

Abstract
A challenge in cancer therapy has been to identify targets whose function is essential for survival of malignant cells but not normal cells. This Perspective discusses recent evidence that novel inhibitors of the kinase TOR can provide an unprecedented balance of anti-cancer efficacy and tolerability.
INTRODUCTION
Dysregulation of cell growth and proliferation have been commonly linked to phosphoinositide 3-kinase (PI3K) and its downstream signaling effectors, which include the serine/threonine kinases AKT and target of rapamycin (TOR). Through activating mutations in PI3K or AKT, inactivating mutations in the PTEN tumor suppressor, or other mechanisms, there are many routes to augment PI3K/AKT/TOR signaling that promote cardinal hallmarks of malignant transformation [1]. A worldwide effort in academic and biopharma laboratories has resulted in a number of new therapeutic strategies to target one of more components of this complex signaling network [2-7]. Several small molecule inhibitors have shown impressive preclinical efficacy and are now in clinical trials. However, it has not been clear which of these approaches will best suppress oncogenic signaling while sparing normal cell homeostasis.
TOR is a conserved Ser/Thr kinase that integrates both extracellular and intracellular signals to regulate cell growth, protein translation and metabolism [8-10]. Mammalian TOR (often termed mTOR) exists in two functionally distinct multi-protein complexes, TOR complex 1 (TORC1) and TOR complex 2 (TORC2). TOR kinase interacts with RAPTOR, LST8, FKBP38, DEPTOR and PRAS40 to form TORC1, or with RICTOR, LST8, SIN1, DEPTOR and PROTOR to form TORC2. The complexity of the signaling network is illustrated by the fact that TORC1 functions downstream of AKT, whereas TORC2 functions upstream (Fig. 1). Recent evidence indicates that both TORC1 and TORC2 function to orchestrate and maintain the excessive proliferative demands of tumorigenic cells [11-14].
Fig. 1.
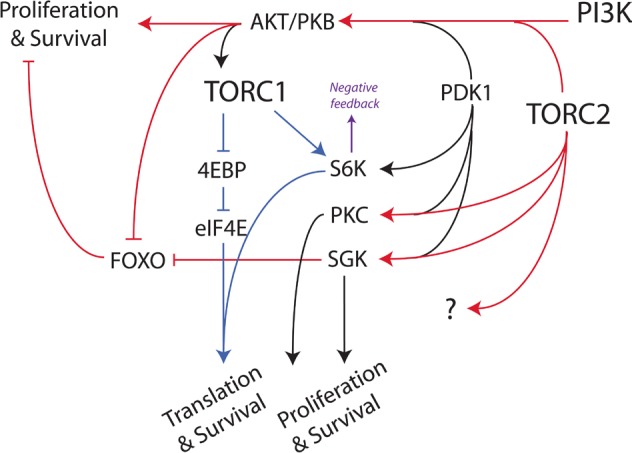
Simplified diagram of the PI3K/AKT/TOR signaling network. Red indicates TORC2-dependent steps. Blue indicates TORC1-dependent steps. The arrow between AKT and TORC1 represents a multistep process, in which activated AKT and other inputs from growth factor signaling pathways and nutrients are integrated to control TORC1 activity. Activated S6K mediates feedback inhibition of upstream signaling through several mechanisms.
Within the last year, a series of ATP-competitive catalytic site TOR inhibitors (TORC1/2 kinase inhibitors) have been developed, and compared to rapamycin (and “rapalogs”) that use an allosteric-based mechanism to inhibit TOR [15-21]. These reports strongly support the conclusion that TORC1/2 kinase inhibitors provide an improved strategy to target the PI3K/AKT/TOR network for therapeutic benefit in cancer.
Mechanistic differences of TORC1/2 kinase inhibitors and rapalogs
TORC1 is an essential sensor for amino acids, oxygen, energy, and growth factor signaling [8-10]. When conditions are favorable for cell growth and division, TORC1 integrates these signals to promote mRNA translation, ribosome biogenesis and glycolytic metabolism. Two notable TORC1 substrates are S6K1 (on Thr389) and 4EBP1 (on several sites) (Fig. 1). Phosphorylation of S6K1 activates the enzyme, leading to increased phosphorylation of the S6 ribosomal protein and other substrates that regulate translation. Phosphorylation of 4EBP1 blocks its function as a suppressor of the initiation factor eIF4E. Rapamycin disrupts the TORC1 complex and partially inhibits TORC1 activity, with greater effects on phosphorylation of S6K than 4EBP1 [22-24]. This is an important distinction because of emerging evidence that 4EBP1 inhibition is a crucial gatekeeper of regulated mRNA translation and is more important than S6K for cellular transformation [12, 14]. TORC2 is activated through unknown mechanisms, and is insensitive to nutrients, energy or acute rapamycin treatment. TORC2 regulates a subgroup of AGC family kinases (Fig. 1), which include AKT, SGK (serum– and glucocorticoid–induced protein kinase), and PKC (protein kinase C), by phosphorylating the hydrophobic and turn motifs [25-28]. Genetic ablation of TORC2 (via deletion of rictor or Sin1) has significant impact on metabolic tissues [29-31] but seems to be selectively toxic to cancer cells compared to normal cells [11, 16, 17, 19, 26].
Rapamycin and rapalogs (everolimus, temsirolimus) can slow the proliferation of cancer cell lines and have achieved some success in specific malignancies [23, 32]. Unfortunately, however, their overall efficacy as cancer therapeutics has been limited. The major drawbacks of rapalogs are: 1) S6K is exquisitely inhibited, yet the control of 4EBP and mRNA translation is far less sensitive [23, 24]; 2) TORC2 activity is not acutely blocked (though it can be suppressed upon sustained exposure [33]); 3) the loss of a feedback inhibition pathway mediated by S6K results in amplified PI3K signaling, with potential to amplify RAS, MAPK, and TORC2 itself [34-38]. In addition to these drawbacks, cell-extrinsic factors have been reported to prompt rapalog resistance in the clinical setting of recurrent PTEN-deficient glioblastomas [39]. To overcome these drawbacks, the pursuit of selective TOR kinase inhibitors has been a strong priority [23, 40]. ATP-competitive TOR kinase inhibitors that also inhibit PI3K and other enzymes have been studied for decades, exemplified by the highly nonselective compound LY294002 and the more refined panPI3K/TOR inhibitors PI-103 and BEZ-235 [3, 6, 7]. These compounds generally have stronger anti-cancer activity than rapalogs, but strong PI3K inhibition might be a liability when considering toxicity to normal cells (see below).
A report from the Shokat group was the first to describe ATP-competitive inhibitors that selectively inhibit TOR, and to document the mechanistic differences between TOR kinase inhibitors and rapamycin [16]. Soon after, several other groups confirmed these findings using TOR kinase inhibitors with distinct chemical scaffolds [15, 17, 19-21]. In each case, the active-site inhibitor completely blocked TORC1 signaling (both S6KThr389 and 4EBP1Thr37/46 and Ser65) and TORC2 signaling (AKTSer473). In fibroblasts, muscle cells and solid tumor cell lines, the inhibition of rapamycin-resistant TOR outputs was associated with stronger suppression of protein synthesis and cell proliferation, and greater impact on cell size and metabolism. Possible mechanisms for differential effects on mRNA translation and metabolism were reviewed recently [40]. Supporting the selectivity of TORC1/2 kinase inhibitors, none of the compounds strongly reduced phosphorylation of AKT on Thr308, the activation loop site that is phosphorylated in a PI3K-dependent manner by phosphoinositide-dependent kinase-1 (PDK-1).
In fibroblasts, the anti-proliferative effects of TORC1/2 kinase inhibitors were entirely attributable to TORC1 inhibition. Thus, cells lacking TORC2 components proliferated normally yet retained full sensitivity to TORC1/2 kinase inhibitors [16, 17, 19]. The relative importance of TORC1 vs. TORC2 inhibition for suppression of cancer cell proliferation and survival is not yet clear, and might be dependent on cell context. If TORC2 inhibition contributes to the mechanism, it will be important to determine which TORC2 substrates are the relevant mediators of cancer cell growth and survival. Although much attention has focused on TORC2-dependent phosphorylation of AKT (on Ser473), regulation of SGK and PKC might be of crucial importance in some contexts. With PDK-1-dependent phosphorylation of AKTT308 intact, AKT remains partially active in TORC2-deficient cells [41, 42]. In contrast, TORC2-dependent phosphorylation of the SGK hydrophobic loop is a prerequisite for SGK phosphorylation on the activation loop [26]. The SGK family (SGK1, SGK2, SGK3) of AGC kinases share strong (>50%) homology with the AKT kinase domain. SGK1 shares substrate preference for FOXO transcription factors (Fig. 1), a well-described target of AKT [43]. SGK3 plays a role in AKT-independent survival in a variety of cancers driven by activating mutations in PI3K [44]. Therefore, SGK may be a key target of TORC2 and the potency attributed to TORC1/2 kinase inhibitors could be due in part to diminished SGK activation. PKC activity might also contribute to tumor growth [45].
An interesting twist to TOR biology was the discovery of DEPTOR as an endogenous inhibitor of both mTOR complexes [46]. Peterson, Sabatini and colleagues tested whether overexpression of DEPTOR could phenocopy cells treated with TORC1/2 kinase inhibitors. They observed that a prominent effect of DEPTOR was to augment AKT activity through disabling of the negative feedback loop mediated by S6K. The rebound activation of PI3K was strong enough to override DEPTOR's inhibitory effect on TORC2, such that AKT phosphorylation on Ser473 was maintained. They further showed that prolonged treatment of cells with suboptimal doses of TORC1/2 kinase inhibitors also elevated AKT phosphorylation. These findings emphasize that when TORC1 activity is reduced, any remaining TORC2 activity can phosphorylate AKT and other substrates that are co-regulated via PI3K/PDK-1. In other words, a complete and optimal amount of TORC1/2 inhibition might need to be achieved and maintained to prevent rebound activation of PI3K signaling. Consequently, it will be important to monitor the degree of PI3K function and downstream signaling in cells and tissues exposed to different doses of TORC1/2 inhibitors.
Efficacy of TORC1/2 kinase inhibitors in vivo
Several TORC1/2 kinase inhibitors have been tested in cancer models in vivo. The first publication, from a group at Wyeth, showed that the compound WYE-354 could delay growth of U87MG tumors in nude mice [21]. Lead optimization by this group led to the discovery of the compound WYE-125132 (abbrev. WYE-132), which showed strong single-agent activity in a range of xenograft models representing various solid tumor types [20]. The anti-tumor effect of WYE-132 was markedly stronger than that achieved by the rapalog CCI-779. In some models, regression was observed. Similarly, the TORC1/2 kinase inhibitor AZD8055 (developed by AstraZeneca) demonstrated growth inhibition and/or regression in xenograft models [15]. In each of these studies, in vitro experiments confirmed that TORC1/2 kinase inhibitors block rapalog-resistant outputs of TORC1 and TORC2 across a range of cancer cell lines. Interestingly, WYE-354 failed to inhibit protein synthesis in the colon cancer cell lines HCT116 and HT29 and this was associated with absence of pro-apoptotic effects [21, 40]. Further study of cell lines and primary specimens that are resistant to TORC1/2 inhibitors might provide biomarkers that can be used to predict efficacy.
Our group compared the TORC1/2 kinase inhibitor PP242 to rapamycin in acute leukemia models driven by the BCR-ABL fusion tyrosine kinase encoded by the t(9;22) Philadelphia (Ph) chromosomal translocation [18]. As in studies of fibroblasts and solid tumor cell lines, we found that PP242 blocked rapamycin-resistant TORC1 and TORC2 signaling outputs in mouse and human leukemia cells representing either Ph+ B-precursor acute lymphoblastic leukemia (B-ALL) or chronic myeloid leukemia (CML). PP242 did not alter cellular levels of PIP3, a measure of PI3K activity. In vitro, PP242 caused cell cycle arrest similar to rapamycin, but also triggered apoptosis. In a mouse syngeneic model of Ph+ B-ALL, PP242 prolonged survival whereas rapamycin had no protective effect. In these experiments we also compared PP242 to compounds representing the panPI3K/TOR target profile. Notably, we found that the efficacy of PP242 in vitro and in vivo was comparable to PI-103 or BEZ-235. These results indicate that PI3K inhibition is dispensable for strong anti-leukemic efficacy when both TOR complexes are fully suppressed. PP242 also slowed growth and caused apoptosis in vivo when tested in a mouse thymoma model [12]; rapamycin had no protective effect in this model.
In patients with chronic phase CML, impressive therapeutic responses have been achieved with BCR-ABL tyrosine kinase inhibitors (TKIs) [47]. Unfortunately, TKI resistance often develops and these agents fail to achieve durable remission in later phases of CML (i.e. blast crisis) or in patients with Ph+ B-ALL [48]. We compared the ability of various TOR inhibitors to augment the efficacy of BCR-ABL TKIs. Although functional synergy was achieved with all combination approaches, PP242 was more effective than rapamycin when combined with imatinib or dasatinib, both in vitro and in vivo. In mice bearing xenografts of the Ph+ B-ALL cell line SUP-B15, dasatinib plus PP242 caused leukemia regression whereas dasatinib plus rapamycin only slowed expansion. In xenografts of primary human Ph+ B-ALL specimens, dasatinib plus PP242 caused significantly greater inhibition of leukemia cell proliferation compared to dasatinib alone. At the signaling level, PP242 suppressed TOR outputs more thoroughly than rapamycin even in the presence of BCR-ABL TKIs, probably because cell-extrinsic factors provide an alternative oncogene-independent route to TOR activation. These results provide proof-of-concept that encourages further testing of TORC1/2 kinase inhibitors in combination with TKIs in other tumor settings (to avoid confusion, we oppose a recent suggestion that TORC1/2 kinase inhibitors be abbreviated "TKIs" [40]). TORC1/2 kinase inhibitors also have broader anti-angiogenic impact than rapalogs [20] and could enhance the efficacy of existing angiogenesis inhibitors. Indeed, in a renal cell carcinoma model the compound WYE-132 showed greater ability than CCI-779 to synergize with bevacizumab (Avastin), a monoclonal antibody to VEGF-A [20]. These findings highlight the clinical potential of TORC1/2 kinase inhibitors for combinatorial therapies. Careful strategies will need to be employed when developing a dosing regimen to best obtain the full therapeutic benefit of drug combinations.
Tolerability of TORC1/2 kinase inhibitors in vivo
TOR was first discovered as the molecular target of the immunosuppressive drug rapamycin. In fact, much of what we know about mTOR and its roles in immunological function has derived from experiments using rapamycin. It is now clear that rapamycin suppresses T and B cell proliferation, and promotes tolerance induction through at least three mechanisms: induction of T cell anergy, generation of regulatory T cells (Tregs), and impairment of dendritic cell maturation [49-51]. Paradoxically, rapamycin enhances the generation and quality of CD8 T cell memory [52, 53], and also potentiates inflammatory responses of innate immune cells [49-51]. In addition to these immune effects, systemic rapamycin treatment in humans results in other significant toxicities [54].
Given the more complete suppression of TOR signaling by ATP-competitive inhibitors, one might expect these compounds to have even more severe toxicities and immunomodulatory activities. However, the evidence so far indicates that this is not the case. Several TORC1/2 kinase inhibitors have entered clinical trials, indicating that animals tolerate these compounds at doses that produce therapeutic effects. In our leukemia models, we noted that PP242 was not toxic to normal mouse bone marrow cells under conditions where human Ph+ B-ALL cells disappear [18]. In fact, proliferation of endogenous bone marrow cells returned in mice treated with dasatinib plus PP242. In vitro, PP242 caused hematotoxicity only at concentrations much higher than those needed to cause leukemia cell death. Remarkably, PP242 and the structurally unrelated TORC1/2 inhibitor Ku-0063794 had little impact on the proliferation of activated lymphocytes, whereas these responses were strongly suppressed by rapamycin. Mice treated with rapamycin showed a disruption of lymphoid architecture and a near-complete block in T cell-dependent antibody responses. These effects were not observed in mice treated with PP242 at doses showing profound suppression of leukemia. Importantly, the panPI3K/TOR inhibitor PI-103 showed less selectivity for leukemia cells and was reported by another group to be immunosuppressive [55]. Thus, selective TORC1/2 kinase inhibition provides a favorable tolerability profile compared to rapamycin or panPI3K/TOR inhibitors.
It is interesting to speculate on the mechanism of selective anti-cancer effects of TORC1/2 kinase inhibitors. Mouse models of PTEN-deficient prostate cancer support the idea that cancer cells with elevated PI3K signaling are uniquely addicted to TORC2 [11, 13]. One study showed that simultaneously deleting Pten and mTor in the prostate epithelium suppressed prostate cancer development while sparing the morphology and function of normal prostate tissue [13]. The suppression of neoplasia was more pronounced than with 4–week pharmacological treatment with the rapalog RAD001. This observation does not distinguish whether the PTEN-deficient cells relied on rapalog-resistant outputs of TORC1 or TORC2. However, another group obtained similar results by deleting Rictor in the prostate epithelium, showing that Pten-null driven prostate cancer progression requires TORC2 function [11]. In support of this conclusion, knockdown of rictor expression suppressed the development of a PTEN-null PC-3 human prostate cancer xenograft model. The preservation of normal tissue in the absence of TORC2 is consistent with studies of mouse embryo fibroblasts (MEFs). In MEFs, loss of rictor or Sin1 does not affect cell proliferation and the cells remain equally sensitive as wild-type MEFs to growth suppression by TORC1/2 kinase inhibitors [16, 19]. On the other hand, the fact that complete TORC1 inhibition strongly suppresses growth factor-dependent proliferation of MEFs implies that TORC1/2 kinase inhibitors should have negative impacts on mitotic tissues. Indeed, we found that PP242 did suppress hematopoietic colony formation and lymphocyte proliferation at a high concentration (1μM) [18]. We propose a “threshold” model in which normal cells can tolerate a lower output of TORC1 and TORC2 signaling than transformed cells (Fig. 2A). In some cell types, such as prostate epithelium, mTor expression appears completely dispensable [13]. In other cell types, exemplified by T lymphocytes, deletion of mTor delays but does not fully arrest proliferation [56]. In support of the threshold model, mice heterozygous for a kinase-dead allele of mTor show no impairment of T cell proliferation [57].
Fig. 2. Working models to explain selective effects of TORC1/2 kinase inhibitors on leukemia cells (A) and rapalogs on normal lymphocytes (B).

The threshold model (A) posits that leukemia cells depend on a higher output of mTOR signaling for growth and survival. Leukemia cells commit to cell death process when TORC1 and TORC2 are strongly (or transiently) suppressed, whereas normal lymphocytes grow more slowly but do not die. At intermediate levels of mTOR signaling, leukemia cells exhibit slower growth whereas normal cells are unaffected. The scheme in panel (B) proposes that in normal lymphocytes, rapamycin and analogs inhibit TORC1 and TORC2 more effectively than in leukemia cells. Because rapalogs act through an allosteric mechanism, the magnitude and kinetics of their effects might differ depending on the composition and turnover of TOR complexes in distinct cell contexts. We reported experimental evidence to support the threshold model in (A) [18], but further work is necessary to test the model in (B).
Perhaps a greater puzzle is why rapamycin seems more potent at suppressing normal lymphocytes than leukemia cells or solid tumor cell lines. It is possible that lymphocyte signaling complexes are wired differently, such that rapamycin suppresses TOR outputs that are resistant to the drug in other settings (Fig. 2B). There is evidence that the magnitude and kinetics of rapamycin's effect on 4EBP1 phosphorylation and TORC2 function are cell type-dependent [22, 33]. Alternatively, feedback effects of TORC1 inhibition might have severe impact in primary lymphocytes. There is also evidence that rapamycin disrupts noncatalytic scaffolding functions of TOR [58-60], whereas TORC1/2 kinase inhibitors do not affect the complexes [20]. It is worth testing these models in lymphocytes and in cancer cell lines that are particularly sensitive to rapamycin.
FUTURE DIRECTIONS
TORC1/2 kinase inhibitors represent a major breakthrough in targeting the PI3K/AKT/TOR signaling network for cancer therapeutics. In order to realize the full clinical potential of these compounds, more basic research is required. Here we emphasize three objectives of primary importance.
First, we need a greater understanding of the mechanism of action of TORC1/2 kinase inhibitors in cancer cells. Which cellular processes regulated by TORC1 and/ or TORC2 are relevant to the therapeutic effects? These efforts will help identify biomarkers of drug efficacy and resistance. A recent study from the Ruggero group provided important insights in a mouse thymoma model driven by activated AKT [12]. This study showed that TORC1-dependent eIF4E hyperactivation (via 4EBP1 inactivation) was essential for tumor growth, whereas S6K activation was dispensable. In this model, the pro-apoptotic effect of PP242 was entirely dependent on the 4EBP1/eIF4E axis. However, it remains possible that TORC2 inhibition contributes to death of cancer cells that do not express a constitutively active AKT molecule. An interesting finding of Hsieh et al. was that regulated translation of the anti-apoptotic Bcl-2 family member MCL-1 might play a unique pro-survival role. MCL-1 was also found to be modulated by PI3K/TOR signaling in the setting of mutant epidermal growth factor receptor (EGFR) driven non-small cell lung cancers [61]. It will be interesting to determine whether TORC1/2 kinase inhibitors trigger cell cycle arrest also by controlling expression of specific proteins, or through general inhibition of translation.
Second, it will be important to determine the efficacy of TORC1/2 kinase inhibitors across a broad range of tumor types and driving mutations. Human cancers are heterogeneous, and xenograft models have already shown variable responses of solid tumors and primary leukemias [15, 18, 20]. Preclinical screens of drug efficacy can provide valuable information about which populations are most likely to benefit from any targeted agent. Identifying tumor characteristics that correlate with drug resistance will also be valuable, as understanding the molecular basis of resistance can lead to combination approaches that achieve greater efficacy. There might also be heterogeneity within tumors such that subpopulations, for example quiescent cells with stem-like properties, survive independently of TOR activity. A related issue is the need to test TORC1/2 kinase inhibitors in combination with current front-line therapies. It is likely that novel targeted agents will be tested first in clinical settings where standard therapy has failed, but could eventually be used as adjuvant therapy to augment the initial response. Our data provide strong justification for including TORC1/2 kinase inhibitors in clinical protocols that involve TKIs targeting BCR-ABL or other oncogenic kinases.
Third, we need to explore in greater detail how TORC1/2 kinase inhibitors affect immune function. Our published experiments with lymphocyte proliferation and antibody production have only scratched the surface of this important problem. TORC1/2 kinase inhibitors should be evaluated in various settings in which rapalogs have a profound impact: innate immune cell function, T and B cell differentiation, memory, and regulatory T cell induction. It is likely that drug concentration will have an important influence on functional outcomes, as illustrated by our studies of lymphocyte proliferation [18]. Similarly, the impact of TORC1/2 kinase inhibitors on immune function in vivo will likely depend on pharmacokinetics---and may therefore differ among different candidate molecules. An important complementary approach will be to study mice genetically deficient in individual mTOR complexes, to elucidate the separate roles of TORC1 versus TORC2 on innate and adaptive immune responses. One consequence of these studies might be that TORC1/2 kinase inhibitors have therapeutic value in certain immune-mediated diseases. In the setting of high-risk leukemias, it is essential to eradicate minimal residual disease (MRD) following chemotherapy regimens. One strategy to minimize MRD is allogeneic hematopoietic stem cell transplantation (allo-SCT), where the donor marrow attacks recipient MRD, also termed graft versus leukemia (GVL). It is interesting to note that many active clinical trials of rapalogs in cancer are testing their efficacy to suppress graft versus host disease (GVHD), a common allo-SCT complication. In the setting where rapamycin is used to suppress GVHD, will it also affect MRD by modulating GVL or through direct effects on the residual leukemia cells? Will TORC1/2 inhibitors achieve similar immunosuppression of GVHD, yet still achieve effective clearance of MRD? In solid tumor settings, will TORC1/2 kinase inhibitors have different effects than rapalogs on immunological tolerance? As TOR kinase-targeted therapies enter the clinical arena, it will be important to understand how they will best complement or interact with current clinical practices to harness immune responses and eradicate tumor cells.
Acknowledgments
Our studies of TOR inhibitors have been supported by NIH training grant T32-CA009054 (to MRJ), a Discovery Grant from the University of California Industry-University Cooperative Research Program (to DAF), and a sponsored research agreement from Intellikine, Inc. (to DAF). MRJ is an awardee of the Jackie Murphy and Carol Malouf Scholar Achievement Rewards for College Scientists.
Footnotes
The authors have no conflict of interests to declare.
REFERENCES
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Abraham RT, Eng CH. Mammalian target of rapamycin as a therapeutic target in oncology. Expert Opin Ther Targets. 2008;12:209–222. doi: 10.1517/14728222.12.2.209. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 4.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A Pharmacological Map of the PI3-K Family Defines a Role for p110alpha in Insulin Signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 Kinome: From Chemical Tools to Drugs in the Clinic. Cancer Res. 2010;70:2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Abraham RT. Regulation of the mTOR signaling pathway: from laboratory bench to bedside and back again. F1000 Biology Reports. 2009;1:a8. doi: 10.3410/B1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 11.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, Chen Z, Egia A, Fornari A, Fiorentino M, Loda M, Kozma SC, Thomas G, Cordon-Cardo C, Pandolfi PP. Differential Requirement of mTOR in Postmitotic Tissues and Tumorigenesis. Sci Signal. 2009;2:ra2. doi: 10.1126/scisignal.2000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Reilly KE, Warycha M, Davies MA, Rodrik V, Zhou XK, Yee H, Polsky D, Pavlick AC, Rosen N, Bhardwaj N, Mills G, Osman I. Phosphorylated 4E-BP1 is associated with poor survival in melanoma. Clin Cancer Res. 2009;15:2872–2878. doi: 10.1158/1078-0432.CCR-08-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GC, Guichard S, Pass M. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2009;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 16.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009 doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, Kim SK, Conant R, Curran K, Kaplan J, Verheijen J, Ayral-Kaloustian S, Mansour TS, Abraham RT, Zask A, Gibbons JJ. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 2010;70:621–631. doi: 10.1158/0008-5472.CAN-09-2340. [DOI] [PubMed] [Google Scholar]
- 21.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, Nowak P, Zask A. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 22.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 24.Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5 doi: 10.4161/auto.5.5.8504. [DOI] [PubMed] [Google Scholar]
- 25.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 27.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO journal. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. The EMBO journal. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Ruegg MA. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A. 2009;106:9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol. 2008;28:61. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houghton PJ. Everolimus. Clin Cancer Res. 2010;16:1368–1372. doi: 10.1158/1078-0432.CCR-09-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Molecular Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treins C, Warne PH, Magnuson MA, Pende M, Downward J. Rictor is a novel target of p70 S6 kinase-1. Oncogene. 2009;29:1003–1016. doi: 10.1038/onc.2009.401. [DOI] [PubMed] [Google Scholar]
- 39.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–3837. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 41.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 42.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Developmental cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a) Mol. Cell. Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redig AJ, Platanias LC. Protein kinase C signalling in leukemia. Leuk Lymphoma. 2008;49:1255–1262. doi: 10.1080/10428190802007726. [DOI] [PubMed] [Google Scholar]
- 46.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 48.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in the Blast Crisis of Chronic Myeloid Leukemia and Acute Lymphoblastic Leukemia with the Philadelphia Chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 49.Janes MR, Fruman DA. Immune regulation by rapamycin: moving beyond T cells. Sci Signal. 2009;2:pe25. doi: 10.1126/scisignal.267pe25. [DOI] [PubMed] [Google Scholar]
- 50.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weichhart T, Saemann MD. The multiple facets of mTOR in immunity. Trends Immunol. 2009;30:218–226. doi: 10.1016/j.it.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sankhala K, Mita A, Kelly K, Mahalingam D, Giles F, Mita M. The emerging safety profile of mTOR inhibitors, a novel class of anticancer agents. Target Oncol. 2009;4:135–142. doi: 10.1007/s11523-009-0107-z. [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Fauqued M, Gil R, Grueso J, Hernandez J, Pujol A, Moline T, Recio JA. The dual PI3K/mTOR inhibitor (PI-103) promotes immunosupression, in vivo tumor growth and increases survival of sorafenib treated melanoma cells. Int J Cancer. 2009 doi: 10.1002/ijc.24926. [DOI] [PubMed] [Google Scholar]
- 56.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shor B, Cavender D, Harris C. A kinase-dead knock-in mutation in mTOR leads to early embryonic lethality and is dispensable for the immune system in heterozygous mice. BMC Immunol. 2009;10:28. doi: 10.1186/1471-2172-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delgoffe GM, Kole TP, Cotter RJ, Powell JD. Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Mol Immunol. 2009;46:2694–2698. doi: 10.1016/j.molimm.2009.05.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1-S cell cycle progression of T lymphocytes. Nat Immunol. 2007;8:64–73. doi: 10.1038/ni1413. [DOI] [PubMed] [Google Scholar]
- 60.Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P, Arrieumerlou C, Hall MN. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One. 2007;2:e1217. doi: 10.1371/journal.pone.0001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, Garcia-Echeverria C, Wong KK, Engelman JA. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]