

Abstract
Background & Aims
Hepatopulmonary syndrome (HPS) affects 10%–30% of patients with cirrhosis and portal hypertension and significantly increases mortality. Studies in experimental models indicate that pulmonary angiogenesis contributes to the development of HPS, but pathogenesis in humans is poorly understood. We investigated genetic risk factors for HPS in patients with advanced liver disease.
Methods
We performed a multicenter case-control study of patients with cirrhosis being evaluated for liver transplantation. Cases had an alveolar-arterial oxygen gradient ≥15 mm Hg (or ≥20 mm Hg if age > 64 years) and contrast echocardiography with late appearance of microbubbles after venous injection of agitated saline (intrapulmonary vasodilatation); controls did not meet both criteria for case status. The study sample included 59 cases and 126 controls. We genotyped 1086 common single nucleotide polymorphisms (SNPs) in 94 candidate genes.
Results
Forty-two SNPs in 21 genes were significantly associated with HPS after adjustments for race and smoking. Eight genes had at least 2 SNPs associated with disease: CAV3, ENG, NOX4, ESR2, VWF, RUNX1, COL18A1, and TIE1. For example, rs237872 in CAV3 showed an odds ratio of 2.75 (95% confidence interval: 1.65–4.60, P =.0001) and rs4837192 in ENG showed an odds ratio of 0.35 (95% confidence interval: 0.14–0.89, P =.027). Furthermore, variation in CAV3 and RUNX1 was associated with HPS in gene-based analyses.
Conclusions
Polymorphisms in genes involved in the regulation of angiogenesis are associated with the risk of HPS. Further investigation of these biologic pathways might elucidate the mechanisms that mediate the development of HPS in certain patients with severe liver disease.
Keywords: Genetic Polymorphism, Portal Hypertension
The hepatopulmonary syndrome (HPS) occurs when vascular alterations in the pulmonary microvasculature lead to abnormalities in systemic oxygenation in the setting of liver disease or portal hypertension.11 This syndrome is found in 10%–30% of patients with cirrhosis being evaluated for liver transplantation, and we and others have shown that HPS is associated with worse quality of life and increased mortality.2–4 Currently, the only established treatment for HPS is liver transplantation, although postoperative survival may be lower in patients with HPS relative to those without HPS.5 Together, these observations support the need to define the pathogenesis of HPS to develop effective medical therapies. Identification of genetic risk factors for this prevalent and morbid complication of liver disease could suggest novel therapeutic approaches.
The mechanisms for HPS in patients with cirrhosis are unclear. Early experimental and human studies implicated pulmonary microvascular dilation, in part related to excess nitric oxide production and altered estrogen signaling in disease pathogenesis.6–8 Although impaired vasomotor tone contributes to the pathophysiology of HPS, incomplete response to pharmacologic blockade of these pathways implies additional mechanisms.9,10 Observations of increased angiogenesis in the splanchnic and hepatic microvascular beds11 and increased pulmonary capillary density12 in advanced liver disease suggest that vascular remodeling and pulmonary angiogenesis may also play a role. This hypothesis is supported by the recent demonstration of increased in pulmonary microvessels as well as up-regulation of vascular endothelial growth factor-mediated angiogenic pathways in the common bile duct-ligated rat, an animal model of human HPS.13
We therefore hypothesized that variation in genes responsible for vascular phenotype and homeostasis contributes to the risk of developing HPS. We performed a high-throughput candidate gene study in an attempt to identify common genetic variation associated with the risk of HPS in a group of patients undergoing liver transplantation evaluation. This work has been previously published in abstract form.14
Patients and Methods
For other information regarding patients and methods, please see Supplementary Patients and Methods.
Study Cohort and Study Sample
The Pulmonary Vascular Complications of Liver Disease Study enrolled a cohort of 536 patients evaluated for liver transplantation at 7 centers in the United States between 2003 and 2006. The only inclusion criterion was the presence of chronic portal hypertension with or without intrinsic liver disease. We excluded patients with evidence of active infection, recent (<2 weeks) gastrointestinal bleeding, or those who had undergone liver or lung transplantation. The institutional review boards at each of the participating centers approved this study, and informed consent was obtained.
We performed a case-control study. The study sample included new patients from the Pulmonary Vascular Complications of Liver Disease Study cohort evaluated with contrast transthoracic echocardiography, spirometry, and arterial blood gas sampling (routinely performed for pretransplant evaluation) during the study period with available genetic data. We excluded patients with pulmonary function testing showing a significant obstructive or restrictive ventilatory defect (see Supplementary Patients and Methods). We also excluded patients with intracardiac shunting (or with uninterpretable shunt timing) by transthoracic echocardiography as described below.
Contrast transthoracic echocardiography was performed and interpreted at each center. Agitated saline was injected via a peripheral vein during imaging. Appearance of microbubbles in the left heart ≥ 3 cardiac cycles after venous injection of agitated saline was considered “late,” consistent with intrapulmonary shunting. Appearance of microbubbles in the left heart < 3 cardiac cycles after venous injection was considered “early,” consistent with intracardiac shunting.
Case and Control Definitions
Cases and controls were identified from those patients fulfilling the inclusion and exclusion criteria defined above. HPS was defined by (1) contrast echocardiography with late appearance of microbubbles after venous injection of agitated saline and (2) an alveolar-arterial (A-a) oxygen gradient ≥15 mm Hg (or ≥20 mm Hg if age > 64 years), as recommended by the European Respiratory Society Task Force Pulmonary-Hepatic Vascular Disorders Scientific Committee.1 Patients who did not meet both criteria were considered in the “non-HPS” group (controls). Patients with either “early” or indeterminate timing of the appearance of microbubbles in the left heart after agitated saline injection were excluded from the study.
Clinical Variables and Blood Sampling
Data were collected from subjects and the medical record. The Model for End-stage Liver Disease score was calculated, without inclusion of exception points for either hepatocellular carcinoma or HPS.15 Phlebotomy was performed, and blood was collected into EDTA-containing tubes. Plasma and buffy coat layers were stored at −80°C.
Candidate Genes and Single Nucleotide Polymorphism Selection
Ninety-four genes affecting vascular function were selected by the investigators (Table 1). For this study, 1086 single nucleotide polymorphisms (SNP) in the 94 candidate genes were genotyped (Supplementary Table 1). We genotyped an additional set of 61 SNPs (null loci) from a validated list of Ancestry Informative Markers (AIM)16 to detect potential population stratification. (See Supplementary Patients and Methods for details of gene and SNP selection.)
Table 1.
Candidate Genes: Gene Ontology Annotation
| Pathway | Gene | Reference sequence | Chr | SNPs |
|---|---|---|---|---|
| Control of blood circulation GO:0008015 | Angiotensin I converting enzyme (ACE) | NM_152831 | 17q23 | 15 |
| Elastin (ELN) | NM_000501 | 7q11 | 5 | |
| Endothelin 1 (EDN1) | NM_001955 | 6p24 | 7 | |
| Endothelin converting enzyme 1 (ECE1) | NM_001397 | 1p36 | 10 | |
| Endothelin receptor, nonselective type (EDNRB) | NM_000115 | 13q22 | 13 | |
| Endothelin receptor, type A (EDNRA) | NM_001957 | 4q31 | 11 | |
| Heme oxygenase 1 (HMOX1) | NM_002133 | 22q13 | 8 | |
| Natriuretic peptide precursor A (NPPA) | NM_006172 | 1p36 | 13a | |
| Natriuretic peptide precursor B (NPPB) | NM_002521 | 1p36 | 13a | |
| Nitric oxide synthase 2 (NOS2A) | NM_000625 | 17q11 | 15 | |
| Phosphodiesterase 5 (PDE5A) | NM_001083 | 4q26 | 9 | |
| Potassium channel, voltage-gated, shaker, member 5 (KCNA5) | NM_002234 | 12p13 | 9 | |
| Rho-associated protein kinase 2 (ROCK2) | NM_004850 | 2p24 | 15 | |
| Transient receptor potential cation channel, subfamily C, 6 (TRPC6) | NM_004621 | 11q21 | 18 | |
| Cell growth | Activin A receptor, type II-like kinase (ACVRL1) | NM_000020 | 12q11 | 6 |
| Apoptosis GO:0008283 GO:0006915 |
Apolipoprotein E (APOE) | NM_000041 | 19q13 | 4 |
| BCL2-associated X protein (BAX) | NM_138764 | 19q13 | 6 | |
| Bone morphogenetic protein receptor type 1a (BMPR1A) | NM_004329 | 10q22 | 20 | |
| Bone morphogenetic protein receptor type 2 (BMPR2) | NM_001204 | 2q33 | 12 | |
| Caveolin 1 (CAV1) | NM_001753 | 7q31 | 20a | |
| Caveolin 2 (CAV2) | NM_001233 | 7q31 | 20a | |
| Caveolin 3 (CAV3) | NM_033337 | 3p25 | 19 | |
| CD14 molecule (CD14) | NM_000591 | 5q22 | 3 | |
| Cyclin-dependent kinase inhibitor 2A (CDKN2A) | NM_000077 | 9p21 | 13 | |
| Growth differentiation factor 2 (GDF2) | NM_016204 | 10q11 | 5 | |
| Homolog of drosphila mothers against dpp 3 (SMAD3) | NM_005902 | 15q21 | 34 | |
| Homolog of drosphila mothers against dpp 4 (SMAD4) | NM_005359 | 18q21 | 5 | |
| Nitric oxide synthase 3 (NOS3) | NM_000603 | 7q36 | 10 | |
| Nuclear factor κB p100 subunit (NFKB2) | NM_001077493 | 10q24 | 5 | |
| Nuclear factor κB p105 subunit (NFKB1) | NM_003998 | 4q23 | 13 | |
| Nuclear factor κB p65 subunit (RELA) | NM_021975 | 11q13 | 4 | |
| Prostaglandin I2 synthase (PTGIS) | NM_000961 | 20q13 | 13 | |
| Protein kinase C, α (PRKCA) | NM_002737 | 17q22 | 33 | |
| Protein kinase C, β 1(PRKCB1) | NM_002738 | 16p11 | 13 | |
| Protein kinase C, γ (PRKCG) | NM_002739 | 19q13 | 5 | |
| Transforming growth factor, β-1 (TGFβ1) | NM_000660 | 19q13 | 5 | |
| V-AKT murine Thymoma viral oncogene homolog 1 (AKT1) | NM_005163 | 14q32 | 7 | |
| Blood vessel growth and development GO: 0001568 | Angiopoietin 1 (ANGPT1) | NM_001146 | 8q22 | 37 |
| Calcium-binding protein A4 (S100A4) | NM_019554 | 1q21 | 6 | |
| Endoglin (ENG) | NM_000118 | 9q34 | 15 | |
| Hypoxia-inducible factor 1, α subunit (HIF1A) | NM_001530 | 14q21 | 8 | |
| Plasminogen (PLG) | NM_000301 | 6q26 | 21 | |
| Runt-related transcription factor 1 (RUNX1) | NM_001754 | 21q22 | 58 | |
| Thrombospondin-1 (THBS1) | NM_003246 | 15q15 | 5 | |
| Tyrosine kinase with Ig and EGF Factor homology domains (TIE1) | NM_005424 | 1p34 | 8 | |
| Vascular endothelial growth factor (VEGF) | NM_00125366 | 6p12 | 7 | |
| Inflammation GO:0006954 | Complement component 4A (C4A) | NM_007293 | 6p21 | 4 |
| C-reactive protein (CRP) | NM_000567 | 1q21 | 8 | |
| Cytochrome b-245, NADPH Oxidase 2, NOX2 (CYBB) | NM_000397 | Xp21 | 6 | |
| Lipopolysaccharide binding protein (LBP) | NM_004139 | 20q11 | 7 | |
| Tumor necrosis factor (TNF) | NM_000594 | 6p21 | 5 | |
| Oxidation reduction GO: 0006979 | Dual oxidase 1 (DUOX1) | NM_017434 | 15q15 | 15a |
| Dual oxidase 2 (DUOX2) | NM_014080 | 15q15 | 15a | |
| NADPH Oxidase 1 (NOX1) | NM_007052 | Xq22 | 7 | |
| NADPH Oxidase 4 (NOX4) | NM_016931 | 11q14 | 19 | |
| Superoxide dismutase 1, soluble (SOD1) | NM_000454 | 21q22 | 3 | |
| Superoxide dismutase 2, mitochondrial (SOD2) | NM_00636 | 6q25 | 3 | |
| Xanthine dehydrogenase (XDH) | NM_00379 | 2p23 | 24 | |
| Tissue development GO:0009888 | Homolog of drosphila mothers against dpp 2 (SMAD2) | NM_005901 | 18q21 | 10 |
| Ikaros (IKZF1) | NM_006060 | 7p12 | 7 | |
| Peroxisome proliferator activated receptor, γ (PPARG) | NM_005037 | 3p25 | 13 | |
| Recombination signal-binding protein 1 for J-κ (RBPSUH) | NM_005349 | 4p15 | 13 | |
| Steroid hormone GO:0008202 GO:0030518 |
Aromatase (CYP19A1) | NM_000103 | 15q21 | 24 |
| Estrogen receptor 1 (ESR1) | NM_000125 | 6q25 | 36 | |
| Estrogen receptor 2 (ESR2) | NM_001437 | 14q24 | 14 | |
| Farnesoid × receptor (NR1H4) | NM_005123 | 12q | 7 | |
| Pregnane × receptor (NR1I2) | NM_003889 | 3q13 | 13 | |
| Sex hormone binding globulin (SHBG) | NM_001040 | 17p13 | 6 | |
| Small heterodimer partner (NR0B2) | NM_021969 | 1p36 | 5 | |
| Extracellular matrix structure and regulation GO:0043062 GO:0006508 |
Collagen, type XVIII, α-1 (COL18A1) | NM_130445 | 21q22 | 29 |
| Elastase 1 (ELA1) | NM_001971 | 12q13 | 8 | |
| Elastase 2 (ELA2) | NM_001972 | 19p13 | 4 | |
| Matrix metalloproteinase 2 (MMP2) | NM_004530 | 16q13 | 11 | |
| Matrix metalloproteinase 3 (MMP3) | NM_002422 | 11q23 | 6 | |
| Matrix metalloproteinase 9 (MMP9) | NM_004994 | 20q11 | 6 | |
| Proteinase inhibitor 3; elafin (PI3) | NM_002638 | 20q12 | 4 | |
| Tenascin C (TNC) | NM_002160 | 9q33 | 16 | |
| Coagulation GO:0050817 | Plasminogen activator inhibitor 1 (SERPINE1) | NM_000602 | 7q21 | 9 |
| Thrombomodulin (THBD) | NM_000361 | 20p11 | 4 | |
| Thromboplastin (HEMB) | NM_000133 | Xq27 | 11 | |
| Von Willebrand factor (VWF) | NM_000552 | 12p13 | 39 | |
| Serotonin GO:0006587 GO:0007210 |
Serotonin 2B receptor (HTR2B) | NM_000867 | 2q36 | 8 |
| Serotonin transporter (SLC6A4) | NM_001045 | 17q11 | 7 | |
| Tryptophan hydroxylase (TPH1) | NM_004179 | 11p15 | 8 | |
| Tryptophan hydroxylase 2 (TPH2) | NM_173353 | 12q21 | 16 | |
| Na/bile acid transporter GO:0008508 | Solute carrier family 10, member 1 (SLC10A1) | NM_003049.1 | 14q24 | 5 |
| Solute carrier family 10, member 2 (SLC10A2) | NM_000452.1 | 13q33 | 12 | |
| Metabolism GO:0008152 | 5,10-Methylenetetrahydrofolate reductase (MTHFR) | NM_005957 | 1p36 | 7 |
| Betaine-homocysteine methyltransferase (BHMT) | NM_001713 | 5q13 | 4 | |
| Cystathionine-β-synthase (CBS) | NM_000071 | 21q22 | 6 | |
| Peroxisome proliferator activated receptor, α (PPARA) | NM_005036 | 22q12 | 9 | |
| Retinoic acid signaling GO:0048384 | Retinoic acid receptor, α (RARA) | NM_000964 | 17q21 | 4 |
| Retinoic acid receptor, β (RARB) | NM_016152 | 3p24 | 29 | |
| Retinoic acid receptor, γ (RARG) | NM_000966 | 12q13 | 6 |
Chr, chromosome; SNP, single nucleotide polymorphism.
Indicates adjacent genes which were defined by a single genomic region and tagging SNPs. Thus the number of SNPs indicated refers to the total number of SNPs assayed in the region containing both genes.
Genotyping
Genomic DNA was isolated from peripheral leukocytes using standard procedures (Gentra Puregene; Qiagen, Valencia, CA). SNP genotyping was performed using the GoldenGate Assay (Illumina, Inc, San Diego, CA). SNP assays that failed to generate results in >10% of subjects were considered to have failed and not used for analyses.
Statistical Analysis
Continuous data were summarized using mean ± standard deviation or median (interquartile range), as appropriate. Categorical variables were summarized using number and percentage. To test for differences in covariates between cases and controls, Student t tests, Wilcoxon rank-sum tests, χ2 tests, and Fisher exact tests were used, as appropriate.
Genotype distributions were tested for consistency with expected Hardy–Weinberg equilibrium (HWE) proportions in controls. Single locus association analyses were assessed assuming an additive genetic model using multivariable logistic regression, with adjustment for race and smoking (previously associated with case status4). The association of genotype with case/control status was expressed with odds ratios (ORs). Potential population stratification within our sample was tested using multidimensional scaling using AIM.17 These analyses were performed in PLINK v1.02 (http://pngu.mgh.harvard.edu/purcell/plink/).18
For genes in which more than 1 SNP was associated with HPS, we identified linkage disequilibrium blocks containing 3 or more SNPs using Haploview 4.0.19 We used an expectation-maximization algorithm to estimate haplotypes. Association between disease status and haplotypes was assessed using a generalized linear model approach via the R package Haplo.stats.20 Both global tests of haplotype association and haplotype-specific analysis (providing ORs with respect to a referent haplotype) were conducted.
Principal component (PC) regression analysis was used to synthesize information across several SNPs within a gene in a gene-based approach.21,22 Each SNP was assigned a score based on the per-allele model, and PCs were constructed to be linear combinations of these scores. We used the PCs in a logistic regression model to investigate the association between each gene and case status. For each gene, we calculated PCs using the pcreg procedure in R.23
In a second gene-based approach, we used classification and regression trees (CART) to help select a small initial subset of interesting markers with high probability for further investigation.24 In the CART analysis, we specified a minimum group size of 7 and minimum splitting size of 20 in R. Furthermore, we conducted a Random Forests analysis, which creates an ensemble of CART trees using random two-thirds samples of the data then tests the tree with the remaining one third of the data.25 Missing data were replaced using the multiple imputation algorithm and the Random Forests algorithm.
There was 80% power to detect ORs of ≥ 1.91–3.92 (or ≤0.26–0.52), depending on the minor allele frequency of the SNP (0.05–0.45). Power analysis was performed using QUANTO 1.2.26 Because the main goal of this study was hypothesis generation, adjustment for multiple comparisons was not performed. P <.05 was considered significant for all analyses.
Results
There were 59 cases and 126 controls included in this analysis (Table 2). The mean age of the subjects was 53 ± 10 years, 39% were female, and the majority (83%) was non-Hispanic. The majority of subjects in both groups had liver disease because of hepatitis C infection (44%) or alcohol (41%). Subjects with HPS had a mean PaO2 of 75 ± 13 mm Hg and a median alveolar-arterial oxygen gradient of 25 mm Hg (interquartile range, 19–35 mm Hg).
Table 2.
Demographic and Clinical Data
| Variable | HPS (n = 59) | No HPS (n = 126) | P value |
|---|---|---|---|
| Age (y), mean ± SD | 53 ± 9 | 53 ± 10 | .71 |
| Female, n (%) | 28 (48) | 46 (37) | .23 |
| Race/ethnicity, n (%) | |||
| Non-Hispanic white | 53 (90) | 101 (80) | .02 |
| Hispanic white | 2 (3) | 16 (13) | |
| Non-Hispanic black | 1 (2) | 8 (6) | |
| Other | 3 (5) | 1 (1) | |
| Etiology of liver disease, n(%) | |||
| Alcohol | 23 (39) | 54 (43) | .62 |
| Hepatitis C infection | 26 (44) | 55 (44) | .96 |
| Nonalcoholic steatohepatitis | 8 (14) | 16 (13) | .87 |
| Cryptogenic cirrhosis | 7 (12) | 9 (7) | .29 |
| Autoimmune hepatitis | 2 (3) | 8 (6) | .72 |
| Primary scelrosing cholangitis | 2 (3) | 8 (6) | .51 |
| Hepatitis B infection | 0 (0) | 9 (7) | .06 |
| Primary biliary cirrhosis | 2 (3) | 4 (3) | 1 |
| Smoking, n (%) | 28 (48) | 81 (64) | .03 |
| MELD score, mean ± SD | 14 ± 4 | 13 ±5 | .7 |
| Intrapulmonary shunt, n (%) | 59 (100) | 56 (44) | <.0001 |
| Arterial blood gas | |||
| pH, mean ± SD | 7.44 ± 0.03 | 7.43 ± 0.04 | .05 |
| pCO2 (mm Hg), mean ± SD | 34 ± 4 | 35 ± 5 | .32 |
| pO2 (mm Hg), mean ± SD | 75 ± 13 | 90 ± 15 | <.0001 |
| Alveolar-arterial O2 gradient, mm Hg, median (IQR) | 25 (19–35) | 10 (4–16) | .0001 |
HPS, hepatopulmonary syndrome; MELD, Model for End-Stage Liver Disease; DLCOcorr, diffusing capacity of the lung for carbon monoxide corrected for hemoglobin (% predicted).
Of the 1086 SNPs genotyped in candidate genes, 3 assays failed, 13 SNPs were monomorphic, and 65 SNPs did not conform to HWE (P <.05), leaving 1005 SNPs in the analysis. Of the 61 AIM SNPs, 3 were out of HWE (P <.05) and were thus not used for assessment of population stratification. There was no evidence of population stratification in our study population based on these AIMs.
Single SNP Analysis
Forty-two SNPs in 21 genes were significantly associated with HPS after adjustment for race and smoking (Table 3). Thirty-two of these SNPs were clustered in 8 genes: Caveolin 3 (CAV3); Endoglin (ENG); NADPH Oxidase 4 (NOX4); Estrogen receptor 2 (ESR2); von Willebrand Factor (VWF); Runt-related transcription factor 1 (RUNX1); Collagen, type XVIII, α-1 (COL18A1); and Tyrosine kinase with immunoglobulin g and EGF Factor homology domains (TIE1). Polymorphisms associated with an increased risk of HPS included 2 CAV3 SNPs, rs237872 (OR, 2.75; 1.65–4.60, P =.0001) and rs237875 (OR, 2.11; 1.29–3.45, P =.003). In addition, a missense variant (R126C) in spermidine/spermine N1-acetyltrans-ferase family member 2 (SAT2), a regulator of Hypoxia-inducible factor 1, α subunit (HIF1A) activity, was associated with HPS (OR, 3.65; 1.43–9.31, P =.007).
Table 3.
Multivariable Logistic Regression Models for SNPs and the Risk of HPS, Adjusted for Race and Smoking
| Chr | Gene | SNP | Risk allele | Risk allele frequency | Per-allele OR | 95% CI | P value | ||
|---|---|---|---|---|---|---|---|---|---|
| Identification | Location | Cases | Controls | ||||||
| 1 | NPP | rs198388 | 3UTR flank | A | 0.37 | 0.49 | 0.58 | 0.35–0.94 | .027 |
| 1 | TIE1 | rs7527092 | Intron 1 | A | 0.51 | 0.39 | 1.72 | 1.05–2.82 | .030 |
| 1 | TIE1 | rs2991990 | Intron 14 | A | 0.36 | 0.46 | 0.58 | 0.35–0.97 | .039 |
| 1 | TIE1 | rs1199039 | Exon 18 | G | 0.36 | 0.47 | 0.60 | 0.36–0.98 | .041 |
| 1 | TIE1 | rs11210834 | Intron 22 | G | 0.34 | 0.23 | 1.83 | 1.06–3.15 | .029 |
| 3 | CAV3 | rs237872 | Intron 1 | A | 0.61 | 0.41 | 2.75 | 1.65–4.60 | .0001 |
| 3 | CAV3 | rs237875 | Intron 1 | G | 0.57 | 0.40 | 2.11 | 1.29–3.45 | .003 |
| 6 | ESR1 | rs1543403 | 3UTR flank | G | 0.32 | 0.44 | 0.59 | 0.37–0.94 | .027 |
| 8 | ANGPT1 | rs1283695 | Intron 1 | G | 0.13 | 0.22 | 0.52 | 0.28–0.99 | .046 |
| 9 | ENG | rs4836585 | Intron 1 | C | 0.06 | 0.16 | 0.38 | 0.15–1.00 | .049 |
| 9 | ENG | rs4837192 | Intron 1 | G | 0.05 | 0.15 | 0.35 | 0.14–0.89 | .027 |
| 11 | RELA | rs1466462 | 3UTR flank | G | 0.45 | 0.36 | 1.69 | 1.05–2.70 | .029 |
| 11 | NOX4 | rs2164521 | Intron 2 | A | 0.05 | 0.15 | 0.30 | 0.12–0.77 | .012 |
| 11 | NOX4 | rs585197 | 5UTR flank | G | 0.15 | 0.25 | 0.53 | 0.29–0.98 | .043 |
| 11 | TRPC6 | rs7931676 | Intron 1 | G | 0.36 | 0.25 | 1.66 | 1.02–2.72 | .043 |
| 12 | VWF | rs4764478 | Intron 45 | A | 0.25 | 0.17 | 1.86 | 1.06–3.27 | .030 |
| 12 | VWF | rs216902 | Exon 35 | A | 0.32 | 0.46 | 0.54 | 0.34–0.87 | .011 |
| 12 | VWF | rs216312 | Intron 27 | A | 0.56 | 0.41 | 1.68 | 1.06–2.66 | .028 |
| 12 | VWF | rs11609815 | Intron 24 | G | 0.35 | 0.24 | 1.75 | 1.03–2.97 | .039 |
| 12 | VWF | rs216330 | Intron 18 | C | 0.47 | 0.35 | 1.67 | 1.04–2.67 | .032 |
| 12 | VWF | rs11614912 | Intron 18 | G | 0.34 | 0.21 | 1.77 | 1.04–2.99 | .034 |
| 12 | VWF | rs10849378 | Intron 18 | A | 0.38 | 0.25 | 1.71 | 1.03–2.82 | .037 |
| 12 | VWF | rs11064004 | Intron 18 | C | 0.40 | 0.25 | 1.79 | 1.10–2.94 | .020 |
| 12 | VWF | rs1063856 | Exon 18 | G | 0.50 | 0.31 | 2.18 | 1.35–3.52 | .002 |
| 12 | VWF | rs980130 | Intron 13 | A | 0.46 | 0.31 | 1.85 | 1.15–2.97 | .011 |
| 12 | VWF | rs980131 | Intron 13 | A | 0.55 | 0.38 | 2.04 | 1.25–3.32 | .004 |
| 12 | ELA1 | rs4762041 | 3UTR flank | C | 0.37 | 0.26 | 1.66 | 1.02–2.70 | .043 |
| 14 | HIF1A | rs2301113 | Intron 9 | C | 0.15 | 0.27 | 0.53 | 0.29–0.97 | .039 |
| 14 | ESR2 | rs1256061 | Intron 7 | A | 0.55 | 0.41 | 1.74 | 1.09–2.78 | .020 |
| 14 | ESR2 | rs1256059 | Intron 7 | A | 0.35 | 0.45 | 0.60 | 0.38–0.96 | .032 |
| 14 | ESR2 | rs1256049 | Exon 6 | A | 0.08 | 0.03 | 3.20 | 1.08–9.46 | .036 |
| 14 | ESR2 | rs1256030 | Intron 2 | A | 0.37 | 0.49 | 0.58 | 0.36–0.93 | .024 |
| 15 | SMAD3 | rs6494636 | Intron 6 | C | 0.38 | 0.52 | 0.51 | 0.31–0.84 | .008 |
| 17 | SAT2 | rs13894 | Exon 6 | A | 0.12 | 0.04 | 3.65 | 1.43–9.31 | .007 |
| 17 | SHBG | rs6258 | Exon 4 | A | 0.04 | 0.00 | 10.35 | 1.15–93.02 | .037 |
| 17 | ACE | rs4311 | Intron 9 | G | 0.57 | 0.46 | 1.64 | 1.06–2.55 | .028 |
| 20 | PTGIS | rs6091000 | Intron 5 | G | 0.07 | 0.03 | 3.93 | 1.21–12.78 | .023 |
| 21 | RUNX1 | rs2248720 | Intron 4 | C | 0.56 | 0.43 | 1.83 | 1.13–2.96 | .015 |
| 21 | RUNX1 | rs2834726 | Intron 1 | G | 0.02 | 0.06 | 0.19 | 0.04–0.87 | .032 |
| 21 | COL18A1 | rs2838920 | Intron 2 | A | 0.07 | 0.16 | 0.42 | 0.18–0.98 | .044 |
| 21 | COL18A1 | rs7278425 | Intron 37 | A | 0.21 | 0.14 | 2.05 | 1.05–4.03 | .036 |
| 22 | PPARA | rs11090819 | Intron 6 | A | 0.14 | 0.10 | 2.23 | 1.02–4.86 | .044 |
Chr, chromosome; UTR, untranslated region; SNP, single nucleotide polymorphism; OR, odds ratio.
Other variants that were associated with the risk of HPS included 2 tightly linked (D′ = 0.969, r2 = 0.912) intron 1 SNPs in ENG, rs4836585 (OR, 0.38; 0.15-1.00, P =.49) and rs4837192 (OR 0.35, 0.14–0.89, P =.027). In addition, 2 NOX4 SNPs (rs585197 and rs2164521) were associated with case status in our subjects. Among genes in steroid hormone signaling pathways, 4 of 14 tested SNPs in ESR2 affected the risk for HPS, as did a steroid hormone binding globulin (SHBG) missense variant in exon 4 (P184L).
Finally, 10 of 11 associated SNPs in VWF conferred an increased risk for HPS (OR, 1.66–2.18). One of these SNPs, rs1063856, encodes a missense variant (T789A) in exon 18 previously demonstrated to associate with higher circulating levels of VWF.27 In our cohort, possession of the alanine allele was significantly associated with case status (OR, 2.18; 95% confidence interval [CI]: 1.35–3.52, P =.002).
Haplotype Analyses
The haplotype block 3 of CAV3, (Table 4, Supplementary Figure 1) was significantly associated with the risk for HPS (Global, P =.003). Haplotype-specific analyses demonstrated that possession of haplotype AGAAA confers the greatest increase in HPS risk (OR, 5.28; 95% CI: 2.02-13.82, P =.0009) in comparison with the most common haplotype. In VWF, haplotype block 6 (Table 4, Supplementary Figure 2) was significantly associated with HPS (Global, P =.008). In the individual haplotype analysis, the possession of the rare haplotype CGAGG was associated with a significantly lower risk of HPS (OR, 0.21; 95% CI: 0.06–0.76, P =.02).
Table 4.
Distribution of Haplotypes in CAV3 and VWF Associated With HPS
| Frequency | Case frequency | Control frequency | Odds ratio | 95% Confidence interval | |||
|---|---|---|---|---|---|---|---|
| Lower | Upper | P value | |||||
| Caveolin 3 | |||||||
| Block 3 (Global, P =.003) | |||||||
| A-G-A-A-G | 0.47 | 0.36 | 0.52 | Referent group | — | — | — |
| A-A-A-A-G | 0.05 | 0.03 | 0.06 | 0.81 | 0.20 | 3.17 | .76 |
| A-G-A-A-A | 0.07 | 0.12 | 0.04 | 5.28 | 2.02 | 13.82 | .0009 |
| A-G-G-C-A | 0.28 | 0.30 | 0.27 | 2.03 | 1.12 | 3.68 | .02 |
| T-G-A-A-A | 0.11 | 0.16 | 0.09 | 2.89 | 1.33 | 6.28 | .01 |
| Rare | 0.03 | 0.03 | 0.02 | 4.17 | 0.79 | 22.09 | .09 |
| VWF | |||||||
| Block 6 (Global, P =.008 | |||||||
| G-A-G-G-A | 0.43 | 0.46 | 0.41 | Referent group | — | — | — |
| C-G-A-A-G | 0.25 | 0.27 | 0.25 | 0.90 | 0.52 | 1.54 | .69 |
| C-G-A-G-A | 0.07 | 0.03 | 0.08 | 0.29 | 0.08 | 1.01 | .05 |
| C-G-A-G-G | 0.08 | 0.03 | 0.11 | 0.21 | 0.06 | 0.76 | .02 |
| C-G-G-G-A | 0.15 | 0.21 | 0.12 | 1.78 | 0.90 | 3.52 | .10 |
| Rare | 0.02 | 0.01 | 0.03 | 0.38 | 0.04 | 3.30 | .38 |
NOTE. Caveolin 3 haplotype block 3 is composed of the following 5 SNPs: rs13061909, rs4686300, rs237870, rs237871, rs237872. VWF haplotype block 6 is composed of the following 5 SNPs: rs216891, rs216893, rs216902, rs216905, rs216805.
VWF, von Willebrand Factor.
Gene-Based Analyses
Elastase 1 (ELA1) (P <.005), CAV3 (P <.04), BMPR2 (P <.03), and NFKB1 (P <.03) were all significantly associated with HPS in the PC analysis. In the CART analysis, the following SNPs were identified as being most predictive of HPS phenotype: rs2834650 (RUNX1), rs1800472 (TGFB1), rs11224779 (TRPC6), rs429342 (PRKCB1), and rs237870 (CAV3) (Figure 1). The Random Forests algorithm was run 1000 times, and the 3 most important SNPs identified in each iteration were recorded. The following SNPs were most frequently identified as being influential: rs2834650 (RUNX1), rs2274751 (TNC), rs3729904 (PRKCB1), rs1800472 (TGFB1), rs237872 (CAV3).
Figure 1.
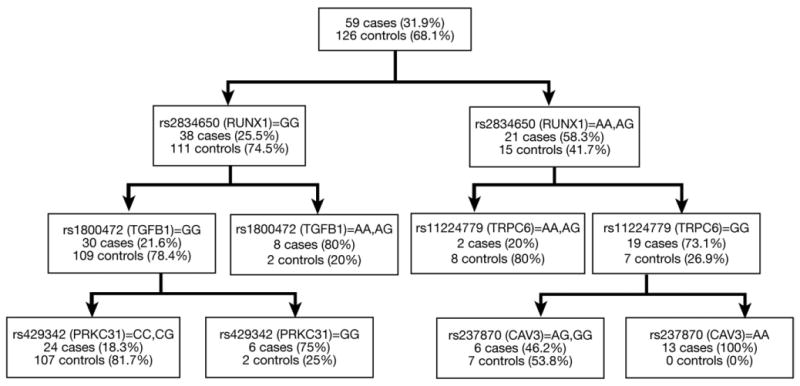
Classification and regression tree (CART). Each split in the tree maximizes the separation of cases and controls based on SNP genotypes.
Discussion
Using a hypothesis-generating approach, we have identified that the possession of common genetic variation in genes associated with vascular growth and development and estrogen action and signaling was associated with HPS in this case-control study. In contrast, we did not find any association between HPS and vasoregulatory genes such as nitric oxide, heme oxygenase, and the endothelin-B receptor, which have been specifically implicated in HPS.28–31 Our findings are in line with recent experimental results that demonstrate an important role for pulmonary angiogenesis in HPS.13
We have identified a number of genetic risk factors for HPS that modulate angiogenesis or vascular development. For example, endostatin, the proteolytic fragment of the C-terminus COL18A1, inhibits angiogenesis.32,33 In addition to the genetic association reported here, we have recently demonstrated that overexpression of endostatin in an animal model of HPS blocks the expansion of pulmonary microvessels as well as the oxygen diffusion impairment characteristic of that model.13 Endoglin is a transmembrane auxillary receptor for transforming growth factor (TGF)-β that is predominantly expressed on proliferating endothelial cells. Mutations in endoglin and activin receptor-like kinase 1 (ALK1), an endothelial specific TGF-β type I receptor, have been linked to hereditary hemorrhagic telangiectasia, an autosomal dominant vascular dysplasia characterized by telangiectasias and arteriovenous malformations.34,35 Interestingly, among patients with hereditary hemorrhagic telangiectasia, pulmonary arteriovenous malformations are significantly more likely in subjects with endoglin mutations.36 Last, TIE1, an endothelial specific receptor tyrosine kinase, is essential for the activation of TIE2 by vascular endothelial growth factor (VEGF), thus modulating vascular remodeling and blood vessel development.37
Low oxygen tension (hypoxia) is a potent stimulator of vascular growth and remodeling, and, in the pulmonary vasculature, oxygen sensing is critical for maintenance of normal gas exchange via adjustments in vascular tone. Four of the genes implicated here–HIFA1, SAT2, RUNX1 and NOX4–play central roles in oxygen-dependant vascular phenotypes. HIF1A stimulates endothelial cell angiogenesis under hypoxic conditions by activating the transcription of numerous transcription and growth factors38 and is regulated by SAT2.39 Variation in both genes was associated with HPS case status. RUNX1 is a hematopoetic transcription factor that contributes to the angio- and vasculogenic phenotype via its interaction with other transcription factors such as HIF1A and insulin growth factor binding protein 3.40–42 Last, NOX4 is one of the enzymes responsible for generation of reactive oxygen species in endothelial cells that modulate angiogenesis and has been implicated in hypoxia-induced proliferation.43 These results identify variation in specific genes that may contribute to susceptibility in HPS and be candidates for future studies.
Three specific signaling pathways–carbon monoxide, nitric oxide, and endothelin–have been implicated in pulmonary vasodilatation in experimental and human HPS. Increased production of the gaseous vasodilators nitric oxide and carbon monoxide has also been associated with vascular dilatation in HPS,30,44,45 and, thus, we tested variants in the inducible and endothelial forms of nitric oxide synthase (NOS) as well as heme oxygenase 1 (HMOX1), the rate-limiting enzyme in the production of carbon monoxide. A recent report found that the Glu298Asp (rs1799983) variant in NOS3 was associated with risk of HPS in 20 subjects with pediatric (predominately anatomic or metabolic) liver disease. We did not replicate this observation in our cohort (OR, 0.75; 95% CI: 0.43–1.31, P =.31). Altered endothelin signaling has been implicated in experimental HPS, with the liver producing increased circulating ET-1, which signals through up-regulated ET-B receptors on pulmonary endothelial cells.46 We analyzed SNPs in endothelin converting enzyme as well as both endothelin A and B receptors. Germ-line variation in none of these genes was associated with risk of HPS in our study population.
In addition to our single SNP analyses, we undertook gene- and pathway-based approaches to provide additional insight into the relationship between genotype and disease phenotype. Two genes with single SNP associations–CAV3 and RUNX1–were also identified in these analyses. CAV3 gene had an overall association with HPS using PC analysis, and SNPs from CAV3 were found in the CART and Random Forests approaches. A SNP from RUNX1 was identified as the most discriminating polymorphism (first split) in the CART tree, and this was confirmed by the Random Forests algorithm. Because these 2 genes were shown to be important using multiple methodologies, this provides stronger evidence that CAV3 and RUNX1 are associated with HPS. In addition to supporting these associations, these analyses also indicated 3 genes not found in the single SNP analysis—TGFB1, TNC, and TRPC6–may actually be associated with the disease.
There are several limitations to this study. First, the sample size was small, limiting our ability to find genetic alleles associated with HPS that were rare, had small effect sizes, or whose effect depended on gene-gene or gene-environment interaction. However, this is the largest reported epidemiologic study of HPS with strict case and control phenotypes and the first in HPS to employ high-throughput genotyping.
A fundamental challenge in high-throughput genetic analyses is the control of type I error. Given that we analyzed multiple SNPs for each of more than 90 genes, we can reasonably expect a certain number of statistically significant associations because of chance alone. We attempted to minimize the chance of “false-positives” by using a curated candidate gene list, thusly increasing the prior probability that one or more of these genes has mechanistic importance in HPS. There are commonly utilized frequentist methods to adjust for multiple comparisons in high-throughput studies, such as the Bonferroni correction and false discovery rate.47 Both methodologies assume that the association of each individual SNP with case status is entirely independent of those of the other SNPs. We have documented patterns of linkage disequilibrium between genotyped SNPs (data not shown). Because most accepted methods to account for multiple comparisons do not consider such relatedness, they are overly conservative for this purpose. We have therefore presented the results without adjustment and consider these results to be hypothesis generating. Whereas replication would be important, the biologic plausibility of our findings, the multiple gene “hits” in certain pathways, and the demonstration of association via both single loci and gene-based approaches is reassuring that type I error does not explain the findings.
In conclusion, our results implicate common genetic variation in the pathogenesis of HPS. Future studies should focus on replication in other populations and the mechanisms that explain the associations between the SNPs of interest and HPS.
Supplementary Material
Supplementary Table 1. Genotyped Single Nucleotide Polymorphisms
Supplementary Table 2. Pair-Wise Linkage Disequilibrium Between SNPs in HPS Candidate Genes
Supplementary Figures 1 and 2. Linkage disequilibrium structure of genes associated with heppatopulmonary syndrome. Pair-wise linkage disequilibrium (LD) between loci (D′ and r2) and haplotype structure were measured using Haploview 4.0.11 Here, LD was measured using all SNPs genotyped in the controls in this study. The strength of LD is depicted graphically for each pair-wise comparison (squares), such that white and blue represent low levels of LD, and red indicates high levels of LD (see color key). The SNPs are identified by their RS numbers and displayed relative to the candidate gene region. The display range of the chromosome (black line) corresponds to the genomic region of the candidate gene (roughly coding sequence ± 5–10 kilobases) targeted by this study. Exon/intron structure of the genes is indicated by thick/thin purple lines according to genome assembly hg17/May 2004. Annotated graphical images were generated using into LocusView 2.0.20
Acknowledgments
The authors thank May Huang; John Schlatterer; and John O'Connor, PhD, from the Irving Institute for Clinical and Translational Research at Columbia University for their technical assistance.
A listing of additional members of the Pulmonary Vascular Complications of Liver Disease Study Group can be found in Appendix 1.
Funding: Supported by NIH grants DK064103, DK065958, RR00645, RR00585, RR00046, RR00032, HL67771, HL089812 and, in part, under a grant with the Pennsylvania Department of Health, which specifically disclaims responsibility for any analysis, interpretations, or conclusions.
Abbreviations used in this paper
- 95% Cl
95% confidence interval
- AIM
Ancestry Informative Marker
- CART
classification and regression trees
- CAV3
Caveolin 3
- COL18A1
collagen, type XVIII, α-1
- ENG
endoglin
- ESR2
Estrogen receptor 2
- HIF1A
Hypoxia-inducible factor 1, α subunit
- HPS
hepatopulmonary syndrome
- HWE
Hardy-Weinberg equilibrium
- MELD
Model for End-stage Liver Disease
- NOX4
NADPH Oxidase 4
- OR
odds ratio
- PC
principal component regression analysis
- RUNX1
Runt-related transcription factor 1
- SAT2
Spermidine/spermine N1-acetyltransferase family member
- SHBG
Steroid hormone binding globulin
- SNP
single nucleotide polymorphism
- TIE1
Tyrosine kinase with Ig and EGF factor homology domains 1
- VWF
von Willebrand factor
Appendix: 1
Additional members of the Pulmonary Vascular Complications of Liver Disease Study Group are as follows: Columbia University College of Physicians and Surgeons: Evelyn M. Horn, MD; Jeffrey Okun, BA; Sonja Olsen, MD; Daniel Rabinowitz, PhD; Jenna Reinen, BS; Lori Rosenthal, NP; Debbie Rybak, BS. Mayo Clinic: Russell Wiesner, MD; Linda Stadheim, RN. University of Alabama: Raymond Benza, MD; J. Stevenson Bynon, MD; Devin Eckhoff, MD; Dorothy Faulk; Harpreet Singh; Rajasekhar Tanikella; Keith Wille, MD. University of Colorado: David Badesch, MD; Lisa Forman, MD; Ted Perry. The University of North Carolina at Chapel Hill: Roshan Shrestha, MD; Carrie Nielsen, RN. University of Pennsylvania School of Medicine: Vivek Ahya, MD; Michael Harhay, BS; Sandra Kaplan, RN; Harold Palevsky, MD; Rajender Reddy, MD; Darren Taichman, MD, PhD. University of Southern California: Neil Kaplowitz, MD.
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2010
References
- 1.Rodriguez-Roisin R, Krowka MJ, Herve P, et al. Pulmonary-hepatic vascular Disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 2.Fuhrmann V, Madl C, Mueller C, et al. Hepatopulmonary syndrome in patients with hypoxic hepatitis. Gastroenterology. 2006;131:69–75. doi: 10.1053/j.gastro.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Swanson KL, Wiesner RH, Krowka MJ. Natural history of hepatopulmonary syndrome: impact of liver transplantation. Hepatology. 2005;41:1122–1129. doi: 10.1002/hep.20658. [DOI] [PubMed] [Google Scholar]
- 4.Fallon MB, Krowka MJ, Brown RS, et al. Impact of hepatopulmonary syndrome on quality of life and survival in liver transplant candidates. Gastroenterology. 2008;135:1168–1175. doi: 10.1053/j.gastro.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: implications for liver transplantation. Clin Chest Med. 2005;26:587–597. doi: 10.1016/j.ccm.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Cremona G, Higenbottam TW, Mayoral V, et al. Elevated exhaled nitric oxide in patients with hepatopulmonary syndrome. Eur Respir J. 1995;8:1883–1885. doi: 10.1183/09031936.95.08111883. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Ling Y, Luo B, et al. Analysis of pulmonary heme oxygenase-1 and nitric oxide synthase alterations in experimental hepatopulmonary syndrome. Gastroenterology. 2003;125:1441–1451. doi: 10.1016/j.gastro.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Yol S, Erikoglu M, Toprak SS, et al. The effects of serum estrogen levels on hypoxemia and blood nitric oxide levels in experimental hepatopulmonary syndrome. Hepatol Res. 2005;33:7–13. doi: 10.1016/j.hepres.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Schenk P, Madl C, Rezaie-Majd S, et al. Methylene blue improves the hepatopulmonary syndrome. Ann Intern Med. 2000;133:701–706. doi: 10.7326/0003-4819-133-9-200011070-00012. [DOI] [PubMed] [Google Scholar]
- 10.Gomez FP, Barbera JA, Roca J, et al. Effects of nebulized N(G)-nitro-L-arginine methyl ester in patients with hepatopulmonary syndrome. Hepatology. 2006;43:1084–1091. doi: 10.1002/hep.21141. [DOI] [PubMed] [Google Scholar]
- 11.Lee JS, Semela D, Iredale J, et al. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 12.Schraufnagel DE, Malik R, Goel V, et al. Lung capillary changes in hepatic cirrhosis in rats. Am J Physiol. 1997;272:L139–L147. doi: 10.1152/ajplung.1997.272.1.L139. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Luo B, Tang L, et al. Pulmonary angiogenesis in a rat model of hepatopulmonary syndrome. Gastroenterology. 2009;136:1070–1080. doi: 10.1053/j.gastro.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fallon MB, R KE, Krowka MJ, et al. A multi-center case-control study of genetic predictors for hepatopulmonary syndrome (HPS) Boston, MA: American Association for the Study of Liver Diseases; 2007. [Google Scholar]
- 15.Lazaridis KN, Frank JW, Krowka MJ, et al. Hepatic hydrothorax: pathogenesis, diagnosis, and management. Am J Med. 1999;107:262–267. doi: 10.1016/s0002-9343(99)00217-x. [DOI] [PubMed] [Google Scholar]
- 16.Seldin MF, Shigeta R, Villoslada P, et al. European population substructure: clustering of northern and southern populations. PLoS Genet. 2006;2:e143. doi: 10.1371/journal.pgen.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Q, Yu K. Improved correction for population stratification in genome-wide association studies by identifying hidden population structures. Genet Epidemiol. 2008;32:215–226. doi: 10.1002/gepi.20296. [DOI] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 20.Schaid DJ, Rowland CM, Tines DE, et al. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauderman WJ, Murcray C, Gilliland F, et al. Testing association between disease and multiple SNPs in a candidate gene. Genet Epidemiol. 2007;31:383–395. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Abbott D. A principal components regression approach to multilocus genetic association studies. Genet Epidemiol. 2008;32:108–118. doi: 10.1002/gepi.20266. [DOI] [PubMed] [Google Scholar]
- 23.Team RDC. R: A language and environment for statistical computing. 2009 [Google Scholar]
- 24.Foulkes AS. Applied statistical genetics with R. New York: Springer; 2009. [Google Scholar]
- 25.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 26.Gauderman W, Morrison J. QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. Available at: http://hydra.usc.edu/gxe. Accessed.
- 27.Klemm T, Mehnert AK, Siegemund A, et al. Impact of the Thr789Ala variant of the von Willebrand factor levels, on ristocetin co-factor and collagen binding capacity and its association with coronary heart disease in patients with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes. 2005;113:568–572. doi: 10.1055/s-2005-872896. [DOI] [PubMed] [Google Scholar]
- 28.Fallon MB, Abrams GA, Luo B, et al. The role of endothelial nitric oxide synthase in the pathogenesis of a rat model of hepatopulmonary syndrome. Gastroenterology. 1997;113:606–614. doi: 10.1053/gast.1997.v113.pm9247483. [DOI] [PubMed] [Google Scholar]
- 29.Degano B, Mittaine M, Herve P, et al. Nitric oxide production by the alveolar compartment of the lungs in cirrhotic patients. Eur Respir J. 2009;34:138–144. doi: 10.1183/09031936.00148008. [DOI] [PubMed] [Google Scholar]
- 30.Carter EP, Hartsfield CL, Miyazono M, et al. Regulation of heme oxygenase-1 by nitric oxide during hepatopulmonary syndrome. Am J Physiol Lung Cell Mol Physiol. 2002;283:L346–L353. doi: 10.1152/ajplung.00385.2001. [DOI] [PubMed] [Google Scholar]
- 31.Luo B, Liu L, Tang L, et al. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: a potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556–563. doi: 10.1016/s0168-8278(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 32.O'Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 33.Digtyar AV, Pozdnyakova NV, Feldman NB, et al. Endostatin: current concepts about its biological role and mechanisms of action. Biochemistry (Mosc) 2007;72:235–246. doi: 10.1134/s0006297907030017. [DOI] [PubMed] [Google Scholar]
- 34.McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 35.Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 36.Sabba C, Pasculli G, Lenato GM, et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007;5:1149–1157. doi: 10.1111/j.1538-7836.2007.02531.x. [DOI] [PubMed] [Google Scholar]
- 37.Singh H, Milner CS, Aguilar Hernandez MM, et al. Vascular endothelial growth factor activates the Tie family of receptor tyrosine kinases. Cell Signal. 2009;21:1346–1350. doi: 10.1016/j.cellsig.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 39.Baek JH, Liu YV, McDonald KR, et al. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1α. J Biol Chem. 2007;282:23572–23580. doi: 10.1074/jbc.M703504200. [DOI] [PubMed] [Google Scholar]
- 40.Peng ZG, Zhou MY, Huang Y, et al. Physical and functional interaction of Runt-related protein 1 with hypoxia-inducible factor-1α. Oncogene. 2008;27:839–847. doi: 10.1038/sj.onc.1210676. [DOI] [PubMed] [Google Scholar]
- 41.Iwatsuki K, Tanaka K, Kaneko T, et al. Runx1 promotes angiogenesis by down-regulation of insulin-like growth factor-binding protein-3. Oncogene. 2005;24:1129–1137. doi: 10.1038/sj.onc.1208287. [DOI] [PubMed] [Google Scholar]
- 42.Suda T, Takakura N. Role of hematopoietic stem cells in angiogenesis. Int J Hematol. 2001;74:266–271. doi: 10.1007/BF02982059. [DOI] [PubMed] [Google Scholar]
- 43.Ismail S, Sturrock A, Wu P, et al. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{β}1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rolla G, Brussino L, Colagrande P, et al. Exhaled nitric oxide and oxygenation abnormalities in hepatic cirrhosis. Hepatology. 1997;26:842–847. doi: 10.1053/jhep.1997.v26.pm0009328302. [DOI] [PubMed] [Google Scholar]
- 45.Arguedas MR, Drake BB, Kapoor A, et al. Carboxyhemoglobin levels in cirrhotic patients with and without hepatopulmonary syndrome. Gastroenterology. 2005;128:328–333. doi: 10.1053/j.gastro.2004.11.061. [DOI] [PubMed] [Google Scholar]
- 46.Ling Y, Zhang J, Luo B, et al. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of hepatopulmonary syndrome in the rat. Hepatology. 2004;39:1593–1602. doi: 10.1002/hep.20244. [DOI] [PubMed] [Google Scholar]
- 47.Benjamini Y, Yekutieli D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 2005;171:783–790. doi: 10.1534/genetics.104.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Genotyped Single Nucleotide Polymorphisms
Supplementary Table 2. Pair-Wise Linkage Disequilibrium Between SNPs in HPS Candidate Genes
Supplementary Figures 1 and 2. Linkage disequilibrium structure of genes associated with heppatopulmonary syndrome. Pair-wise linkage disequilibrium (LD) between loci (D′ and r2) and haplotype structure were measured using Haploview 4.0.11 Here, LD was measured using all SNPs genotyped in the controls in this study. The strength of LD is depicted graphically for each pair-wise comparison (squares), such that white and blue represent low levels of LD, and red indicates high levels of LD (see color key). The SNPs are identified by their RS numbers and displayed relative to the candidate gene region. The display range of the chromosome (black line) corresponds to the genomic region of the candidate gene (roughly coding sequence ± 5–10 kilobases) targeted by this study. Exon/intron structure of the genes is indicated by thick/thin purple lines according to genome assembly hg17/May 2004. Annotated graphical images were generated using into LocusView 2.0.20