

Abstract
Nonalcoholic fatty liver disease (NAFLD) is characterized by insulin resistance, which results in elevated serum concentration of free fatty acids (FFAs). Circulating FFAs provide the substrate for triacylglycerol formation in the liver, and may also be directly cytotoxic. Hepatocyte apoptosis is a key histologic feature of NAFLD, and correlates with progressive inflammation and fibrosis. The molecular pathways leading to hepatocyte apoptosis are not fully defined; however, recent studies suggest that FFA-induced apoptosis contributes to the pathogenesis of nonalcoholic steatohepatitis. FFAs directly engage the core apoptotic machinery by activating the proapoptotic protein Bax, in a c-jun N-terminal kinase-dependent manner. FFAs also activate the lysosomal pathway of cell death and regulate death receptor gene expression. The role of ER stressand oxidative stress in the pathogenesis of nonalcoholic steatohepatitis has also been described. Understanding the molecular mediators of liver injury should promote development of mechanism-based therapeutic interventions.
Keywords: Bim, c-jun N-terminal kinase, oleic acid, palmitic acid, hepatocyte injury
Lipotoxicity is loosely defined as cellular toxicity observed in the presence of an abnormal accumulation of fat.1 Fat accumulation in the liver or hepatic steatosis is a morphologic feature of myriad conditions. This review focuses on the known mechanisms of cellular toxicity secondary to obesity-related fatty liver. Adipose tissue serves as the primary repository of caloric excess and undergoes numerous functional changes that characterize the obese,2,3 insulin resistant, adipocyte; starkly absent among these changes, especially in comparison to other target tissues, is adipocyte apoptosis. These adipocyte changes include enhanced lipolysis with release of free fatty acids [also known as nonesterified fatty acids (NEFA)],4 release of adipokines5,6 and inflammatory cytokines.7 These perturbations along with hepatocellular insulin resistance promote the development of hepatocyte steatosis, a condition recognized as nonalcoholic fatty liver disease (NAFLD). In NAFLD progressive triacylglycerol deposition exceeds the livers capacity to export very low-density lipoprotein particles (VLDL) and β-oxidation of free fatty acids (FFAs). Furthermore, a proportion of patients with NAFLD develop hepatic inflammation, known as nonalcoholic steatohepatitis (NASH), and can progress to cirrhosis with all its attendant complications.8 Possible mechanisms of liver injury in this syndrome include, but are not limited to, increased fatty acid oxidation and oxidative stress,9 alteration of cellular membrane fatty acid and phospholipid composition,10,11 alteration of cellular cholesterol content,12 disturbances in ceramide signaling,13 and direct free fatty acid toxicity.14 Recent developments have elucidated some of the cellular mechanisms mediating liver injury in NASH, and these are discussed in detail.
FREE FATTY ACIDS
There is unequivocal data supporting the role of circulating FFAs in the development of hepatic steatosis.15 FFAs, here and throughout this article, refer to long chain (16 carbons or more), saturated and unsaturated fatty acids. Elevated circulating FFAs occur in patients with NAFLD.16 Hepatic uptake of FFAs is unregulated, and in the face of elevated circulating levels, relentless hepatocyte uptake of these toxins occurs.17 FFAs are hydrophobic and can diffuse across cell membranes or be transported by fatty acid transport proteins (FATP) or the fatty acid transporter CD36.18,19 In particular FATP5 is expressed by hepatocytes and aids in the uptake of long chain FFAs.20 Mice lacking FATP5 partition lipids away from the liver to other tissues. Similarly, liver-fatty acid binding protein (FABP) is present in the cytosol of hepatocytes and in its absence hepatocytes are protected from saturated fat-induced triacylglycerol deposition.21 It is possible that alterations in expression of these proteins promote hepatocyte lipotoxicity in humans. Although these patterns have not been clarified in human NAFLD, the contribution of circulating FFAs to liver triacylglycerol composition has been studied elegantly by Donnelly et al. Hepatocyte triacylglycerol and circulating triacylglycerol-rich lipoprotein particles contain esterified fatty acids that can arise from three sources, including circulating FFAs, indirectly from dietary carbohydrates and amino acids via de novo lipogenesis, and dietary fatty acids. In this study, adipose tissue derived circulating FFAs contributed to 82% and 62% of the total circulating pool in the fasted and fed states, respectively, which, in turn, provided 59% and 62% of FFAs in liver and circulating triacylglycerol-rich lipoprotein particles, respectively.15 Thus, the bulk of hepatic triacylglycerol is derived from circulating FFAs, generated by adipocyte lipolysis, a feature of the insulin resistant, metabolic syndrome.
HEPATIC LIPIDS
The hepatic lipid composition of patients with nonalcoholic steatohepatitis and nonalcoholic fatty liver has recently been comprehensively profiled by Puri et al.12 In this study, consisting of 9 subjects in each group, total hepatic lipid content and triacylglycerol were markedly elevated compared with age, sex, and body mass index matched controls. Hepatic FFAs were unchanged across the spectrum of liver injury. Within triacylglycerol and diacylglycerol, the saturated fatty acid palmitic acid and the monounsaturated fatty acid oleic acid were elevated, reflecting, perhaps, their greater availability in the circulating FFA pool. In this study, circulating FFAs were not profiled. However, others have demonstrated elevated levels of circulating oleic acid and palmitic acid in patients with nonalcoholic fatty liver.22 In another study that profiled hepatic FFAs in patients with NASH, both oleic acid and palmitoleic acid were elevated.23 Taken together, these studies have demonstrated the marked increase in triacylglycerol in livers of patients with nonalcoholic fatty liver and NASH, and the contribution of circulating FFAs toward deposition of hepatic triacylglycerol. Liver FFAs were not found to be consistently elevated, perhaps, secondary to their efficient esterification by the hepatocyte to form triacylglycerol. Of note, no differences were observed in the lipidomic profile of patients with NAFLD compared with patients with NASH; one can conclude from this that other pathogenic mechanisms exist to account for the development of liver injury, that are independent of the metabolic alterations leading to steatosis.
Finally, considerable data now indicate that FFAs and not their esterified products mediate lipotoxicity. In some experimental systems, diversion of palmitic acid to triacylglycerol formation—such as by cotreatment with oleic acid or by overexpression of the enzyme stearoyl CoA desaturase-1 (SCD1) that catalyzes formation of oleic acid from stearic acid and palmitoleic acid from palmitic acid—saturated fatty acid toxicity can be abrogated.24,25 In fact, the absence of SCD1 led to diminished hepatic lipid accumulation, supporting the fact that monounsaturated fatty acids are preferentially incorporated into triacylglycerol. Genetic deletion of diacylglycerol acyltransferase 2, an enzyme responsible for intracellular free fatty esterification prevents cellular steatosis, and accentuates rather than attenuates FFA cytotoxicity.26
LIPOAPOPTOSIS
Apoptosis is a key morphologic and pathogenic feature of human NASH27-29; secondary to its association with excess lipid deposition, it is a form of lipoapoptosis. Apoptosis, or programmed cell death, is a form of highly regulated cell death, increasingly recognized for its pathogenic role in liver diseases. The readers are referred to recent reviews for detailed information on apoptotic pathways and their relevance in diverse liver diseases.30 Briefly, hepatocytes can undergo apoptosis via an extrinsic pathway activated by death ligands, Fas and tumor necrosis factor related apoptosis inducing ligand (TRAIL) or via an intrinsic pathway, which can be activated by intracellular stress of membrane-bound organelles, such as lysosomes, endoplasmic reticulum (ER), and mitochondria. On activation of apoptotic cascades, mitochondrial permeabilization occurs, leading to activation of caspases 3 and 7, cleavage of cellular targets, such as cytokeratin 18, and the characteristic apoptotic morphology. The M30 neoantigen is one protein released from apoptotic cells, on cleavage of cytokeratin 18 by caspase 3 at aspartate 396 position.31,32 Upstream of mitochondria, members of the Bcl-2 family of proteins serve as pro- and antiapoptotic regulators.33,34 At a mitochondrial level, Bak and Bax, the multidomain proapoptotic members of the Bcl-2 family, are required for mitochondrial permeabilization and release of proapoptotic proteins from the intermembrane space. Other proapoptotic proteins of the Bcl-2 family are Bid, Noxa, Puma, Bim, Bmf, Bik, Hrk, and Bad (known as BH-3 domain only, based on their structure), and antiapoptotic proteins are Bcl-2, Bcl-xL, A1, and Mcl-1.
Apoptosis was first reported in patients with NASH in liver biopsy specimens.27-29 The magnitude of hepatocyte apoptosis correlates with liver injury, with higher rates of apoptosis in patients with aspartate aminotransferase/alanine aminotransferase ratio >1. Furthermore, apoptosis was higher in patients with NASH, in comparison to patients with alcoholic steatohepatitis.27 This study went on to demonstrate that apoptosis also correlates with histologic inflammatory activity as well as fibrosis, as subjects with higher grades of inflammation and higher stages of fibrosis had higher apoptosis rates. In a serologic assay designed to specifically measure a marker of epithelial cell apoptosis (serum cytokeratin 18 fragments), it was demonstrated that this test reliably distinguished patients with NASH from patients with simple steatosis from controls.32 Thus, apoptosis is a key pathophysiologic feature of human nonalcoholic steatohepatitis. The factors underlying the progression of steatosis to steatohepatitis are not known; however, it is recognized that apoptosis correlates with steatohepatitis. It is possible that susceptibility to lipoapoptosis may be what separates patients with simple steatosis from steatohepatitis. Known cellular mediators of this lipoapoptosis are discussed here.
FREE FATTY ACID-INDUCED APOPTOSIS
Long chain FFAs (containing 16 carbons or more) of biologic relevance are further divided into saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids based on the presence of double bonds. The most abundant and well-studied FFAs in the context of apoptosis are palmitic acid, a 16 carbon length saturated fatty acid (C16:0), oleic acid, an 18 carbon length monounsaturated fatty acid (C18:1), stearic acid, a 18 carbon length saturated fatty acid (C18:0), and palmitoleic acid, a 16 carbon length monounsaturated fatty acid (C16:1).12,16,22 The polyunsaturated fatty acids are altered in NASH,23 and alter hepatic gene expression especially lipid metabolism and inflammatory gene expression; however, they have not proved to be directly toxic to hepatocytes and are not discussed further. In fact, when analyzed in detail, hepatocyte apoptosis and injury were independent of dietary polyunsaturated fatty acid content in an animal model.35
Saturated fatty acids can induce apoptosis in many different cell types. Palmitic acid induces apoptosis in pancreatic β cells,36,37 cardiac myocytes,38 microvascular endothelial cells,39 and hepatocytes, among many cell types. When individually studied in a reductionist model, the monounsaturated fatty acids, oleic acid, and palmitoleic acid lead to apoptosis as well,14 but this is minimal, compared with the saturated fatty acids. Indeed, in hepatocyte- and several nonhepatocyte cell lines oleic acid treatment or increased expression of SCD1 favors triacylglycerol deposition, which is associated with decreased toxicity, due to decreased cellular levels of the toxic saturated fatty acids.25,40
MITOCHONDRIAL PATHWAY
In hepatocytes, saturated fatty acids, palmitic acid, and stearic acid lead to a concentration- and time-dependent lipoapoptosis14 (Fig. 1). These toxic fatty acids activate the intrinsic apoptotic pathway, leading to Bax activation, mitochondrial permeabilization, release of cytochrome c and activation of caspases 3 and 7. Bax activation in this model is regulated by the proapoptotic protein Bim and C-jun N-terminal kinase (JNK) an intracellular stress kinase. JNK is activated by FFAs in proportion to their toxicity and indeed FFA-induced Bax activation and lipoapoptosis were JNK-dependent. Furthermore, in cell free systems, it has been demonstrated that palmitic acid promotes Bax-induced permeabilization of liposomes.41 Whether the altered and elevated fatty acids observed in NASH directly activate Bax is an interesting question, and not easily addressed in the complex in vivo milieu.
Figure 1.
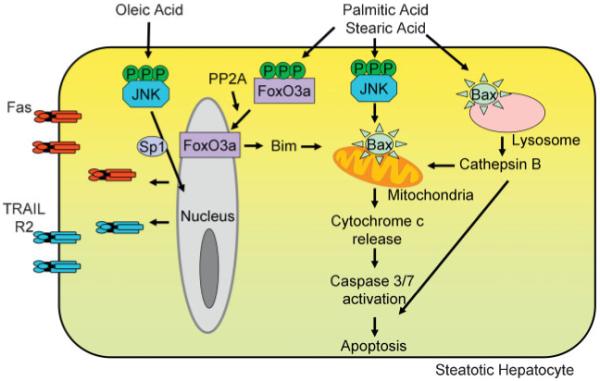
Free fatty acids (FFAs) induce hepatocyte apoptosis. FFAs can modulate both the extrinsic and the intrinsic pathways of hepatocyte apoptosis. The saturated fatty acids, palmitic acid and stearic acid, lead to c-jun N-terminal kinase (JNK) dependent activation of the proapoptotic protein Bax, which then leads to mitochondrial permeabilization with release of cytochrome c, activation of effector caspases, and apoptosis. Palmitic acid can also activate the lysosomal pathway of apoptosis, via Bax activation and Bax-dependent lysosomal permeabilization. Furthermore, palmitic acid and stearic acid activate protein phosphatase 2A (PP2A) leading to activation of FoxO3a, and transcriptional activation of the proapoptotic protein Bim. The monounsaturated fatty acid, oleic acid, which is minimally toxic per se, imparts sensitivity to the death receptor mediated extrinsic pathway of apoptosis. Fas and TRAIL-R2 expression is induced by oleic acid treatment. TRAIL-R2 expression is under the transcriptional control of JNK in oleic acid treated cells. This sensitizes fatty hepatocytes to circulating Fas or TRAIL.
BH-3 ONLY PROAPOPTOTIC PROTEINS
Bim expression was induced by the saturated fatty acids, palmitic acid and stearic acid, and was independent of JNK.14,42 Palmitic acid and stearic acid led to protein phosphatase 2A mediated dephosphorylation and activation of the transcription factor FoxO3a, a member of the Forkhead box-containing proteins, class O.42 Indeed, FoxO3a not only induced Bim expression, but palmitic acid and stearic acid toxicity could be abrogated by inhibition of FoxO3a activation or expression. Oleic acid, which was found to be minimally toxic, led to only very modest expression of Bim and FoxO3a activation, consistent with its lower toxicity. In neuronal cortical cells, oleic acid induces apoptosis via dephosphorylation of Bad, another potential mechanism for FFA-induced hepatocyte lipoapoptosis.43 The mechanism for protein phosphatase 2A activation was not elucidated in these studies; however, this phosphatase can be activated by ER stress in hepatocytes,44 and this is a likely mechanism for its activation during lipoapoptosis (vide infra).
LYSOSOMAL PATHWAY
FFAs can also activate the lysosomal pathway of apoptosis.45,46 Lysosomes are membrane-bound organelles that maintain an acidic intravesicular pH and contain hydrolytic enzymes that are active at an acidic to neutral range of pH. Cathepsin B is one of the family of lysosomal proteases known as cathepsins that is abundant and active at neutral pH. It is released into the cytosol on lysosomal permeabilization and can mediate downstream mitochondrial permeabilization and caspase activation.47 A combination of oleic acid and palmitic acid (2:1), which was minimally toxic per se, led to lysosomal permeabilization and release of cathepsin B secondary to FFA-induced Bax activation and translocation to lysosomes.46 Furthermore, in patients with NASH, lysosomal permeabilization and cathepsin B release correlate with the degree of inflammatory activity, and cathepsin B deficient mice were protected from diet-induced fatty liver despite development of obesity. Furthermore, lysosomal permeabilization was associated with nuclear factor κB (NF-κB) activation, which resulted in hepatocyte generation of tumor necrosis factor α (TNF-α). Thus, this study provides significant insight into the source of TNF-α in NASH. In a follow-up study, it was demonstrated that palmitic acid treatment alone could lead to Bax activation and lysosomal permeabilization.45 Palmitic acid treatment was also associated with decreased expression of the antiapoptotic protein Bcl-xL. Forced overexpression of Bcl-xL abrogated lysosomal permeabilization and apoptosis in palmitic acid treated cells.
ENDOPLASMIC RETICULUM
The ER is an intracellular membranous network that performs key cellular functions. Protein synthesis and processing (in association with ribosomes), lipid synthesis, carbohydrate metabolism, calcium sequestration, drug detoxification, and protein glycosylation are some of the functions of the ER. In situations of “ER stress,” key adaptive functions are activated by membrane sensors of ER stress and are collectively termed the unfolded protein response.48,49 These adaptive processes serve to overcome the stress stimulus; however, when faced with prolonged ER stress, apoptotic pathways are activated, prevail, and lead to cell death. The membrane transducers of ER stress are inositol-requiring protein-1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase RNA-like ER kinase (PERK). Their downstream mediators are mentioned here, as they serve as markers of ER stress. IRE1 activation can lead to JNK activation and endoribonucleolytic cleavage of X-box binding protein-1 (uXBP1) mRNA to its spliced form (sXBP-1), both of which are markers of ER stress. PERK activation phosphorylates eukaryotic translation initiation factor-2α (eIF2α), leading to selective translation of activating transcription factor-4 (ATF-4), transcription of C/EBP-homologous protein (CHOP) and activation of NF-κB, and growth arrest and DNA damage 34 (GADD34).
Early work in mouse models of genetic and dietary obesity demonstrated that ER stress was induced in the liver leading to JNK activation, along with other markers of ER stress.50 JNK activation led to serine phosphorylation of insulin receptor substrate 1 (IRS-1) resulting in insulin resistance. In an experimental model, mice fed a diet enriched in saturated fatty acids activated the ER stress pathway characterized by sXBP-1, increased BiP/GRP 78 (immunoglobulin binding protein), and CHOP protein had higher rates of apoptosis compared with mice fed a control diet or a diet enriched in polyunsaturated fatty acids.35 In a follow-up study, by dissecting the toxicity of individual fatty acids, it was demonstrated that the saturated fatty acids, palmitic acid and stearic acid activated the ER stress pathway and led to apoptosis in vitro; however, this was not observed in oleic acid or linoleic acid treated cells.51 Indeed, palmitic acid induced ER stress and toxicity were abrogated in oleic acid or linoleic acid treated cells. In human subjects with NAFLD and NASH, robust activation of PERK, as measured by phosphorylation of eukaryotic translation initiation factor-2α (p-eIF2α) was observed.52 However, downstream of this, ATF4, CHOP, and GADD34 activation was not uniformly observed. BiP/GRP 78, a major ER chaperone protein and regulator of the transducers of ER stress, mRNA levels were elevated in subjects with NASH, uXBP-1 mRNA was increased in NAFLD and sXBP-1 mRNA was decreased in NASH. These changes demonstrate the activation of ER stress pathways in human fatty liver; however, no consistent pattern of alterations was identified and actual protein content of these markers was not evaluated. Recognizing the limitations of human studies, this opens an area for future research, with detailed experimental studies to address the role of ER stress in the pathogenesis of NASH. Indeed, the patchy activation of some markers, poses the question, “Is NASH a heterogeneous group of disorders, at least at the mechanistic and molecular level?”
C-JUN N TERMINAL KINASE
JNK belongs to a family of intracellular mitogen activated protein (MAP) kinases; on activation it leads to phosphorylation and activation of transcription factors leading to the regulation of gene expression, such as immediate-early genes.53 It can also phosphorylate and regulate other proteins. For example, JNK mediated phosphorylation of Bcl-2 inactivates it and promotes apoptosis.54 JNK may also directly activate Bax, leading to apoptosis.55 In cellular models of saturated fatty acid induced toxicity, JNK mediates Bax activation and apoptosis, both of which are abrogated in the presence of JNK inhibitors.14 Of three known JNK genes, two are expressed in the liver, JNK1 and JNK2. In experimental murine dietary and genetic models of obesity, JNK is activated in the liver, along with adipose tissue and skeletal muscle,56 and JNK1 mediates obesity-induced insulin resistance. In murine methionine and choline deficient dietary model of steatohepatitis, JNK1 mediates the development of liver injury.57 JNK2 may also be involved in steatohepatitis, but its function is not obvious in the presence of JNK1.58 JNK activation is observed in liver samples from subjects with NASH.52 Subjects with NAFLD demonstrated JNK phosphorylation levels that were similar to control subjects.
DEATH RECEPTORS
Death receptors are cell surface receptors that belong to the tumor necrosis factor receptor gene superfamily.30 They are transmembrane proteins that bind their cognate ligands on the cell surface and signal cell death through conserved cytoplasmic death domains by recruitment of adaptor molecules. Tumor necrosis factor receptor 1 (TNFR-1) and tumor necrosis factor receptor 1 (TNFR-2) are expressed on hepatocytes, and are activated by tumor necrosis factor α. Only TNFR-1 is a complete receptor and can initiate intracellular apoptosis. On activation, TNFR-1 first activates NF-κB leading to transcriptional activation of its target genes, including proinflammatory genes and survival signals. Later, the receptor-ligand complex leads to initiation of apoptotic signaling if NF-κB mediated survival signals are inhibited. TNF-α can also lead to JNK activation, which, if sustained can promote cell death. Primary hepatocytes are resistant to the apoptotic effects of TNF-α; however, in the presence of protein synthesis inhibitors, or NF-κB inhibitors, hepatocytes will undergo TNF-α mediated cell death. Fas is expressed on hepatocytes, is activated on binding Fas ligand, and signals apoptotic cell death. Unlike TNF-α, Fas activation in itself leads to apoptotic cell death. Tumor necrosis factor related apoptosis inducing ligand (TRAIL) and two of its receptors TRAIL receptor 2 [TRAIL-R2/death receptor 5 (DR5)] and TRAIL receptor 1 [TRAIL-R1/death receptor 4 (DR4)] have been implicated in the pathogenesis of NASH.
TNF-α is a proinflammatory cytokine that can also induce cell death in susceptible cells. Obesity is a chronic inflammatory state characterized by adipose tissue macrophage infiltration and the release of many inflammatory mediators,59 including but not limited to TNF-α.7 Adipose tissue and circulating TNF-α levels are elevated in dietary obesity and lead to insulin resistance. TNF-α promotes the development of hepatic steatosis by increasing lipogenic gene expression,60 and pharmacologic or genetic inhibition of TNF-α signaling is associated with decreased hepatic steatosis.61,62 An apoptotic role for TNF-α in human NASH has not been observed. Fas expression is enhanced in livers of patients with NASH.27 In dietary murine models, such as the methionine and choline deficient diet, and the high sucrose diet, the induction of steatosis is accompanied by an increase in hepatic Fas expression.63,64 Increase in Fas expression confers sensitivity to Fas ligand mediated apoptosis. In a genetic murine model of obesity and fatty liver, steatotic hepatocytes were sensitized to Fas toxicity, however, this phenomenon occurred in the absence of enhanced Fas expression.65 In cell culture systems, steatotic cells induce the expression of Fas and are sensitized to its apoptosis inducing effect.63 Similarly, TRAIL-R2 expression is enhanced in free fatty acid treated cell lines.66 Even the minimally toxic fatty acid, oleic acid induces TRAIL-R2 expression and sensitizes cells to TRAIL-induced apoptosis. Steatotic cells are not sensitized to TNF-α toxicity.66 JNK activation transcriptionally regulates the oleic acid induced TRAIL-R2 upregulation and sensitization to TRAIL. In human samples, TRAIL-R2 and TRAIL-R1 expression was upregulated in steatotic livers.66,67 Though healthy human hepatocytes are thought to be resistant to TRAIL, steatosis sensitizes hepatocytes to TRAIL-induced apoptosis.67
CERAMIDE
Ceramides are complex lipids composed of sphingosine and fatty acid, synthesized in the ER from sphingosine and a fatty acid moiety, usually palmitoyl CoA.13 Long chain saturated fatty acid availability is the rate-limiting step in ceramide synthesis; therefore, in nutritional obesity with associated elevations of palmitic acid and stearic acid, excess ceramide synthesis is possible. Ceramide further combines with phosphorylcholine forming sphingomyelin, the major sphingophospholipid in humans. Ceramide can also be rapidly generated from sphingomyelin by sphingomyelinase induced hydrolysis. The role of ceramides in insulin resistance has been reviewed elsewhere.68 In a hematopoietic precursor cell line, palmitic acid and stearic acid treatment induced de novo ceramide synthesis, which was associated with apoptosis.69 Palmitic acid induced apoptosis was abrogated by pharmacologic inhibitors of de novo ceramide synthesis, and enhanced by promoting ceramide synthesis by preventing mitochondrial uptake of palmitoyl CoA. Though TNF-α and Fas can both activate sphingomyelinases leading to rapid accumulation of ceramide, and its regulation of apoptosis, the role of ceramide in the pathogenesis of human fatty liver is unclear. In a murine model, saturated fat diet induced obesity led to liver injury via ER stress and apoptosis, independent of ceramide.35 Palmitic acid induced lysosomal permeabilization was also ceramide independent, as was the induction of Bim expression by FoxO3a by palmitic acid and stearic acid.42,46 Basal sphingomyelin content was unchanged across the range of normal liver to fatty liver to steatohepatitis, though it cannot be excluded that in the Fas-induced apoptotic signaling cascade, ceramide may play a role.12,70
CHOLESTEROL
Analyses of lipid composition of human liver samples demonstrates a progressive increase in free cholesterol content in subjects with NAFLD and subjects with NASH, achieving a significant, almost 2-fold, increase in free cholesterol compared with controls.12 This is accompanied by an increase in serum total cholesterol in both conditions. Liver cholesterol ester levels are comparable in all three groups. In one rodent study designed to evaluate the role of dietary free cholesterol loading, rats fed a high cholesterol diet developed a microvesicular steatosis and were sensitized to the apoptotic effect of TNF-α, whereas, control-fed (Lombardi-diet) rats developed macrovesicular steatosis with no sensitization to TNF-α71. Furthermore, free cholesterol loading of the endoplasmic reticulum in this model was insufficient to induce ER stress. The observed sensitization to TNF-α in this study was secondary to reduced mitochondrial glutathione stores and could be corrected by S-adenosyl-L-methionine, which repletes mitochondrial glutathione levels. In atherosclerotic macrophages, free cholesterol accumulation leads to calcium depletion and ER stress with activation of the unfolded protein response and ER stress induced apoptosis.72,73 The role of free cholesterol in human nonalcoholic steatohepatitis, and its ability to induce ER stress requires further investigation.
OXIDATIVE STRESS
Oxidative stress, due to increased generation of reactive oxygen species and decreased antioxidant defenses is observed in human and experimental models of steatohepatitis.10,74,75 Enhanced mitochondrial and microsomal fatty acid β oxidation, cytochrome P450 (CYP) 2E1 (CYP2E1) induction, and leukocyte infiltration can all lead to oxidative stress. Several groups have studied markers of oxidative stress in liver samples as well as plasma samples from subjects with NAFLD and NASH. A significant increase in oxidative stress measured by oxidized cellular components, such as 3-nitrotyrosine was demonstrated in patients with both fatty liver and steatohepatitis; although patients with steatohepatitis had higher levels than steatosis alone.10 Similarly, HNE (4-hydroxy-2-noneal, a marker of lipid peroxidation) and 8-hydroxydeoxyguanosine (a marker of oxidative DNA damage) both progressively increased in subjects from simple steatosis to steatohepatitis.75 Thiobarbituric acid-reacting substrate (TBARS) or malondialdehyde levels, also a marker of lipid peroxidation are elevated in liver samples from subjects with NAFLD.76-78 Hepatic CYP2E1 levels are increased in patients with NAFLD,79 and in one study evaluating liver expression and plasma activity of CYP2E1 no difference was found between steatosis and steatohepatitis.80,81 In a large retrospective study of 167 patients (79 with simple steatosis and 24 with steatohepatitis), antibody levels against lipid peroxidation derived antigens were significantly elevated compared with controls.82 However, within the NAFLD cohort, antibody levels did not correlate either with extent of steatosis or with the presence and grade of inflammatory activity. Thus, oxidative stress may contribute to the development of both steatosis and steatohepatitis, though in the later condition in some studies the level of oxidative stress markers is higher than simple steatosis alone.
TOLL-LIKE RECEPTORS
Toll-like receptors (TLR) are a family of pattern recognition receptors that recognize microbial pathogens and respond by activating the innate immune system. In macrophage cell culture systems, the medium chain fatty acid, lauric acid (C12:0) can activate TLR4 and dimerized TLR2, whereas the polyunsaturated fatty acid, docosahexaenoic acid inhibits activation of all TLRs tested.83 TLR4 can also be activated by palmitic acid leading to activation of NF-κB, and upregulation of its target genes, TNF-α and interleukin (IL-) 6 in macrophages and adipocytes.84 Mice lacking TLR4 demonstrate greater obesity when fed a high fat diet, compared with wild-type controls, but are still partially protected against development of insulin resistance.85 Liver tissue from these mice demonstrates the absence of inflammatory gene expression induction by high fat diet, pointing to the role of TLR4 in the pathogenesis of obesity-related hepatic inflammation. In the methionine and choline-deficient dietary model of murine steatohepatitis, TLR4 activation results in greater systemic levels of TNF-α and IL-6 and IL-12.86 Mice lacking TLR4 demonstrate decreased liver injury, triacylglycerol accumulation, and inflammatory gene expression. Hepatocyte apoptosis and FFA levels were not determined in these studies.
CONCLUSIONS
NAFLD is characterized by insulin resistance that leads to the deposition of fat, predominantly triacylglycerol in the liver. The steatotic liver exhibits low-grade liver injury, hepatocyte apoptosis, and oxidative stress. However, a proportion of patients develops progressive liver injury, hepatocyte apoptosis, exhibits greater oxidative stress, and develops liver inflammation. Similar to other environmental disorders, such as lung cancer in smokers or alcoholic steatohepatitis, the factors that lead to the progression of steatosis to steatohepatitis, are likely multiple and complex. We propose this model of hepatocyte injury in fatty liver: in the susceptible steatotic hepatocyte, circulating FFA can activate myriad intracellular responses (Fig. 2). These include JNK activation, TLR4 activation, Bax activation, lysosomal permeabilization, and ER stress. If these changes are of sufficient magnitude, mitochondrial permeabilization and hepatocyte apoptosis ensue. In other hepatocytes, circulating FFA uptake can regulate gene expression, enhancing susceptibility to Fas and TRAIL induced apoptosis. In patients with NAFLD, hepatocyte apoptosis is significantly higher in patients with steatohepatitis as opposed to simple steatosis. Enhanced susceptibility to apoptosis in fatty hepatocytes such as by regulation of Bcl-2 family proteins or alterations in JNK activation is a possible explanation for the development of progressive steatohepatitis. Alternatively, the ER stress pathway may be preferentially activated in patients that develop progressive steatohepatitis. Better understanding of the molecular pathways of liver injury should promote development of diagnostic and therapeutic interventions aimed at reducing the morbidity and mortality associated with NAFLD.
Figure 2.
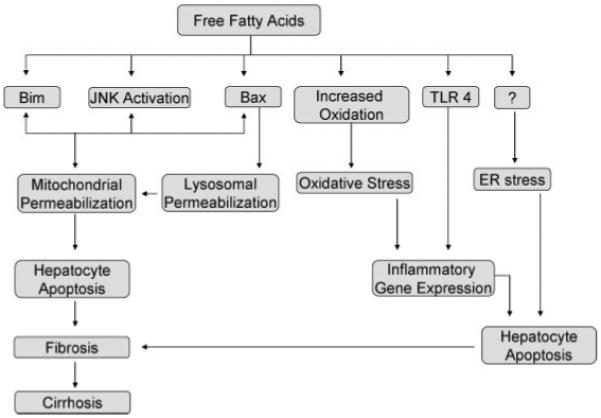
Free fatty acids (FFAs) and their molecular targets. FFA toxicity occurs at multiple molecular levels. Palmitic acid and stearic acid induce Bim expression, under transcriptional control of FoxO3a. Free fatty acids can activate c-jun N-terminal kinase (JNK), and the magnitude of induction correlates with their toxicity. JNK activation leads to Bax-induced mitochondrial permeabilization. Bax can also lead to lysosomal permeabilization and apoptosis. Increased oxidation of fatty acids promotes the formation of reactive oxygen species, resulting in antioxidant depletion and oxidative stress. Toll-like receptor 4 is activated by palmitic acid and oleic acid, thus regulating inflammatory gene expression. Endoplasmic reticulum stress is also a feature of human nonalcoholic fatty liver disease and can be induced by saturated fatty acids, palmitic acid and stearic acid; however, the mechanisms are unclear.
ACKNOWLEDGMENTS
Support for the work is provided by NIH grant DK 41876 and the Mayo Foundation.
The authors are grateful to Ms. Erin Nystuen-Bungum for excellent secretarial support.
ABBREVIATIONS
- ATF4
activating transcription factor-4
- ATF6
activating transcription factor 6
- CHOP
C/EBP-homologous protein
- CYP2E1
cytochrome P450 2E1
- eIF2α
eukaryotic translation initiation factor-2α
- ER
endoplasmic reticulum
- FATP
fatty acid transport protein
- FABP
fatty acid binding protein
- FFA
free fatty acid
- GADD 34
growth arrest and DNA damage 34
- HNE
4-hydroxy-2-noneal
- IN
interleukin
- IRE1
inositol-requiring protein-1
- IRS-1
insulin receptor substrate 1
- JNK
c-jun N-terminal kinase
- NAFLD
nonalcoholic fatty liver disease
- NEFA
nonesterified fatty acid
- NF-κB
nuclear factor κB
- PERK
protein kinase RNA-like ER kinase
- SCD1
stearoyl CoA desaturase-1
- TBARS
thiobarbituric acid-reacting substrate
- TLR
toll like receptor
- TNF-α
tumor necrosis factor α
- TNFR-1
tumor necrosis factor receptor 1
- TNFR-2
tumor necrosis factor receptor 2
- TRAIL
tumor necrosis factor related apoptosis inducing ligand
- VLDL
very low-density lipoprotein particle
- XBP
X-box binding protein-1
Footnotes
Fatty Liver Disease; Guest Editor, Arun J. Sanyal, M.B.B.S., M.D., Semin Liver Dis 2008;28:360–369., 333 Seventh Avenue, New York, NY 10001, USA. Tel: +1(212) 584-4662.
REFERENCES
- 1.Unger RH. Minireview: weapons of lean body mass destruction—the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 3.Gregor MF, Hotamisligil GS. Thematic review series: adipocyte biology. adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48(9):1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M, Sul HS. Regulation of triglyceride metabolism: IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol. 2007;293(1):G1–G4. doi: 10.1152/ajpgi.00554.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baranova A, Gowder SJ, Schlauch K, et al. Gene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistance. Obes Surg. 2006;16(9):1118–1125. doi: 10.1381/096089206778392149. [DOI] [PubMed] [Google Scholar]
- 6.Chitturi S, Farrell G, Frost L, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36(2):403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 7.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95(5):2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42(1):132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Kohjima M, Enjoji M, Higuchi N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20:351–358. [PubMed] [Google Scholar]
- 10.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Non-alcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 11.Bakan E, Yildirim A, Kurtul N, et al. Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006;43(4):109–113. doi: 10.1007/s00592-007-0224-4. [DOI] [PubMed] [Google Scholar]
- 12.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 13.Mari M, Fernandez-Checa JC. Sphingolipid signalling and liver diseases. Liver Int. 2007;27(4):440–450. doi: 10.1111/j.1478-3231.2007.01475.x. [DOI] [PubMed] [Google Scholar]
- 14.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281(17):12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 15.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci. 2001;46(11):2347–2352. doi: 10.1023/a:1012338828418. [DOI] [PubMed] [Google Scholar]
- 17.Bradbury MW. Lipid metabolism and liver inflammation: I. Hepatic fatty acid uptake—possible role in steatosis. Am J Physiol Gastrointest Liver Physiol. 2006;290(2):G194–G198. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- 18.Pohl J, Ring A, Hermann T, Stremmel W. Role of FATP in parenchymal cell fatty acid uptake. Biochim Biophys Acta. 2004;1686(1-2):1–6. doi: 10.1016/j.bbalip.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Pohl J, Ring A, Korkmaz U, Ehehalt R, Stremmel W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol Biol Cell. 2005;16(1):24–31. doi: 10.1091/mbc.E04-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doege H, Baillie RA, Ortegon AM, et al. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology. 2006;130(4):1245–1258. doi: 10.1053/j.gastro.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Newberry EP, Xie Y, Kennedy S, et al. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J Biol Chem. 2003;278(51):51664–51672. doi: 10.1074/jbc.M309377200. [DOI] [PubMed] [Google Scholar]
- 22.de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002;21(3):219–223. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 23.Allard JP, Aghdassi E, Mohammed S, et al. Nutritional assessment and hepatic fatty acid composition in non-alcoholic fatty liver disease (NAFLD): a cross-sectional study. J Hepatol. 2008;48(2):300–307. doi: 10.1016/j.jhep.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Wei Y, Wang D, Pagliassotti MJ. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol Cell Biochem. 2007;303(1-2):105–113. doi: 10.1007/s11010-007-9461-2. [DOI] [PubMed] [Google Scholar]
- 25.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100(6):3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with non-alcoholic steatohepatitis. Hepatology. 2007;45(6):1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 27.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 28.Susca M, Grassi A, Zauli D, et al. Liver inflammatory cells, apoptosis, regeneration and stellate cell activation in non-alcoholic steatohepatitis. Dig Liver Dis. 2001;33(9):768–777. doi: 10.1016/s1590-8658(01)80694-0. [DOI] [PubMed] [Google Scholar]
- 29.Ribeiro PS, Cortez-Pinto H, Sola S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99(9):1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 30.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43(suppl 1):S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 31.Grassi A, Susca M, Ferri S, et al. Detection of the M30 neoepitope as a new tool to quantify liver apoptosis: timing and patterns of positivity on frozen and paraffin-embedded sections. Am J Clin Pathol. 2004;121(2):211–219. doi: 10.1309/UK62-1LFJ-4FX0-7KDE. [DOI] [PubMed] [Google Scholar]
- 32.Wieckowska A, Zein NN, Yerian LM, et al. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44(1):27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 33.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121(5):671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 34.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147(2):943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 36.Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998;95(5):2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karaskov E, Scott C, Zhang L, et al. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology. 2006;147(7):3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 38.Kong JY, Rabkin SW. Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: prevention by cyclosporin A. Biochim Biophys Acta. 2000;1485(1):45–55. doi: 10.1016/s1388-1981(00)00028-7. [DOI] [PubMed] [Google Scholar]
- 39.Chai W, Liu Z. p38 mitogen-activated protein kinase mediates palmitate-induced apoptosis but not inhibitor of nuclear factor-kappaB degradation in human coronary artery endothelial cells. Endocrinology. 2007;148(4):1622–1628. doi: 10.1210/en.2006-1068. [DOI] [PubMed] [Google Scholar]
- 40.Miller TA, LeBrasseur NK, Cote GM, et al. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem Biophys Res Commun. 2005;336(1):309–315. doi: 10.1016/j.bbrc.2005.08.088. [DOI] [PubMed] [Google Scholar]
- 41.Epand RF, Martinou JC, Montessuit S, Epand RM. Fatty acids enhance membrane permeabilization by pro-apoptotic Bax. Biochem J. 2004;377(Pt 2):509–516. doi: 10.1042/BJ20030938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barreyro FJ, Kobayashi S, Bronk SF, et al. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282(37):27141–27154. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, Schwarz S, Ahlemeyer B, et al. Oleic acid causes apoptosis and dephosphorylates Bad. Neurochem Int. 2005;46(2):127–135. doi: 10.1016/j.neuint.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Christen V, Treves S, Duong FH, Heim MH. Activation of endoplasmic reticulum stress response by hepatitis viruses upregulates protein phosphatase 2A. Hepatology. 2007;46(2):558–565. doi: 10.1002/hep.21611. [DOI] [PubMed] [Google Scholar]
- 45.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1339–G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40(1):185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 47.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23(16):2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 48.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 49.Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol. 2006;45(2):321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 51.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291(2):E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 52.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 53.Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773(8):1341–1348. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19(12):8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281(30):21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 56.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 57.Schattenberg JM, Singh R, Wang Y, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43(1):163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 58.Tuncman G, Hirosumi J, Solinas G, et al. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2006;103(28):10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Solomon SS, Usdan LS, Palazzolo MR. Mechanisms involved in tumor necrosis factor-alpha induction of insulin resistance and its reversal by thiazolidinedione(s) Am J Med Sci. 2001;322(2):75–78. doi: 10.1097/00000441-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 61.Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37(2):343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 62.Barbuio R, Milanski M, Bertolo MB, Saad MJ, Velloso LA. Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J Endocrinol. 2007;194(3):539–550. doi: 10.1677/JOE-07-0234. [DOI] [PubMed] [Google Scholar]
- 63.Feldstein AE, Canbay A, Guicciardi ME, et al. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39(6):978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 64.Inoue Y, Asanuma T, Smith N, et al. Modulation of Fas-FasL related apoptosis by PBN in the early phases of choline deficient diet-mediated hepatocarcinogenesis in rats. Free Radic Res. 2007;41(9):972–980. doi: 10.1080/10715760701447322. [DOI] [PubMed] [Google Scholar]
- 65.Siebler J, Schuchmann M, Strand S, et al. Enhanced sensitivity to CD95-induced apoptosis in ob/ob mice. Dig Dis Sci. 2007;52(9):2396–2402. doi: 10.1007/s10620-006-9148-7. [DOI] [PubMed] [Google Scholar]
- 66.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56(8):1124–1131. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volkmann X, Fischer U, Bahr MJ, et al. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology. 2007;46(5):1498–1508. doi: 10.1002/hep.21846. [DOI] [PubMed] [Google Scholar]
- 68.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45(1):42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Paumen MB, Ishida Y, Muramatsu M, Yamamoto M, Honjo T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J Biol Chem. 1997;272(6):3324–3329. doi: 10.1074/jbc.272.6.3324. [DOI] [PubMed] [Google Scholar]
- 70.Tepper AD, Cock JG, de Vries E, Borst J, van Blitterswijk WJ. CD95/Fas-induced ceramide formation proceeds with slow kinetics and is not blocked by caspase-3/CPP32 inhibition. J Biol Chem. 1997;272(39):24308–24312. doi: 10.1074/jbc.272.39.24308. [DOI] [PubMed] [Google Scholar]
- 71.Mari M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4(3):185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 72.Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5(9):781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 73.Devries-Seimon T, Li Y, Yao PM, et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol. 2005;171(1):61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chalasani N, Deeg MA, Crabb DW. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99(8):1497–1502. doi: 10.1111/j.1572-0241.2004.30159.x. [DOI] [PubMed] [Google Scholar]
- 75.Seki S, Kitada T, Sakaguchi H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol Res. 2005;33(2):132–134. doi: 10.1016/j.hepres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 76.Madan K, Bhardwaj P, Thareja S, Gupta SD, Saraya A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD) J Clin Gastroenterol. 2006;40(10):930–935. doi: 10.1097/01.mcg.0000212608.59090.08. [DOI] [PubMed] [Google Scholar]
- 77.Yesilova Z, Yaman H, Oktenli C, et al. Systemic markers of lipid peroxidation and antioxidants in patients with non-alcoholic fatty liver disease. Am J Gastroenterol. 2005;100(4):850–855. doi: 10.1111/j.1572-0241.2005.41500.x. [DOI] [PubMed] [Google Scholar]
- 78.Bonnefont-Rousselot D, Ratziu V, Giral P, et al. Blood oxidative stress markers are unreliable markers of hepatic steatosis. Aliment Pharmacol Ther. 2006;23(1):91–98. doi: 10.1111/j.1365-2036.2006.02719.x. [DOI] [PubMed] [Google Scholar]
- 79.Chalasani N, Gorski JC, Asghar MS, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37(3):544–550. doi: 10.1053/jhep.2003.50095. [DOI] [PubMed] [Google Scholar]
- 80.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27(1):128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 81.Chtioui H, Semela D, Ledermann M, Zimmermann A, Dufour JF. Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007;27(6):764–771. doi: 10.1111/j.1478-3231.2007.01524.x. [DOI] [PubMed] [Google Scholar]
- 82.Albano E, Mottaran E, Vidali M, et al. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005;54(7):987–993. doi: 10.1136/gut.2004.057968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee JY, Zhao L, Youn HS, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279(17):16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- 84.Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005;25(10):2062–2068. doi: 10.1161/01.ATV.0000183883.72263.13. [DOI] [PubMed] [Google Scholar]
- 85.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29(suppl):140S–145S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]