

Summary
Central to the epigenetic regulation of chromatin remodeling are the histone-modifying enzymes which catalyze reversible lysine acetylation and methylation. From the early discovery of histone deacetylase inhibitors to the more recent identification of histone demethylase blockers, chemical approaches offer increasingly sophisticated tools for the interrogation of the structure and function of these lysine-modifying enzymes. This review will summarize progress to date on compounds identified from screens or by design that can modulate the activity of classical histone deacetylases, sirtuins, histone acetyltransferses, histone methyltransferases, and histone demethylases. We will highlight applications of compounds to mechanistic and functional studies involving these enzymes and discuss future challenges regarding target specificity and general utility.
Background
Ever since it was recognized that our DNA is packaged in complex nucleosomal structures, containing an octamer of histones H2A, H2B, H3, and H4, there has been great interest in elucidating the factors which govern DNA accessibility to transcription, replication, and repair.1 One of the factors that regulates chromatin remodeling is covalent modification of histones. The reversible post-translational modifications (PTMs) of histones have emerged as critical to the regulation of gene expression and the field of epigenetics.2 Although histones are subject to a myriad of PTMs including phosphorylation, ubiquitination, glycosylation on various residues, there has been a focus in the chromatin remodeling community on lysine acetylation and methylation (Figs. 1 and 2). Initial histone acetylation studies were concentrated on amino-terminal modifications.3 However, the discovery of histone εN-Lys methylation4 and εN-acetylation5 in the 1960s has led to steadily increasing interest in the structural and functional implications of these epigenetic marks.
Figure 1.

Reversible histone acetylation catalyzed by histone acetyltransferases (HATs), classical histone deacetylases (HDACs), and sirtuins (Sir2s). Transferred acetyl group is highlighted in blue. R = 3',5'-adenosine diphosphate; R1 = adenosine 5'-diphosphate.
Figure 2.

Reversible histone methylation catalyzed by histone methyltransferases, LSD1 demethylase, and Jmj demethylases. Transferred methyl group highlighted in red. R = methyl or hydrogen; R1 = ribose-adenosine 5'-diphosphosphate.
During the 70's, 80's, and early 90's, efforts to understand the ramifications of specific PTMs localized to the histone tails were pursued, and site-specific antibody reagents were developed to attempt to elucidate the function of the `histone code' using chromatin immunoprecipitation (CHIP).6 In general terms, histone acetylation has been associated with transcriptional activation whereas methylation appears to be more dependent on the modification site involved. For example, within histone H3, Lys4 methylation is associated with gene activation whereas Lys9 and Lys27 methylation are associated with gene repression.6 Histone H3 Lys9 acetylation is a common mark for transcriptional activation.6
Over the past twelve years, many of the specific enzymes that catalyze reversible lysine acetylation and methylation have been molecularly identified. There is intense interest in understanding the structures, functions, and regulatory mechanisms of these enzymes and their potential as drug targets for a range of diseases. Chemical tools and concepts have played key roles in the analysis. In this review, we discuss some of the exciting advances made over the past decade in the chemical biology of histone lysine acetylation and methylation enzymes with a special emphasis on the development and application of synthetic modulators of their catalytic functions.
Histone lysine acetylation and methylation enzyme overview
After decades of searching, the first nuclear histone acetyltransferase (HAT) and histone deacetylase were reported in 1996.7,8 The nuclear HAT, GCN5, was identified by purification of this activity from Tetrahymena.7 Use of an in-gel HAT assay furnished sufficient material for protein identification, revealing the enzyme to be GCN5.7 GCN5 catalyzes the transfer of the acetyl group from acetyl-CoA directly to Lys side chains (Fig. 1). GCN5 was already known as a transcriptional coactivator so this discovery was very exciting to the field. GCN5's enzymatic activity could be understood as the effector function of its gene regulatory action. It also turned out that the HAT domain of GCN5 shows low, but detectable homology to a large superfamily of acetyltransferases that include other HATs (HAT1, Myst), now known as the GNAT superfamily, involved in antibiotic resistance, melatonin regulation, and polyamine metabolism.9 These GNATs are conserved structurally with respect to CoA binding but utilize a range of catalytic residues for acetyl transfer.9 There are more than 50 GNATs thought to be present in the human genome but the function of many has not been elucidated.
Simultaneous with the experimental linkage of GCN5 to HAT activity, it was shown that yeast Rap5 possesses histone deacetylase activity (Fig. 1).8 This work was based on prior discoveries that the actions of two cancer cell killing natural products, trapoxin B and trichostatin A (see Fig. 3), involved blocking histone deacetylase activity, the former irreversibly presumably by epoxide-induced enzyme alkylation.10,11 Using trapoxin B as bait, it was demonstrated that the histone deacetylase (HDAC) activity was associated with the Rap5 protein,8 a Zn-dependent hydrolase, mechanistically akin to the large class of metalloproteases.12 As Rap5 was a component of a transcriptional silencing complex, this discovery, like that of GCN5, was well-received by the molecular biology community as such an acetyl-cleaving enzymatic activity would be predicted to show a gene repression phenotype. Humans encode eleven classical (Rap5-related, class I and class II) HDACs13 which have been identified based on significant sequence homology in the catalytic domains.
Figure 3.

Selected classical histone deacetylase inhibitors. Warhead features highlighted in red.
Four years after the discovery of the classical HDACs, it was demonstrated in 2000 that the yeast gene silencer Sir2 catalyzed histone deacetylation, in an NAD-dependent mechanism (Fig. 1).14 The Sir2 protein, which demonstrated weak homology to NAD-utilizing enzymes, was initially thought to be as an ADP ribosyltransferase.15 However, it was ultimately revealed that nicotinamide displacement from NAD was coupled to N-acetyl cleavage in a mechanistically unprecedented fashion (Fig. 1).14 Unlike classical HDACs, no metal is required for the Sir2 (sirtuin) enzymes. As with classical HDACs, the linkage of a deacetylase action to a general transcriptional repressive function has been consonant with the general understanding of histone acetylation enhancing gene expression.16 There are seven sirtuin (class III HDACs) human homologs known.16
The first histone lysine methyltransferase Su(var)3–9 was reported in 2000.17 The action of this enzyme was elucidated based on its homology to plant methyltransferases in its SET domain. These SET methyltransferases utilize S-adenosylmethionine (SAM) as a co-substrate and catalyze the transfer of one or more methyl groups to the Lys side chain amino groups (Fig. 2). Its ability to target histone H3 Lys9 methylation along with its transcriptional repressor function led to general acceptance of Su(var)3–9 as an epigenetic modifier. A range of more than 50 SET domain containing histone methyltransferases are thought to exist in mammals with differing site specificities and distinctiveness from the arginine methyltransferase proteins.18
The reversibility of the lysine methylation mark was uncertain until 2004 when the first histone demethylase, LSD1 was described.19 LSD1, a component of the CoREST/HDAC transcriptional repressor complex, was initially thought to be a polyamine oxidase based on homology to this flavin-dependent family. After failing to detect enzymatic processing of polyamine substrates, LSD1 was shown to catalyze the oxidative demethylation of histone H3 Lys4-Me (Fig. 2).19 As H3 Lys4 methylation is a gene activation mark, the LSD1 demethylase activity could be understood as linked to its gene regulatory function. Following the discovery of LSD1, in 2006 a new class of non-heme Fe, α–ketoglutarate-dependent histone demethylases, the Jmj family, was reported (Fig. 2).20 Mechanistically related to the prolyl-hydroxylase enzymes, the Jmjs, have been suggested to have distinct site-specificities, although their biochemical characterization remains sparse. At least eleven mammalian Jmjs have been reported to be histone demethylases.21
Histone deacetylase chemical probes
As mentioned, the natural products trapoxin B and trichostatin A (Fig. 3) played a key role in the discovery of the metallohydrolase (classical) HDACs.8,10,11 We now know that most or all of the eleven classical human HDACs studied biochemically appear to be potently inhibited by trapoxin B and TSA.13,22 Insights into the structural basis of inhibition of classical HDACs by small molecules have come from crystallographic studies on a bacterial HDAC homolog and more recently HDAC8 and HDAC7.12,23,24 These X-ray co-crystal structures have shown that the hydroxamic acid functionalities of trichostatin A and the clinically used SAHA coordinate the Zn ion in the active site.12,23,24
Various synthetic inhibitors of classical HDACs have been widely used as biological tools for epigenetic studies and some have advanced to clinical studies.13,22,25,26 In particular, many of these compounds show anti-tumor properties.13,22,25,26 The precise basis for the anti-neoplastic effects are not well-understood. In some cases, the HDAC inhibitor effects may be inducing cellular re-expression of tumor suppressor genes.25 Consistent with this idea, DNA methyltransferase inhibitors, which also often stimulate transcription of silenced genes, can in some cases synergize with HDAC inhibitors to kill cancer cells.25
Generally speaking, analogs of trichostatin A, trapoxin B, and SAHA have lacked specificity among the HDACs; however, there are some synthetic derivatives (Fig. 3) which display at least modest selectivity.26 For example, the hydroxamic acid compound tubacin (Fig. 3) shows 70-fold selectivity for blocking the tubulin deacetylase HDAC 6 versus inhibiting histone deacetylation in A549 cells.26 In contrast, the aminoanilide histacin (Fig. 3) and related derivatives show marked selectivity for inhibiting histone deacetylation versus tubulin deacetylation.26 Given the rather highly conserved active sites of these enzymes, it may be very difficult to identify compounds that demonstrate exquisite specificity for each classical HDAC homolog.27 However, obtaining specificity may ultimately prove to be clinically important as the functions of individual HDAC isoforms are revealed. For example, RNAi studies suggest that HDAC 5 and HDAC 9 are especially important in cardiac development whereas HDAC4 is proposed to play an important role in skeletal remodeling.22 The specificity of tubacin has made this a valuable reagent in molecular biology research. Consistent with the suggested role for HDAC6 in the aggresome function in degrading misfolded proteins, tubacin was shown to synergize with the proteasome inhibitor bortezomib in enhancing apoptosis of multiple myeolma cells.28 In a different study, HDAC6 inhibition by tubacin revealed a role for tubulin acetylation in the dynamics of cellular adhesion.29 Recent studies on HDAC inhibitors have been directed at their use in activity-based proteomic studies and have been able to probe the activity of HDAC1 in cancer after chemotherapeutic treatment.30
Sirtuin Modulators
The Sir2 proteins have been implicated in the regulation of aging since overexpression of yeast Sir2 increases longevity in model organisms.31 Different theories have been proposed to account for the Sir2 enzymes in modulating the aging phenotype.31,32 It has been hypothesized that the redox/energetic state of the cell, which correlates with the ratio of NAD/NADH, will govern the cellular Sir2 deacetylase activity, which in turn will modulate gene expression.31,32 Other Sir2 regulatory models have considered the rate of biosynthesis of the NAD cofactor and how this might affect deacetylase action and aging.31,32
Regardless of the precise mechanism, there has been a search for small molecule activators of Sir2 within an atmosphere of exaggerated claims that such compounds may offer admission to the fountain of youth.33 In general, small molecules that stimulate rather than inhibit enzymes are hard to identify because many enzymes have evolved to be excellent catalysts. Initial studies that have received a great deal of press attention reported that the natural product resveratrol (Fig. 4) can stimulate Sir2 deacetylase activity, inducing a 35-fold Km reduction for peptide substrate, as the mechanism for extending the lifespan of a variety of organisms.34 Since resveratrol is found in small amounts in some wines and dietary supplements, these studies were electrifying to members of the non-scientific community. However, this stimulation was later shown by two groups to be an artifact of the fluorescent deacetylase assay rather than authentic changes in activity with natural substrates.35,36 Despite these counter-findings, subsequent reports still attribute the anti-aging and metabolic effects of resveratrol to its allosteric activation of Sir2.37 Resveratrol is known to have a myriad of effects on cellular proteins38 so experiments relying on the pharmacologic actions of this natural product should be interpreted cautiously.
Figure 4.

Activators and inhibitors of Sir2 enzymes.
A more extensive study identifying small molecule Sir2 activators from high throughput screening of synthetic libraries has recently been reported.39 The maximal 8-fold activation of Sir2 with heterocyclic compound SRT1720 (Fig. 4) was documented in several complementary assays. The particularly potent compound SRT1720, with EC50=160 nM, displayed the ability to stimulate Sir2 by lowering the acetylated peptide substrate Km.39 An allosteric model is proposed in which SRT1720 and analogs interact with the protein's N-terminus, based on studies analyzing a series of Sir2 deletion mutants.39 Given the plethora of crystal structures of the Sir2 catalytic domain that have been obtained,40 it will hopefully prove possible to confirm the proposed binding interactions of SRT1720 using X-ray crystallography. One pharmacologic application of this Sir2 activating compound regarding glucose homeostasis was reported.39 It was demonstrated that compound SRT1720 could induce insulin sensitivity and glucose lowering in several mouse model systems, and a molecular mechanism involving reversible TORC2 acetylation is now proposed.41
Separate efforts have been focused on the identification of Sir2 antagonists which might be expected to synergize with classical HDAC inhibitors. The product nicotinamide (Fig. 1) and analogs show modest Sir2 inhibitory activity,42 and nicotinamide has been used in gene regulation experiments.43 Moreover, peptide acetyl-Lys analogs have been shown to have in vitro inhibitory potential and thiocetyl-Lys peptide has the ability to block Sir2 but not classical HDACs.44,45 A high-throughput yeast screen with a chemical library designed to phenocopy the loss of function of Sir2 led to the discovery of the naphthyl-lactone splitomycin (Fig. 4).46 Splitomycin treatment of yeast cells induces gene expression changes similar to those found in yeast Sir2 knockout cells.46 An analogous and concurrent effort led to the discovery of sirtinol (Fig. 4) which has a related structure to splitomycin.47 These compounds show low micromolar potency for blocking Sir2 deacetylase activity.46,47 Although the structural basis for inhibition has not yet been elucidated, modeling studies, structure-activity analysis, and enzyme resistance mutants have suggested a mode of action for these compounds.48
Sirtinol was used in a chemical genetics screen in the discovery of a plant protein Sir1 that is important in auxin gene regulation.49 Sirtinol has also been employed to elucidate the role of Sir2 in axonal protection following nerve injury.50 In studies on the role of deacetylase activity in cardiac myocytes, Sir2 was demonstrated to play a cardioprotective role which could be abrogated by sirtinol treatment.51 These studies highlight the diverse functions of Sir2 in physiologic pathways.
A recent effort to identify a SirT2 selective inhibitor culminated in the report of vinyl nitrile AGK2 (Fig. 4).52 While the mechanism of enzyme inhibition with this potentially electrophilic compound has not yet been established, AGK2 showed IC50 of 3.5 μM for SirT2 and was more than 10-fold selective vs. SirT1 and SirT3. AGK2 displayed the ability to block alpha-synuclein-mediated toxicity in a Parkinson's disease model, possibly by modulating tubulin acetylation.52
Histone acetyltransferase inhibitors
Unlike the classical HDACs or sirtuins, the many families of HATs that have been reported show minimal sequence and rather modest structural homology.9 This fact may be related to the chemical simplicity of the acetyl transfer reaction which involves lysine aminolysis of the acetyl-CoA thioester (Fig. 1). In principle, this lack of homology suggests that identifying specific HAT inhibitors should be possible. The potential clinical impact of HAT inhibitors cuts across a wide range of indications and includes multiple cancers, HIV, diabetes mellitus, and cardiac disease.53–57 There have been several reported approaches to developing HAT inhibitors: rational design, high throughput screening of synthetic libraries, and enzymatic screens with natural products.53–57 In general, the chemical screening approaches have led to some inhibitors with low micromolar potencies such as some isothiazolone derivatives, exemplified by CCT077791 (Fig. 5), but without great specificity demonstrated.55 Some of the natural products that have emerged from these screens include curcumin, anacardic acid, and garcinol (Fig. 5) and derivatives of these have led to moderately potent and selective HAT inhibitors.53,56 The precise enzymatic inhibitory mechanisms of these natural products have not been elucidated but these compounds and related derivatives have been applied to investigate the role of acetylation in cardiac development and HIV.56,57
Figure 5.

Natural product and synthetic histone acetyltransferase inhibitors.
The design of bisubstrate analog HAT inhibitors (Fig. 5) was rooted in the landmark 1969 studies on carnitine O-acetyltransferase, an enzyme which uses a ternary complex kinetic mechanism, which demonstrated that CoA linked to the acylated substrate via an acetyl bridge could potently inhibit the partner enzyme.58 Like peptide-ATP conjugates which block protein kinases,59 peptide-CoA conjugates including the simplest analog, Lys-CoA (Fig. 5), have proved to be potent and selective HAT inhibitors.60 For example, H3-CoA-20, a 20 amino acid tail peptide derived from the histone H3 tail linked to CoA (Fig. 5), showed a Ki of 28 nM versus PCAF/GCN5 HAT and high selectivity (>100-fold) versus p300/CBP.61 The potency was readily understood from the in vitro specificity that PCAF/GCN5 for acetylation of histone H3 on Lys14, the point of attachment to CoA. An X-ray crystal structure of an H3-CoA-20 derivative complexed to the GCN5 HAT domain revealed the molecular basis for the potent inhibition and the unexpected role of loop dynamics in the catalytic mechanism.62,63
The high potency (Ki = 19 nM) and specificity of Lys-CoA inhibition of p300/CBP were unexpected.60,64 While p300/CBP preferentially acetylates longer peptide substrates versus the Lys moiety of Lys-CoA, it is less potently inhibited by the corresponding longer peptide-CoA conjugates.60,64 Recent structural and kinetic studies have revealed the probable basis for this unconventional behavior.65 The-X-ray crystal structure of p300 HAT in complex with Lys-CoA shows that the inhibitor is enclosed by a narrow tunnel with the Lys α-terminus surrounded by enzyme.65 Product inhibition experiments suggest that p300/CBP HAT follows a Theorell-Chance or `hit-and-run' catalytic mechanism.65 Taken together with the structure, the catalytic model suggests that the histone peptide binds very weakly to the p300/CBP-acetyl-CoA complex, allowing sufficient time for the Lys side chain to snake through the enzyme tunnel and receive the acetyl group.
Both Lys-CoA and H3-CoA-20 have been applied in in vitro and cellular studies that have investigated the role of p300/CBP or PCAF/GCN5 HAT activity in biochemical processes. For example, using Lys-CoA, roles for p300/CBP HAT activity have been suggested in muscle cell differentiation, p73 acetylation, C. elegans development, cyclooxygenase-2 gene regulation, melanocyte growyh, HIV gene regulation, and nuclear hormone receptor induced gene expression.54 Second generation versions of Lys-CoA and H3-CoA-20 have been prepared in which cell permeablizing peptide sequences have been attached.66,67 The compounds Lys-CoA-Tat and H3-CoA-20-Tat (Fig. 5) maintain HAT inhibition selectivity in cell culture for p300/CBP and PCAF/GCN5, respectively, as judged by luciferase reporter assays.67
Lys-CoA-Tat has been shown to block p300/CBP-mediated acetylation of the promyelocytic zinc finger protein which interrupts its function as a transcriptional repressor.67 This links an acetylation event paradoxically to the reduction of gene expression. It has further been shown to disrupt TORC2 acetylation leading to fasting glucose reduction.41 H3-CoA-20-Tat has been employed to assess the role of PCAF/GCN5 in catalyzing acetylation of the DEK protein and in Myc-mediated regulation of Pol III transcripts.68,69 These studies further stimulate efforts to develop smaller, specific HAT inhibitory compounds.
Histone lysine methyltransferase inhibitors
The SET domain-containing histone lysine methyltransferases, which are suggested to include more than 50 members in the mammalian genome, are considered potential cancer and neurologic disease drug targets.18 Early compounds which were shown to have inhibitory activity against a broad range of SET methyltransferases include S-adenosyl methionine (SAM) related analogs such as methylthio-adenosine, sinefungin, and S-adenosyl-homocysteine (SAH) (Figs. 2 and 6).70 Such compounds have proved useful in histone methyltransferase mechanistic studies.71 Methylthioadenosine has been used in conjunction with other approaches to implicate the role of MLL methyltransferase activity in endothelial cell migration.72
Fig. 6.

Histone methyltransferase inhibitors.
In more recent years, there have been increased efforts to identify specific histone methyltransferase inhibitors.73,74 Two histone lysine methyltransferases that have been targeted thus far include SU(VAR)-3–9 and G9a, both histone H3 Lys9-directed methyltransferases. Inhibitors of these Lys9 methyltransferases would be expected to activate gene expression and act synergistically with HDAC inhibitors and DNA methyltransferase inhibitors.73,74 The discovery efforts related to finding these histone lysine methyltransferase inhibitors have relied largely on enzymatic high throughput screens of chemical libraries. In one study which screened 3000 compounds against SU(VAR)3–9, the fungal mycotoxin chaetocin (Fig. 6) was identified as a potent (IC50 0.6 μM) and selective methyltransferase inhibitor.73 Chaetocin has the unusual presence of two internal disulfide bonds but, as reported, its inhibitory action is surprisingly not influenced by thiol reducing agents like dithiothreitol in vitro or in cell culture.73
Chaetocin proved to be a competitive inhibitor versus the co-substrate SAM, suggesting it is active site-directed.73 Interestingly, the compound was rather selective when tested against other methyltransferases, showing only significant inhibition of G9a and DIM5, other Lys9-targted methyltransferases.73 As predicted, cells treated with chaetocin showed reduction in dimethyl- and trimethyl-modification of Lys9 of histone H3 but no changes at Lys27, Lys36, or Lys79 of H3.73 However, recent experiments investigating the cell killing properties of chaetocin focused on its potential redox rather than its chromatin-modifying properties.75
More extensive high throughput screening (125,000 synthetic compound library) led to the discovery of quinazoline BIX-01294 (Fig. 6) as a selective inhibitor of G9a with IC50 of 1.7 μM.74 Although it inhibited Lys9 H3 GLP methyltransferase with modest affinity (IC50 38 μM), BIX-01294 did not significantly block other methyltransferases tested.74 Based on limited kinetic data, BIX-01294 was suggested to show an uncompetitive pattern versus the co-substrate SAM, suggesting that it only binds to G9a complexed with SAM.74 Based on this kinetic inhibition model, BIX-01294 may be binding to the histone H3 interaction site on G9a. Studies investigating cells treated with BIX-01294 showed selective reduction in dimethylation of Lys9 of histone H3 in bulk histones and of H3 at promoter regions of known G9a regulated genes.74 This suggests that BIX-01294 may prove to be a more generally valuable tool than chaetocin for exploring H3 Lys9-methylation in gene regulation.
Histone demethylase suicide inactivators
The most recently discovered chromatin modifying enzymes, the histone lysine demethylases, employ redox chemistry that makes them candidates for mechanism-based or suicide inactivators.21 This class of enzyme inhibitor is relatively inert until it is processed by the target enzyme which unmasks a chemically reactive warhead that leads to covalent modification of residues or cofactors in the active site.76 Advantages of suicide inactivators can include high specificity and irreversible inhibition of the inactivated enzyme, so that the recovery of catalyst function requires de novo protein synthesis.76
Progress reported to date has focused on the first reported histone demethylase, LSD1. A flavin-dependent monoamine oxidase, LSD1 is believed to operate by abstracting electrons from the nitrogen atom of Lys4 of histone H3 to generate an imine (Fig. 2).21 Subsequent hydrolytic cleavage of the imine occurs to generate formaldehyde and the demethylated histone. By exploiting this mechanism, several substrate analog suicide inactivators have been developed for the clinically important monoamine oxidases (MAOs) including MAO A and MAO B.77 In fact, several MAO inhibitors including pargyline and tranylcypromine which contain propargyl and cyclopropyl functionalities, respectively, (see Fig. 7) have been used therapeutically in the treatment of cardiovascular and neuro-psychiatric diseases.77 Propargyl- and cyclopropyl-amine containing inhibitors are oxidized by the MAOs to generate electrophilic and radical species which covalently modify these enzymes.77
Figure 7.
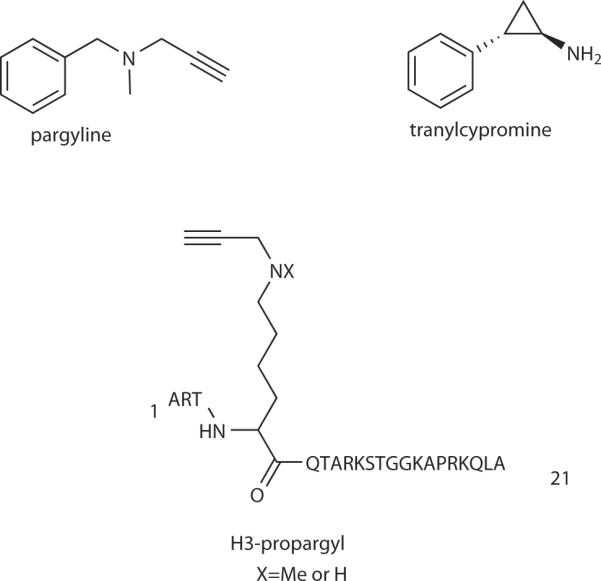
Histone demethylase inhibitors.
Shortly after the discovery of LSD1, several MAO inhibitor analogs were evaluated and suggested to be LSD1 inhibitors, despite the considerable structural differences between MAO substrates norepinephrine, serotonin, and epinephrine and histone H3.78 While further studies have failed to confirm the LSD1 inhibitory properties of the propargyl anti-depressant analogs,79 tranylcypromine has held up as an LSD1 suicide inactivator.80–82
The mechanism of LSD1 inactivation by tranylcypromine has been studied using kinetic and structural approaches. Tranylcypromine shows a Ki of 500 μM and a kinact of 0.67 min−1.81,82 Given that the kcat of LSD1 for substrate turnover is 3.1 min−1, the rate of LSD1 inactivation by tranylcypromine is impressive, although the kinact/Ki is still 20-fold lower in efficiency compared to MAO B inactivation by tranylcypromine. Spectrophotometric analysis suggested FAD (Fig. 2) modification and mass spectrometry confirmed a covalent adduct with the flavin.81,82 X-ray crystallography analysis has led to two models for the adduct.82,83 In both cases, radical ring opening of the cyclopropyl group is proposed, however, differential adduct formation involves either C4a or N5 attack of the flavin on the radical species.82,83
In contrast to expectation based on these data, a Lys4-cyclopropyl (or aziridinyl) 21 amino acid residue derivative of the H3 N-tail peptide substrate are reversible rather than time-dependent inhibitors of LSD1.84,85 Instead, propargylamine analogs of the H3 tail peptide (H3-propargyl, Fig. 7) have been shown to behave as suicide inactivators of LSD1.84–86 Both the propargylamine and N-methyl-propargylamine analogs have been shown to be very potent LSD1 inhibitors, about 500-fold more potent than tranylcypromine, with Ki in the range of 0.1–0.6 μM and kinact ~0.25 min−1.85–86 Covalent modification of the LSD1 flavin by the H3-propargylamines occurs and the flavin adduct has been analyzed by NMR spectrometry and demonstrated to involve an N5-linkage.84–86 In order to capture the structure of the inactivated LSD1 using X-ray crystallography, the H3-propargyl inactivated enzyme was reacted with sodium borohydride to reduce the linker double bonds, prior to crystallization.86 This structure revealed the potential basis for Lys4-selectivity which involved a series of three gamma turns over the first seven H3 amino acid residues.86 It is possible that seven-membered ring gamma turn mimics may offer a new direction in designing LSD1 inhibitors.87
In addition to mechanism-based inactivators, potent reversible peptide and polyamine analogs have been developed.88,89 The potential for re-expression of silenced genes has been demonstrated with the polyamine compounds, providing evidence for potential synergistic therapeutic anti-cancer activity with HDAC inhibitors.89
Emerging directions in chemical biology and histone modifications
The studies reviewed above confirm the rapid progress in synthetic modulators for many of the key enzymes regulating acetylation and methylation of histones. In addition to creating compounds which alter the activity of lysine-modifying enzymes, a series of chemical approaches are influencing the broader area of epigenetics. Synthetic inhibitors of arginine methyltransferases and peptide arginine deiminases have been reported which are important in chromatin regulation.90,91 Several novel activity-based proteomic tools have been described to investigate the action of chromatin modifying enzymes in dynamic conditions including histone deacetylases, histone acetyltransferases, and peptide arginine deiminases.30,92,93 Small molecule inhibitors of bromo domain acetylated peptide binding offer the promise of specific analysis of the consequences of adaptor interactions.94
The application of expressed protein ligation and other semisynthetic strategies have allowed for more precise investigation of the impact of specific histone modifications on nucleosome structure and remodeling.95–98 For example, a recent study has shown that a specific ubiquitylation of a site on histone H2b can directly influence nucleosomal methylation on another histone in a purified in vitro reaction.98 Fluorescent reporters for measuring lysine methyltransferase activity imaging in cells may be applied to enhance spatiotemporal resolution of nucleosomal remodeling.99 One can anticipate the development of small molecule-mutation complementation strategies like those applied in functional analysis of protein kinases100,101 to be used in dissecting actions of closely related family members of histone-modifying enzymes. In summary, the exciting opportunities for chemical biologists investigating chromatin structure and function have been growing for over a decade and are only likely to intensify in years to come.
Acknowledgments
I am grateful to members of my group past and present as well as many collaborators in this field for many helpful discussions and for their key roles in the work cited. I also thank the NIH, FAMRI, and Kaufman foundations for support.
References
- 1.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray K. The occurrence of ε-N-methyl lysine in histones. Biochemistry. 1964;3:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- 5.Gershey EL, Vidali G, Allfrey VG. Chemical studies of histone acetylation. The occurrence of ε-N-acetyllysine in the f2a1 histone. J. Biol. Chem. 1968;243:5018–5022. [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;282:15040–15047. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Brownell JE, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 8.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 9.Vetting MW, et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 11.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 12.Finnin MS, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 13.Itoh Y, Suzuki T, Miyata N. Isoform-selective histone deacetylase inhibitors. Current Pharm. Des. 2008;14:529–544. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- 14.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longetivity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 15.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 16.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 17.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 18.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Tsukada Y, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 21.Culhane J, Cole PA. LSD1 and the chemistry of histone demethylation. Curr. Op. Chem. Biol. 2007;11:561–568. doi: 10.1016/j.cbpa.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paris M, Porcelloni M, Binaschi M, Fattori D. Histone deacetylase inhibitors: from bench to clinic. J. Med. Chem. 2008;51:1505–1529. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- 23.Vannini A, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuetz A, et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong JC, Hong R, Schreiber SL. Structural basis for in-cell histone deacetylase paralog selectivity. J. Am. Chem. Soc. 2003;125:5586–5587. doi: 10.1021/ja0341440. [DOI] [PubMed] [Google Scholar]
- 27.Estiu G, et al. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem. 2008;51:2898–2906. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- 28.Hideshima T, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Nat. Acd. Sci. USA. 2005;102:8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran AD, et al. DAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J. Cell Sci. 2007;120:1469–1479. doi: 10.1242/jcs.03431. [DOI] [PubMed] [Google Scholar]
- 30.Salisbury CM, Cravatt BF. Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J. Am. Chem. Soc. 2008;130:2184–94. doi: 10.1021/ja074138u. [DOI] [PubMed] [Google Scholar]
- 31.Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 32.Guarente L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- 33.Wade N. New hints seen that red wine may slow aging. New York Times. 2008 June 4;:A1. [Google Scholar]
- 34.Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 35.Borra MT, Smith BC, Denu JM. Mechanism of human SirT1 activation by resveratrol. J. Biol. Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 36.Kaeberlein M, et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 37.Lagouge M, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Szewczuk LM, Lee SH, Blair IA, Penning TM. Vinferin formation by COX-1: Evidence for radical intermediates during co-oxidation of resveratrol. J. Nat. Prod. 2005;68:36–42. doi: 10.1021/np049702i. [DOI] [PubMed] [Google Scholar]
- 39.Milne JC, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure. 2006;14:1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, et al. A fasting inducible acetylase/deacetylase switch modulates gluconeogenesis through activator-coactivator exchange. Nature. 2008 doi: 10.1038/nature07349. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J. Biol. Chem. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- 43.Luo J, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 44.Fatkins DG, Monnot AD, Zheng W. Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg. Med. Chem. Lett. 2006;16:3651–3656. doi: 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 45.Smith BC, Denu JM. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J. Biol. Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- 46.Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA. 2001;98:15113–15118. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 48.Hirao M, et al. Identification of selective inhibitors of NAD-dependent deacetylases using phenotypic screens in yeast. J. Biol. Chem. 2003;278:52773–52782. doi: 10.1074/jbc.M308966200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Dai X, Blackwell HE, Schreiber SL, Chory J. Sir1, an upstream component in auxin signaling identified by chemical genetics. Science. 2003;301:1107–1110. doi: 10.1126/science.1084161. [DOI] [PubMed] [Google Scholar]
- 50.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SirT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 51.Alcendor RR, Kirshenbaum LA, Imai S.-i., Vatner SF, Sadoshima J. Silent information regulator 2a, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circulation Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 52.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 53.Swaminathan V, et al. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell. Biochem. 2007;41:397–428. [PubMed] [Google Scholar]
- 54.Zheng Y, et al. Selective HAT inhibitors as mechanistic tools for protein acetylation. Methods Enzymol. 2004;376:188–199. doi: 10.1016/S0076-6879(03)76012-1. [DOI] [PubMed] [Google Scholar]
- 55.Stimson L, et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 2005;4:1521–1532. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- 56.Mantelingu K, et al. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem. Biol. 2007;14:645–657. doi: 10.1016/j.chembiol.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 57.Morimoto T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J. Clin. Invest. 2008;118:868–78. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chase JFA, Tubbs PK. Conditions for the self-catalysed inactivation of carnitine acetyltransferase. Biochem. J. 1969;111:225–235. doi: 10.1042/bj1110225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parang K, et al. Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol. 2001;8:37–41. doi: 10.1038/83028. [DOI] [PubMed] [Google Scholar]
- 60.Lau OD, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 61.Lau OD, et al. PCAF Histone acetyltransferase processing of a peptide substrate: kinetic analysis of the catalytic mechanism. J. Biol. Chem. 2000;275:21953–21959. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- 62.Poux AN, Cebrat M, Kim CM, Cole PA, Marmorstein R. Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc. Natl Acad. Sci. USA. 2002;99:14065–14070. doi: 10.1073/pnas.222373899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng Y, et al. Fluorescence analysis of a dynamic loop in the PCAF/GCN5 histone acetyltransferase. Biochemistry. 2005;44:10501–10509. doi: 10.1021/bi050776i. [DOI] [PubMed] [Google Scholar]
- 64.Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J. Biol. Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 65.Liu X, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. (2008) [DOI] [PubMed] [Google Scholar]
- 66.Zheng Y, et al. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J. Am. Chem. Soc. 2005;127:17182–17183. doi: 10.1021/ja0558544. [DOI] [PubMed] [Google Scholar]
- 67.Guidez F, et al. Histone acetyltransferase activity of p300 is required for transcriptional repression by the promyelocytic zinc finger protein. Mol. Cell. Biol. 2005;25:5552–5566. doi: 10.1128/MCB.25.13.5552-5566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cleary J, et al. Acetylation by P/CAF drives DEK into interchromatin granule clusters. J. Biol. Chem. 2005;280:31760–31767. doi: 10.1074/jbc.M500884200. [DOI] [PubMed] [Google Scholar]
- 69.Kenneth NS, et al. Activation of Pol III transcription by c-Myc involves selective acetylation of histone H3 and recruitment of TRAPP, GCN5, and TFIIIB. Proc. Natl Acad. Sci. USA. 2007;104:14917–14922. doi: 10.1073/pnas.0702909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang S. Histone methyltransferases, diet nutrients and tumour suppressors. Nat. Rev. Cancer. 2002;2:469–476. doi: 10.1038/nrc819. [DOI] [PubMed] [Google Scholar]
- 71.Dirk LM, et al. Kinetic manifestation of processivity during multiple methylations catalyzed by SET domain protein methyltransferases. Biochemistry. 2007;46:3905–3915. doi: 10.1021/bi6023644. [DOI] [PubMed] [Google Scholar]
- 72.Diehl F, Rössig L, Zeiher AM, Dimmeler S, Urbich C. The histone methyltransferase MLL is an upstream regulator of endothelial-cell sprout formation. Blood. 2007;109:1472–1478. doi: 10.1182/blood-2006-08-039651. [DOI] [PubMed] [Google Scholar]
- 73.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 74.Kubicek S, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 75.Isham CR, et al. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walsh CT. Suicide substrates, mechanism-based enzyme inactivators: recent developments. Annu. Rev. Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- 77.Edmondson DE, Mattevi A, Binda C, Li M, Hubálek F. Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 2004;11:1983–1993. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 78.Metzger E, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 79.Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- 80.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhatter R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 81.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 82.Yang M, et al. Structural basis for inhibition of the LSD1 histone demethylase by the antidepressant trans-2-Phenylcyclopropylamine. Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 83.Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochem. Biophys. Res. Commun. 2008;366:15–22. doi: 10.1016/j.bbrc.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 84.Culhane JC, et al. A mechanism-based inactivator for histone demethylase LSD1. J. Am. Chem. Soc. 2006;128:4536–4537. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 85.Szewczuk LM, et al. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang M, et al. Structural basis for histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 2007;14:535–539. doi: 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- 87.Ramanathan SM, et al. Modular synthesis of cyclic peptidomimetics inspired by γ-turns. Org. Lett. 2005;7:1059–1062. doi: 10.1021/ol047323a. [DOI] [PubMed] [Google Scholar]
- 88.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 89.Huang Y, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. USA. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Osborne T, Roska RL, Rajski SR, Thompson PR. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1. J. Am. Chem. Soc. 2008;130:4574–4575. doi: 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]
- 91.Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J. Am. Chem. Soc. 2006;128:14468–14469. doi: 10.1021/ja0656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu M, de Carvalho LP, Sun G, Blanchard JS. Activity-based substrate profiling for Gcn5-related N-acetyltransferases: the use of chloroacetyl-coenzyme A to identify protein substrates. J. Am. Chem. Soc. 2006;128:15356–15357. doi: 10.1021/ja066298w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hwang Y, et al. selective chemical probe for coenzyme A-requiring enzymes. Angew. Chem. Int. Ed. Engl. 2007;46:7621–7624. doi: 10.1002/anie.200702485. [DOI] [PubMed] [Google Scholar]
- 94.Sachchidanand, et al. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006;13:81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 95.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 96.Thompson PR, et al. Regulation of p300 HAT domain via a novel activation loop. Nature Struct. Mol. Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 97.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 2008;453:812–816. doi: 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin CW, Jao CY, Ting AY. Genetically encoded fluorescent reporters of histone methylation in living cell. J. Am. Chem. Soc. 2004;126:5982–5983. doi: 10.1021/ja038854h. [DOI] [PubMed] [Google Scholar]
- 100.Shogren-Knaak MA, Alaimo PJ, Shokat KM. Recent advances in chemical approaches to the study of biological systems. Ann. Rev. Cell. Biol. 2001;17:405–433. doi: 10.1146/annurev.cellbio.17.1.405. [DOI] [PubMed] [Google Scholar]
- 101.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]