in coiled-coil structures to the straighter conformation preferred by BLUE. (D) Close-up of the BLUE-32 region after 15 ns of MD. Residues that were interacting after 15 ns of MD simulation are shown in space filling form. L7029 and L7053 are interacting in the threaded model and remain in contact throughout the simulation. A network of salt-bridges has formed between the two helices. (D) The peptide sequences for the two helices are shown with the octad repeat below the sequence and the 8-4-2-2 hydrophobic residues indicated. (E) Helical net as in Fig. 6A. The residues that are interacting between the two chains have × over them. The line indicates the interface between the two chains.
The PHYRE server also returned α-actinin as a good match for the first 280 residues of the BLUE region of TTN-1. The threaded structure before MD is shown in Fig. 8A, with two antiparallel helices highlighted. The structure after 21 ns of MD is shown in Fig. 8B with the the lower highlighted helix in the same orientation as in Fig. 8A. Again, the straightening of the helices has caused the bundles to open up resulting in a large change in the structure. The highlighted residues in Fig 8B are expanded in Fig. 8C and the LNL residues where the helices interact as well as several charged residues are show in space filling form. The interacting residues are labeled on the structure in Fig. 8C and on the sequences of the helices (Fig. 8D) and the interacting As residues are marked with an asterisk. In the structures, the helices have pivoted at the LNL interaction site and multiple salt bridges have formed. This type of helix conformation is similar to the packing of helices with a 4-4-4 helical net pattern as seen in globular proteins.25
Fig. 8. BLUE helices in an anti-parallel configuration.

(A) Model of the first 280 residues of BLUE threaded onto α-actinin. Structure shown is before minimization and MD simulation. Residues that are close to anti-parallel and then cross are highlighted in color of the residue type. (B) Structure after minimization and 21 ns of MD simulation. (C) Structure of residues 7158–7183 KQEADAKLQKENDDKLKQEADAKLKK and 7217–7239 NDDKLKQEADAKLQKENDDKLKQ) from α-actinin threading after minimization and 21 ns of MD. MD greatly disrupted the anti-parallel bundle conformation of the actinin based structure and the two helices crossed as shown. The interacting hydrophobic residues are the leucines at opposite ends of two LQKENDDKL repeats with the asparagine residue between the leucines the center of the interaction (asterisked residues in sequences in D). The residues of the LNL interaction and the salt bridges are labeled on the structure and the the sequence in D. (D) Sequences of the peptides with the location of the 8-4-2-2 hydrophobic residues and octad repeat. (E) Helical net showing the interacting residues. Residues on the peptide from 7158 to 7183 are marked with a ×. Residues on the peptide from 7217 to 7239 are marked with a ‘+’. Helical net does not represent 7158 to 7239 only a schematic of the interacting residues on these two peptides.
Overall, the structural modeling suggested the following distinguishing features of the octad-based helical bundles:
The BLUE octads prefer to form straight α-helices and convert to local non-helical structures in helical dimers or oligomers to avoid supercoiling around each other, as in coiled-coils of heptads. The helical crossing interface is dominated by both ion clusters and a few hydrophobic pairs, in contrast to a coiled-coil.
The right-handed charge train on the surface of the BLUE octad helix appears to form both intra- and inter-helical ion pairs, and frequently, ion clusters with two or more ion pairs at the interface.
Hydrophobic residues of the 8-4-2-2 pattern on the surface were found either buried at the interface or exposed at the surface of the bundles, therefore available for further oligomerization.
Discussion
Size and sarcomere distribution of TTN-1 in nematode obliquely striated muscle
Antibodies generated to two different regions of the predicted TTN-1 sequence detected a ~2 MDa polypeptide on western blot of total proteins from C. elegans (Fig. 2). This detected protein is of a size close to that (2.2 MDa) predicted by our previous genomic and cDNA sequence analysis.16 None of the four antibodies, however, detect on westerns the two other predicted isoforms of 1.2 MDa and 0.3 MDa. Either these predictions were incorrect, or these proteins might exist at lower than detectable levels, or do not include segments recognized by our antibodies. The two anti-TTN-1 antibodies, generated to epitopes near the C-terminus, that detect the ~2 MDa protein on immunoblot localize broadly to the I-band (Fig. 3), indicate that a single 2.2 MDa TTN-1 polypeptide could span the half I-band, which is ~1.5 μm long. Because we have found that the predicted N-terminal 96 residues, when fused to GFP, localize to dense bodies,16 this suggests that this polypeptide is oriented with its N-terminus anchored at the dense body. Since our immunostaining data shows that C-terminus of TTN-1 localizes broadly in the I-band and the outer edge of A-band, we hypothesize that TTN-1 molecules span from the dense bodies towards the outer edge of the A-band in an N to C orientation. The broad I band staining suggests that the C-termini of some TTN-1 molecules may reach as far as the thick filaments, while others may fall short. We speculate the possible existence of a family of TTN-1 with internal deletions in each sarcomere; or that the C-termini of TTN-1 molecules may be at different stages of assembly/disassembly in the sarcomere. . Our findings that expressed fragments along the length of TTN-1 interact with F-actin and myosin in vitro (Fig. 4) suggest that in the sarcomere, TTN-1 interacts with key sarcomeric proteins in the thin and thick filaments. This estimated length for TTN-1 polypeptide seems plausible; the 2.2 MDa crayfish claw I-connectin, which also contains several long putative elastic regions, has been positioned with antibodies to various known epitopes and spans the entire half I-bands, from 1.0 μm at rest to 3.5 μm in a stretched state.32 Moreover, the C-terminal portion of I-connectin resides at the very edge of the A-bands (thick filaments). The analog of crayfish I-connectin in Drosophila is encoded by the gene called sallimus (sls), previously known as D-titin. The gene sls actually encodes multiple products, including kettin (527 kDa) and polypeptides of 700 kDa, 1000 kDa, 1700 kDa and 1900 kDa.33 Interestingly, Burkart et al.33 have found by immunogold EM that in Drosophila flight muscle, both the 700 kDa protein and kettin span from Z disk to the outer edge of the A-band, whereas in non-flight muscle, the 1000, 1700 and 1900 kDa proteins span the same location.
Thus, this enormous TTN-1 polypeptide could be anchored at its N-terminus in the dense bodies and extends into outer edge of the A-band where its C-terminal region associates with myosin and possibly twitchin in thick filaments. We have shown elsewhere that the M-line region contains UNC-89 (the worm analog of mammalian obscurin),14,15 and the A-band (main portions, except for the middle) contains twitchin (homolog of fly synchronous muscle projectin).13 Our very speculative model is that the function of one molecule of vertebrate titin is provided by 3 different molecules in nematode muscle, perhaps forming one long (up to 6–7 micrometers) filament. Beginning at the M-line, perhaps one molecule of UNC-89 connects to a polymer of twitchin molecules in the A-band; at the outer edge of the A-band, perhaps a part of one of the twitchin molecules interacts with the C-terminal portion of TTN-1, and then a single TTN-1 molecule can extend through the length of the I-band and connect its N-terminus to the dense body. Molecular interaction studies are in progress to evaluate this hypothetical three section filament model.
Mechanical role of APPLE, PEVT and BLUE regions as force sensors
TTN-1 is likely to play a major structural mechanical role by its close analogy to other titins. It links dense bodies to the thick filaments and as such is expected to be elastic and provide the restoring forces in the sarcomere during passive stretch and active contraction. The elasticity is likely manifested by the structural changes of the four/five malleable segments that are intrinsically disordered as in titin PEVK.8,34,35 PEVT is similar to the proline–rich PEVK regions of vertebrate titins. These PEVK regions in titin-like molecules are similarly proline-rich, but are not homologous, and have different repeat module lengths: 28 residues for mammalian titin and 39 residues for TTN-1, 100 residues for Drosophila titin, and 67 residues for Drosophila stretchin-MLCK.16,36–39 This suggests that in each protein the PEVK region has evolved around proline rich structures to meet specific functional requirements of the titin-like molecules in these different muscle types. Detailed conformational studies of mammalian titin PEVK modules by CD and NMR indicated that the PEVK region is intrinsically disordered with ensemble conformations consisting of short polyproline II helices, random coils and other secondary structures that interconvert easily in aqueous solutions. Thus, the PEVK structure is malleable and responds to environmental changes without cooperativity.24 Larger portions of the PEVK region have been shown to interact in vitro with F-actin and some portions of nebulin, especially nebulin’s SH3 domain.8 There is good evidence that vertebrate titin can act as a force sensor and convey information on muscle activity to the muscle cell nucleus.40 Signaling may also be accomplished through the intrinsically disordered domains when protein binding regions are exposed upon stretching.8
For TTN-1, it is clear from our conformational studies of these repeating modules that if the same general features prevail throughout each segment, at least three of the four segments, namely AAPLE, PEVT and DispRep, are intrinsically disordered and display a characteristic combination of PPII helix and random coil structure that interconvert in aqueous solutions. Interestingly, with the exception of PEVT, the other three segments are not as proline rich and display no linear sequence homology to the vertebrate PEVK or TTN-1 PEVT per se. Our results thus greatly extend the repertoire of protein sequences in titin-like proteins that are intrinsically disordered and modular in construction (Fig. 9). A total of ~4000 residues of TTN-1 are intrinsically disordered in four non-adjoining segments and likely elastic. Mechanically, we predict that these repeats are spring elements that bear the passive tension of the nematode sarcomere. The mechanical sensitivity of these segments may differ significantly, as suggested by the resistance of AAPLE to change in the environment and the facile conversion of conformations in PEVT and DispRep upon change in solvent polarity and temperature (Supplemental Fig. S1). The maximal extension of these domains can be estimated by assuming an all trans extended form at 0.38 nm per peptide: AAPLE 250 residues 95 nm; BLUE 1500 residues 570 nm; PEVT/K 2400 residues 912 nm; DispRep 15 × 30 residues = 450 residues 171 nm.
For force sensing, both vertebrate titin and nematode TTN-1 have an autoinhibited protein kinase domain that is stretch-dependent for full activity. Single-molecule force spectroscopy experiments support the hypothesis that TTN-1 kinase may function as a force sensor.17 We have speculated that small forces occurring with each contraction/relaxation cycle might be sufficient to remove the autoinhibitory sequence from the catalytic pocket but not enough to unwind the kinase domain and thus destroy its activity. Molecular dynamics simulations41 and single-molecule force spectroscopy experiments42 of vertebrate titin kinase support this hypothesis.
The potential role of BLUE in oligomerization of TTN1 and the maintenance of oblique striation
The third potential force sensor segment BLUE, is unique among the titin family of elastic proteins. The ~1500 aa long segment consisting of 92 tandem repeats of 16 residue modules, each with two octads, appears to be structurally distinct from the AAPLE, PEVT and DispRep in that it consists of a mixture of unstable α-helix (not PPII) and unordered structures that converts readily at lower temperatures and in TFE to helical bundles. The extreme temperature sensitivity between 2–40 °C of the α-helix to unordered conversion (Fig. 5) suggested an intriguing possibility that this segment may also sense temperature in this poikilothermic animal whose growth temperature is between 15–25 °C, thus coupling thermal sensing and force sensing into one segment. It is tempting to speculate that such a hybrid sensor may be relevant to thermotaxis in this nematode. The BLUE octad is also distinct from the widely studied heptads and related assemblies. While much remains to be explored for these octad motifs, it is clear that the ion pairs and hydrophobic residues work in concert at the interface when they criss-cross each other in either parallel or antiparallel directions to form higher order assemblies.
We speculate that the oligomerization capability of the tandem repeats of BLUE octads is relevant in the construction and function of the obliquely striated sarcomere. Since the dense bodies are cone shaped (not disc shaped), and the thick filaments in the A-band are also staggered, the population of TTN-1 that span the width from various locations on the dense bodies and to each thick filaments is expected to be already pre-stretched to a different degree and not in register across the sarcomere (Fig. 3N). The extended tandem repeat of the BLUE octads would facilitate the side by side oligomerization of individual octads from neighboring TTN-1 molecules at all degrees of stretch. The oligomerization of parallel TTN-1 molecules would coalesce the elastic filaments near the tip of the A-band, reminiscent of the role of end filaments in vertebrate titin.43
As the nematode grows from L1 to adult stages, the number and size of the body wall muscle sarcomeres increase dramatically, this occurring in about 2.5 days at 20 °C.44,45 Body wall muscle cells in L1 larvae have 2 A-bands (0.5 μm wide, 0.4 μm deep), whereas adults have 10 A-bands (1.1 μm wide and 1.4 μm deep). Perhaps the BLUE region of TTN-1, with its potential for side to side oligomerization may stabilize the increasing size of the A-band and aid this growth process.
In summary, TTN-1 appears to link the dense bodies to the end zones of the thick filaments and perform the structural and mechanical functions analogous to the I-band segment of vertebrate titin. TTN-1 is likely to play a significant role in the later, rather than early, phases of myofibril assembly. As assessed by the EU143 or 9/10 antibodies, TTN-1 is not found in embryonic or L1 larval muscle (data not shown), when functional muscle has already formed. This lack of essential function for myofibril assembly is in agreement with observations on mutations in vertebrate titin. For example, mutations in zebrafish titin result in an embryonic heart in which myofibrils can be detected and then later disappear.46 This is also compatible with the mutations in human titin that result in either cardiomyopathy;47,48 a tibial muscular dystrophy,49 or myopathy with respiratory failure40 where myofibrils are formed and function but later function poorly. Therefore, both nematode and vertebrate titins are most likely involved in the later stages of myofibril assembly, and in physiological functions of already assembled myofibrils.
Materials and Methods
C. elegans strains
Bristol N2 was used as the wild type strain. Strain RB1438 (ketn-1(ok1641) was kindly provided by Shoichiro Ono (Emory University).
TTN-1 fragment production
Six TTN-1 fragments from various regions of this giant polypeptide were produced (see Figs. 1 and 9 for regions used). Each fragment, approximately 300–370 amino acids in length was cloned into pET-19b from Novagen. Primers used for generating fragments 1/2 and 3/4 were described in ref. 16, for fragments 5/6 and 7/8 described in ref. 50. For fragments 9/10 (the 319 residues just preceding the protein kinase domain and contains Ig36-Ig37-Fn10), the primers contain added NdeI (5′) and BamHI (3′) sites: GTACCATATGAGTGAATCAATTGAATGCAAAG and CGATGGATCCCCAACGGATTCTTGTTCTTTCAC. For fragment 11/12 (Ig38-Ig39-Ig40, just C-terminal of the protein kinase domain), the following primers with added NdeI (5′) and BamHI(3′) sites were used: GTACCATATGGACGGAGTATTCGAAAGAAAC and CGATGGATCCTTCTTGCTACGTTTCGATAGTAA. The cDNA was generated by PCR from a cDNA library (RB2, a gift from Robert Barstead) using Triplemaster polymerase (Eppendorf) and primers. The resulting fragments were cloned into pET19b (Novagen), verified to be error free by sequencing and used to transform BL21 DE3 E. coli (Stratagene). Protein expression was induced with 1 mM IPTG at 25 °C and the corresponding 6His tagged proteins purified using His-Bind Ni column chromatography as described by Novagen. GST fusion proteins with either the N-terminal 314 residues or the C-terminal 353 residues of TTN-1 were cloned into pGEX-6P-1 (Amersham Pharmacia Biotech). The GST fusion proteins were expressed and purified as described by Smith & Johnson51 except that IPTG induction was conducted at room temperature.
Antibodies
Four different polyclonal antibodies generated to 4 different regions of the predicted 2.2 MDa TTN-1 polypeptide were employed. Antibodies to the 314 residue N-terminal region (EU145, guinea pig), and to the 353 residue C-terminal region (EU143, rat) have been described in ref. 50. Antibodies generated to a region just N-terminal of the PEVT region (EU102, rabbit) were described in ref. 16. The fourth antibody, 9/10, to fragment 9/10 was generated in rabbits (Spring Valley Laboratories) and affinity purified as described.52 Rabbit polyclonal antibodies to twitchin were described by Benian.12 For the western blots presented in Fig. 2, the following antibodies were used: antibody RT11 to titin PEVK;36 antibody NB2 to nebulin (Abcam; Cambridge, MA); monoclonal antibodies 5–6 and 5–23, directed to C. elegans myosin heavy chain A (MHC A) and paramyosin, respectively;53 monoclonal antibodies to actin (clone C4; Chemicon International, Temecula, CA); and monoclonal MH44.54
Immunofluorescence microscopy
Immunofluorescent staining of C. elegans wild type (N2 strain) adults was performed as described.14 Primary antibodies used included: anti-TTN-1 affinity purified rabbit antibodies 9/10, rat antibodies EU143, and rabbit anti-twitchin antibodies (all at 1:200 dilution), monoclonal antibodies (5–8, kindly provided by Dr. H. F. Epstein) to MHC B at a 1:400 dilution, and anti-nematode α-actinin monoclonal antibody MH35 (kindly provided by Dr. M. Coutu Hresko and Dr. R. H. Waterston) at a 1:100 dilution. Cy3 or fluorescein-conjugated secondary antibodis (Jackson Immunoresearch Laboratories) were diluted to 1:400. Samples were mounted in ProLong antifade reagent (Molecular Probes, Eugene, Oregon), allowed to solidify at 4 °C overnight and images were obtained with a confocal microscope.55
Peptides synthesis and characterization
Five peptides, one from each of the five repeat segments were synthesized with solid phase peptide synthesis using an Applied Biosystems (Foster City, CA) model 433A peptide synthesizer. The standard tBoc was used to synthesize the first four peptides (see ref. 56 for details). Following cleavage and final deprotection, the peptides were purified by preparative gradient RP-HPLC using C18-silica columns developed in 0.1% TFA/acetonitrile solvents. It was difficult to synthesize the CEEEI peptide due to its long sequence, highly negative charge and potential for formation of disulfide bonds. Earlier attempts to assemble this peptide using tBoc failed, due to incomplete couplings during the later stages of synthesis (MH and JP, unpublished). The strategy for synthesizing this “difficult” peptide included the use of Fmoc as well as pseudoproline amino acid building blocks. In order to achieve over 99% coupling yields throughout CEEEI synthesis, it was necessary to incorporate residues Thr-10 and Thr-19, and of Ser-25 and Ser-41 via their respective pseudoproline, Fmoc-protected dipeptide analogs (NovaBiochem, San Diego, CA), an approach known57 to suppress secondary structure formation of the growing peptide chain. Successful purification of CEEEI was achieved by RP-HPLC of the crude, reduced (dithiothreitol) peptide using Poros R2/10 polymeric columns (Applied Biosystems) equilibrated in 50 mM aqueous ammonium acetate buffer, pH 6.8, containing 5% of acetonitrile (Solvent A), and developed using a linear gradient of Solvent B (4.5:4.5:1, acetonitrile/2-propanol/50 mM aqueous ammonium acetate, pH 6.8). The highly acidic CEEEI peptide failed in conventional RP HPLC due both to its poor solubility in acidic pH and to the inability of acetonitrile to elute the peptide from silica-based RP packings. The purified peptides were lyophilized in the form of their TFA or ammonium acetate salts, and their purity was confirmed by analytical RP-HPLC, N-terminal sequence analysis58, and MALDI-TOF mass spectrometry;58 PEVT39 (calc. mass 4246.7 Da, measured mass 4246.6 Da), AAPLE (calc. 4509.8, measured 4509.8), BLUE32 (calc. 3769.2, measured 3769.6), DispRep (calc. 3434.9, measured 3435.2) and CEEEI (calc. 5728.5, measured 5728.6). The sequences of the peptides are:
AAPLE (Ac-LEPTQEDVPKEAAPSGPTQEDVPKE EAPSEPTQEDVPKEAAP-NH2) 42 mer
PEVT (Ac-VPETSAPSVEPTVEKLAPVES KETSE VQQAEIVEQKDVP-NH2) 39 mer
CEEEI (Ac-CEEEIKELLTEVEVELFF SQAEVFSGLELDLLMECSEYVTTSIQKGSTAAP-NH2) 51 mer
BLUE (Ac-LKQEADAKLKKENDDKLKQEADAKLKKENDDK-NH2) 32 mer
DISPREP (Ac-ETVDEKPKKKVLKKKTEKSDSSISQKSETS-NH2) 30 mer
Actin pelleting assay
Rabbit skeletal muscle F-actin was kindly provided by James R. Sellers (NIH-NHLBI). TTN-1 peptides and fragments (10 mM MOPS, 2 mM MgCl2 pH 7.1) were pre-cleared by centrifugation under the same conditions as the pelleting assay below. The supernatant of each TTN-1 containing solution was then incubated with F-actin (10–20 μM in the same buffer) at 25 °C for 30 min. The samples were centrifuged for 20 min at 65,000 rpm (150,000 × g), using a TLA-120 rotor in a Beckman Optima TLX ultracentrifuge. The supernatants were transferred to fresh tubes and the pellets were carefully resuspended in either SDS sample buffer for SDS-PAGE or water for MALDI-MS analysis. TTN-1 peptides were analyzed by a Tricine gel system optimized for small peptides (Invitrogen) and silver staining (BioRad). TTN-1 fragments (which were not well resolved from actin due to similar size) were identified by immunoblots using an anti-6His antibody (Santa Cruz), peroxidase conjugate and ECL detection (GE Health Sciences). Films at various exposures for each blot were scanned and analyzed quantitatively using an Alpha Innotech Corporation model 2.1.3 imaging camera coupled with ChemiImager 5500 analysis software for densitometry.
Myosin-binding ELISA
Conventional myosin from C. elegans was prepared as described by MacLeod59 with the following revision: crude ammonium sulfate precipitated myosin was further purified by gel filtration (Sephacryl S-300 in 0.6 M NaCl, 10 mM sodium phosphate buffer pH 7.4, 2 mM MgCl2, 1 mM sodium pyrophosphate, 1 mM sodium azide and 2 mM DTT) at 4 °C. Pooled myosin fractions (as determined by SDS-PAGE and BioRad assays) were coated at 0.5 μM (50 μl per well on polystyrene microtiter plate, Corning #3591) for 48 hr at 4 °C. To perform the enzyme-linked immunosorbant assay (ELISA), wells were incubated with block (0.2% bovine serum albumin (BSA), 100 mM KCl, 10 mM Tris pH 8.0, 0.05% Tween 20) for 1.5 hr at 20 °C and then washed three times with the same buffer without the BSA. TTN-1 fragments were then incubated at 0 to 500 nM, at 50 μl per well, for one hour at room temperature and washed. Wells were then incubated with 50 μl of anti-6His antibody (Santa Cruz) at a 1:2000 dilution for 45 minutes at 37 °C and 50 μl of anti-rabbit-HRP antibody (Amersham) at a 1:6000 dilution for 45 minutes at 37 °C, with the washing procedure repeated in between and after each incubation. The HRP colorimetric reaction was performed with 70 μl of mixed TMB solution (BD Biosciences), placed in the dark for 20 minutes then read at 650 nm. As a control, ELISAs were performed in tandem with a BSA coating. Net absorbance values were plotted, subjected to hyperbolic (single rectangular) regression to curve fit and Kd values were determined (Systat, SigmaPlot 9.0).
Circular dichroism measurement and secondary structure content
The secondary structure content was estimated by analyzing the CD spectra with the CCA program60 as previously described.24 Briefly, eight CD spectra of each peptide (PEVT, AAPLE, CEEEI, BLUE and DispRep) were collected in 0, 20, 40 and 80% TFE (v/v) at 25 °C in 10 mM K-phosphate, 150 mM KCl, pH 7 and at 2, 25, 50 and 75 °C in 10 mM K-phosphate, 150 mM KCl, pH 7. These spectra were then deconvoluted to determine the residual ellipticity and a minimal set of secondary structure component motifs. To determine the optimal set of components, CD spectra of each peptide at various TFE concentrations and temperatures were first subjected to 2, 3, 4 and 5 component fits and selected for the lowest number of components beyond which the curves converge. It is worth noting that the CCA deconvolution algorithm performs well at demonstrating the trend of changes, rather than the absolute amounts of the secondary structures of protein. With the exception of CEEEI peptide, synthetic TTN-1 peptides were easily dissolved at least to 1.5 mM in pH 7.0 aqueous solutions (in 10 mM potassium phosphate, 150 mM potassium chloride and the pH 7.0) and in 80% trifluoroethanol (TFE, v/v in the same buffer). A soluble stock solution of the CEEEI peptide was made by first dissolving it in water at pH 9.6 (adjusted by minimal amount of 0.1N NaOH) and then lowered to pH 7 by 0.1 N HCl. The effects of temperature were checked from 2 to 80 °C. The effects of TFE were measured at 25 °C and at TFE concentrations of 0, 20, 40 and 80% (v/v) in 10 mM K-phosphate, 150 mM KCl, pH 7.0. The effects of ionic strength were carried out at 25 °C in 10 mM K-phosphate, pH 7.0 with or without 150 mM KCl, or with ionic strength adjusted by addition of NaCl to 4 M. The urea effects were examined at 0, 2, 5 M urea in 10 mM K-phosphate, pH 7.0, 150 mM KCl, pH 7.0.
Predictions of disorder, helices and coiled-coil
For prediction of the order/disorder in the TTN-1 sequence, the PONDR predictor22 at http://www.pondr.com was used. For predicting the probability of coiled-coil formation, the paircoil2 server61 was used at http://groups.csail.mit.edu/cb/paircoil2/. For predicting the temperature dependence of the peptides used for CD measurements, the AGADIR method62 was used at http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html. Secondary structure predictions were carried out using AGADIR at EMBL website (http://www.embl-heidelberg.de/ExternalInfo/serrano/agadir/agadir-start.html). Coiled-coil structures were surveyed using Multicoil (http://nightingale.lcs.mit.edu/cgi-bin/multicoil).
3D Structure prediction of BLUE
Residues 6984 to 8484 of TTN-1, genpept accession number AAN61517.1 GI:24620453, were separated into a nested set of 280 residue peptides and each was submitted to the Protein Homology/analogY Recognition Engine30 (PHYRE, http://www.sbg.bio.ic.ac.uk/phyre/html/index.html). PHYRE produces models by finding a sequence alignment to known structures then replacing side chain coordinates for the threaded sequence. PHYRE can detect remotely homologous structures since it uses profiles generated by position-specific iterative (PSI) BLAST63 for both the target sequence and the known structures. Refined structures from the alignments were generated using the threading program NEST31. NEST uses an artificial evolution algorithm to build models from an alignment and a known structure. Separately, residues 6984–7094 was subjected to ab initio structure prediction through the ROSETTA++ suite of programs for comparison.28
Molecular dynamics simulation
Models based on tropomyosin (PDB:1c1g)64 and α-actinin (PDB:1sjj)65 where selected for further study through molecular dynamics simulations. All structural models where visualized and manipulated with VMD.66 The models were first solvated with a 10 Å water shell with Solvate (Grubmüller, H. (1996) Solvate 1.0, http://www.mpibpc.mpg.de/home/grubmueller/downloads/solvate/index.html). VMD was then used to add water molecules to make a water box with the minimal volume to contain the molecule in the water shell. Ions (K+ and Cl−) ions were added to the hydrated structures to an ionic strength of 150 mM using the meadionize plugin (Balabin, I. (2006). http://www.chem.duke.edu/~ilya/Software/Meadionize/docs/meadionize.html) for VMD. The meadionize plugin places ions at the minima of the electrostatic potential map generated by the Potential utility of the MEAD program suite.67 The hydrated α-actinin based system contained 119536 atoms with a water box dimensioned at 76 Å × 73 Å X 216 Å. The hydrated tropomyosin based system contained 315161 atoms with a water box dimensioned at 90 Å × 84 Å × 408 Å. These box sizes kept the periodic images separated by > 20Å. These systems were minimized for 4000 steps and subjected to molecular dynamics with NAMD 2.629 with the CHARM22/CMAP force field.68 Following minimization, the system was warmed to 300 K in 6 K steps with 500 steps of MD at each temperature increment. After reaching 300 K, an additional 50,000 steps of MD were performed to further equilibrate the system. A constant temperature (300 K) and pressure (1 atm) simulation (NPT) was then performed using the hybrid Nosé-Hoover Langevin piston method with a decay period of 100 fs and a damping time-constant of 50 fs. To enforce constant temperature, Langevin dynamics was utilized with a damping coefficient of 5 ps−1. All MD simulations were done at a 1 fs step size with particle mesh Ewald (PME) calculation of long range electrostatic interactions. The density of grid points for PME was ~ 1/Å3. Nonbonded interactions where calculated every step and full electrostatic interactions were calculated every other step.
Supplementary Material
Acknowledgments
We thank Gustavo Gutierrez and Wanxia L. Tsai for technical assistance, Matthew J. Houser and Jan Pohl for synthesis of peptides, Jim Sellers for providing rabbit skeletal muscle F-actin, and Sho Ono for providing the kettin mutant. This work was supported in part to KW by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH, DHHS and to GB by NIAMS/NIH grant AR051466. This study utilized the high performance computational capabilities of the Biowulf PC/Linux cluster at NIH (http://biowulf.nih.gov). NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
Abbreviations
- tBoc
tert-butyloxycarbonyl
- Fmoc
9-fluorenylmethyloxycarbonyl
- MALDI-TOF MS
matrix assisted laser desorption ionization time-of-flight mass spectrometry
- RP-HPLC
reversed-phase high-performance liquid chromatography
- TFA
trifluoroacetic acid
- Ig
immunoglobulin
- Fn3
fibronectin 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle Giants: Molecular Scaffolds in Sarcomerogenesis. Physiol Rev. 2009;89:1217–1267. doi: 10.1152/physrev.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Labeit S, Kolmerer B. Titins - Giant Proteins in Charge of Muscle Ultrastructure and Elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 3.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximate to 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 4.Mues A, van der Ven PFM, Young P, Furst DO, Gautel M. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. Febs Letters. 1998;428:111–114. doi: 10.1016/s0014-5793(98)00501-8. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, Labeit S, Inagaki N, Gregorio CC. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001;313:775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 6.Nicholas G, Thomas M, Langley B, Somers W, Patel K, Kemp CF, Sharma M, Kambadur R. Titin-cap associates with, and regulates secretion of, Myostatin. J Cell Physiol. 2002;193:120–131. doi: 10.1002/jcp.10158. [DOI] [PubMed] [Google Scholar]
- 7.Schallus T, Feher K, Ulrich AS, Stier G, Muhle-Goll C. Structure and dynamics of the human muscle LIM protein. Febs Letters. 2009;583:1017–1022. doi: 10.1016/j.febslet.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 8.Ma K, Forbes JG, Gutierrez-Cruz G, Wang K. Titin as a giant scaffold for integrating stress and Src homology domain 3-mediated signaling pathways - The clustering of novel overlap ligand motifs in the elastic PEVK segment. J Biol Chem. 2006;281:27539–27556. doi: 10.1074/jbc.M604525200. [DOI] [PubMed] [Google Scholar]
- 9.Linke WA. Sense and stretchability: The role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Udd B. Sarcomere and Skeletal Muscle Disease. Vol. 642. Springer-Verlag Berlin; Berlin: 2008. Third Filament Diseases; pp. 99–115. [DOI] [PubMed] [Google Scholar]
- 11.Benian GM, Kiff JE, Neckelmann N, Moerman DG, Waterston RH. Sequence of an Unusually Large Protein Implicated in Regulation of Myosin Activity in C-Elegans. Nature. 1989;342:45–50. doi: 10.1038/342045a0. [DOI] [PubMed] [Google Scholar]
- 12.Benian GM, Lhernault SW, Morris ME. Additional Sequence Complexity in the Muscle Gene, Unc-22, and Its Encoded Protein, Twitchin, of Caenorhabditis-Elegans. Genetics. 1993;134:1097–1104. doi: 10.1093/genetics/134.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moerman DG, Benian GM, Barstead RJ, Schriefer LA, Waterston RH. Identification and Intracellular-Localization of the Unc-22 Gene-Product of Caenorhabditis-Elegans. Genes Dev. 1988;2:93–105. doi: 10.1101/gad.2.1.93. [DOI] [PubMed] [Google Scholar]
- 14.Benian GM, Tinley TL, Tang XX, Borodovsky M. The Caenorhabditis elegans gene unc-89, required for muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J Cell Biol. 1996;132:835–848. doi: 10.1083/jcb.132.5.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small TM, Gernert KM, Flaherty DB, Mercer KB, Borodovsky M, Benian GM. Three new isoforms of Caenorhabditis elegans UNC-89 containing MLCK-like protein kinase domains. J Mol Biol. 2004;342:91–108. doi: 10.1016/j.jmb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Flaherty DB, Gernert KM, Shmeleva N, Tang XX, Mercer KB, Borodovsky M, Benian GM. Titins in C-elegans with unusual features: Coiled-coil domains, novel regulation of kinase activity and two new possible elastic regions. J Mol Biol. 2002;323:533–549. doi: 10.1016/s0022-2836(02)00970-1. [DOI] [PubMed] [Google Scholar]
- 17.Greene DN, Garcia T, Sutton RB, Gernert KM, Benian GM, Oberhauser AF. Single-molecule force spectroscopy reveals a stepwise unfolding of Caenorhabditis elegans giant protein kinase domains. Biophys J. 2008;95:1360–1370. doi: 10.1529/biophysj.108.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ono K, Yu R, Mohri K, Ono S. Caenorhabditis elegans kettin, a large immunoglobulin-like repeat protein, binds to filamentous actin and provides mechanical stability to the contractile apparatuses in body wall muscle. Mol Biol Cell. 2006;17:2722–2734. doi: 10.1091/mbc.E06-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hakeda S, Endo S, Saigo K. Requirements of Kettin, a giant muscle protein highly conserved in overall structure in evolution, for normal muscle function, viability, and flight activity of Drosophila. J Cell Biol. 2000;148:101–114. doi: 10.1083/jcb.148.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolmerer B, Clayton J, Benes V, Allen T, Ferguson C, Leonard K, Weber U, Knekt M, Ansorge W, Labeit S, Bullard B. Sequence and expression of the kettin gene in Drosophila melanogaster and Caenorhabditis elegans. J Mol Biol. 2000;296:435–448. doi: 10.1006/jmbi.1999.3461. [DOI] [PubMed] [Google Scholar]
- 21.Waterston RH. Muscle. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1988. pp. 281–335. [Google Scholar]
- 22.Romero P, Obradovic Z, Li XH, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Ma K, Kan LS, Wang K. Polyproline II helix is a key structural motif of the elastic PEVK segment of titin. Biochemistry. 2001;40:3427–3438. doi: 10.1021/bi0022792. [DOI] [PubMed] [Google Scholar]
- 24.Ma K, Wang K. Malleable conformation of the elastic PEVK segment of titin: non-co-operative interconversion of polyproline II helix, beta-turn and unordered structures. Biochemical Journal. 2003;374:687–695. doi: 10.1042/BJ20030702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chothia C, Levitt M, Richardson D. Helix to Helix Packing in Proteins. J Mol Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- 26.Grigoryan G, Keating AE. Structural specificity in coiled-coil interactions. Current Opinion in Structural Biology. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parry DAD, Fraser RDB, Squire JM. Fifty years of coiled-coils and alpha-helical bundles: A close relationship between sequence and structure. J Struct Biol. 2008;163:258–269. doi: 10.1016/j.jsb.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 29.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett-Lovsey RM, Herbert AD, Sternberg MJE, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins-Structure Function and Bioinformatics. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- 31.Petrey D, Xiang ZX, Tang CL, Xie L, Gimpelev M, Mitros T, Soto CS, Goldsmith-Fischman S, Kernytsky A, Schlessinger A, Koh IYY, Alexov E, Honig B. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 2003;53:430–435. doi: 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 32.Fukuzawa A, Shimamura J, Takemori S, Kanzawa N, Yamaguchi M, Sun P, Maruyama K, Kimura S. Invertebrate connectin spans as much as 3.5 mu m in the giant sarcomeres of crayfish claw muscle. EMBO J. 2001;20:4826–4835. doi: 10.1093/emboj/20.17.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkart C, Qiu F, Brendel S, Benes V, Haag P, Labeit S, Leonard K, Bullard B. Modular proteins from the Drosophila sallimus (sls) gene and their expression in muscles with different extensibility. J Mol Biol. 2007;367:953–969. doi: 10.1016/j.jmb.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 34.Forbes JG, Jin AJ, Ma K, Gutierrez-Cruz G, Tsai WL, Wang K. Titin PEVK segment: Charge-Driven Elasticity of the Open and Flexible Polyampholyte. J Muscle Res Cell Motil. 2005 doi: 10.1007/s10974-005-9035-4. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linke WA, Ivemeyer M, Mundel P, Stockmeier MR, Kolmerer B. Nature of PEVK-titin elasticity in skeletal muscle. Proc Natl Acad Sci U S A. 1998;95:8052–8057. doi: 10.1073/pnas.95.14.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gutierrez-Cruz G, Van Heerden AH, Wang K. Modular motif, structural folds and affinity profiles of the PEVK segment of human fetal skeletal muscle titin. J Biol Chem. 2001;276:7442–7449. doi: 10.1074/jbc.M008851200. [DOI] [PubMed] [Google Scholar]
- 37.Machado C, Andrew DJ. D-TITIN: a giant protein with dual roles in chromosomes and muscles. J Cell Biol. 2000;151:639–651. doi: 10.1083/jcb.151.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Champagne MB, Edwards KA, Erickson HP, Kiehart DP. Drosophila stretchin-MLCK is a novel member of the Titin/Myosin light chain kinase family. J Mol Biol. 2000;300:759–777. doi: 10.1006/jmbi.2000.3802. [DOI] [PubMed] [Google Scholar]
- 39.Greaser M. Identification of new repeating motifs in titin. Proteins. 2001;43:145–149. doi: 10.1002/1097-0134(20010501)43:2<145::aid-prot1026>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 40.Lange S, Xiang FQ, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edstrom L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 41.Grater F, Shen JH, Jiang HL, Gautel M, Grubmuller H. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys J. 2005;88:790–804. doi: 10.1529/biophysj.104.052423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puchner EM, Alexandrovich A, Kho AL, Hensen U, Schafer LV, Brandmeier B, Grater F, Grubmuller H, Gaub HE, Gautel M. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci U S A. 2008;105:13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Houmeida A, Baron A, Keen J, Khan GN, Knight PJ, Stafford WF, Thirumurugan K, Thompson B, Tskhovrebova L, Trinick J. Evidence for the Oligomeric State of ‘Elastic’ Titin in Muscle Sarcomeres. J Mol Biol. 2008;384:299–312. doi: 10.1016/j.jmb.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie JM, Garcea RL, Zengel JM, Epstein HF. Muscle Development in Caenorhabditis Elegans - Mutants Exhibiting Retarded Sarcomere Construction. Cell. 1978;15:751–762. doi: 10.1016/0092-8674(78)90261-1. [DOI] [PubMed] [Google Scholar]
- 45.Moerman DG, Fire A. Muscle: structure, function and development. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. The Nematode Caenorhabditis elegans. II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 417–470. [PubMed] [Google Scholar]
- 46.Xu XL, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, Fishman MC. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nature Genetics. 2002;30:205–209. doi: 10.1038/ng816. [DOI] [PubMed] [Google Scholar]
- 47.Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S, Frenneaux M, Thierfelder L. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nature Genetics. 2002;30:201–204. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 48.Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, Hiroe M, Marumo F, Imaizumi T, Yasunami M, Kimura A. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 49.Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, de Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500. doi: 10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zastrow MS, Flaherty DB, Benian GM, Wilson KL. Nuclear titin interacts with A- and B-type lamins in vitro and in vivo. J Cell Sci. 2006;119:239–249. doi: 10.1242/jcs.02728. [DOI] [PubMed] [Google Scholar]
- 51.Smith DB, Johnson KS. Single-Step Purification of Polypeptides Expressed in Escherichia-Coli as Fusions with Glutathione S-Transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 52.Mercer KB, Flaherty DB, Miller RK, Qadota H, Tinley TL, Moerman DG, Benian GM. Caenorhabditis elegans UNC-98, a C2H2Zn finger protein, is a novel partner of UNC-97/PINCH in muscle adhesion complexes. Mol Biol Cell. 2003;14:2492–2507. doi: 10.1091/mbc.E02-10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller DM, Ortiz I, Berliner GC, Epstein HF. Differential Localization of 2 Myosins within Nematode Thick Filaments. Cell. 1983;34:477–490. doi: 10.1016/0092-8674(83)90381-1. [DOI] [PubMed] [Google Scholar]
- 54.Francis GR, Waterston RH. Muscle Organization in Caenorhabditis-Elegans - Localization of Proteins Implicated in Thin Filament Attachment and I-Band Organization. J Cell Biol. 1985;101:1532–1549. doi: 10.1083/jcb.101.4.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qadota H, Mercer KB, Miller RK, Kaibuchi K, Benian GM. Two LIM domain proteins and UNC-96 link UNC-97/PINCH to myosin thick filaments in Caenorhabditis elegans muscle. Mol Biol Cell. 2007;18:4317–4326. doi: 10.1091/mbc.E07-03-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shafer WM, Pohl J, Onunka VC, Bangalore N, Travis J. Human Lysosomal Cathepsin-G and Granzyme-B Share a Functionally Conserved Broad-Spectrum Antibacterial Peptide. J Biol Chem. 1991;266:112–116. [PubMed] [Google Scholar]
- 57.Sampson WR, Patsiouras H, Ede NJ. The synthesis of ‘difficult’ peptides using 2-hydroxy-4-methoxybenzyl or pseudoproline amino acid building blocks: a comparative study. Journal of Peptide Science. 1999;5:403–409. doi: 10.1002/(SICI)1099-1387(199909)5:9<403::AID-PSC213>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 58.Hubalek F, Edmondson DE, Pohl J. Synthesis and characterization of a collagen model delta-O-phosphohydroxylysine-containing peptide. Anal Biochem. 2002;306:124–134. doi: 10.1006/abio.2002.5693. [DOI] [PubMed] [Google Scholar]
- 59.Macleod AR, Waterston RH, Fishpool RM, Brenner S. Identification of Structural Gene for Myosin Heavy-Chain in Caenorhabditis-Elegans. J Mol Biol. 1977;114:133–140. doi: 10.1016/0022-2836(77)90287-x. [DOI] [PubMed] [Google Scholar]
- 60.Perczel A, Hollosi M, Tusnady G, Fasman GD. Convex Constraint Analysis - a Natural Deconvolution of Circular-Dichroism Curves of Proteins. Protein Eng. 1991;4:669–679. doi: 10.1093/protein/4.6.669. [DOI] [PubMed] [Google Scholar]
- 61.McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- 62.Munoz V, Serrano L. Development of the multiple sequence approximation within the AGADIR model of alpha-helix formation: Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 63.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitby FG, Phillips GN. Crystal structure of tropomyosin at 7 Angstroms resolution. Proteins. 2000;38:49–59. [PubMed] [Google Scholar]
- 65.Liu J, Taylor DW, Taylor KA. A 3-D reconstruction of smooth muscle alpha-actinin by CryoEm reveals two different conformations at the actin-binding region. J Mol Biol. 2004;338:115–125. doi: 10.1016/j.jmb.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 66.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 67.Bashford D. Scientific Computing in Object-Oriented Parallel Environments. Springer; Berlin: 1997. An object-oriented programming suite for electrostatic effects in biological molecules An experience report on the MEAD project; pp. 233–240. [Google Scholar]
- 68.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.