

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV) is a member of the gammaherpesvirus family. It is the etiological agent of three different human cancers, Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (MCD). The far left-end of the KSHV genome encodes a unique transmembrane glycoprotein called K1. K1 possesses the ability to transform rodent fibroblasts and block apoptosis. K1 has also been shown to activate the PI3K/Akt/mTOR pathway in different cells
Using tandem affinity purification (TAP), we identified heat shock protein 90β (Hsp90β) and endoplasmic reticulum (ER)-associated Hsp40 (Erdj3/DnaJB11), as cellular binding partners of K1. Interactions of K1 with Hsp90β and Hsp40 were confirmed by co-immunoprecipitation in both directions. Furthermore, K1 also interacted with the Hsp90α̣ isoform. We report that siRNAs directed against Hsp90 and Hsp40/Erdj3, as well as pharmacological inhibitors of Hsp90 dramatically reduced K1 expression, suggesting that K1 is a client protein of these chaperones. Additionally, both Hsp90 and Hsp40/Erdj3 were essential for K1’s anti-apoptotic function. Finally, we report that the Hsp90 inhibitors, 17-AAG and 17-DMAG, can suppress the proliferation of KSHV-positive PEL cell lines and exhibited IC50 values of 50nM and below.
Keywords: KSHV, K1, heat shock proteins, Hsp90, Hsp40, lytic replication
INTRODUCTION
Kaposi sarcoma-associated herpesvirus (KSHV), or human herpesvirus-8, is a gammaherpesviruses. This virus has been implicated as the etiological agent of Kaposi sarcoma (KS) (Chang et al., 1994) and lymphoproliferative diseases of B cell origin, namely, primary effusion lymphoma (PEL) (Cesarman et al., 1995) and the plasmablastic variant of multicentric Castleman disease (MCD) (Gessain et al., 1996; Soulier et al., 1995).
The first open reading frame (ORF) of KSHV encodes a viral glycoprotein named K1 (Lagunoff & Ganem, 1997). Both K1 transcript and protein have been detected in KS, PEL, and MCD (Bowser et al., 2002; Lee et al., 2003; Samaniego et al., 2001; Wang et al., 2006) and K1 is expressed at low levels during latency (Chandriani & Ganem, 2010; Wang et al., 2006). We, and others, have shown that K1 is a transforming protein of KSHV capable of transforming rodent fibroblasts (Damania et al., 1999; Prakash et al., 2002). Additionally, K1 transgenic mice develop tumors with features resembling spindle-cell sarcomas and malignant plasmablastic lymphoma (Prakash et al., 2002).
K1 is a 46 kDa type-I transmembrane glycoprotein, which structurally and functionally resembles a B cell receptor (BCR). In its cytoplasmic tail, K1 contains an immunoreceptor tyrosine-based activation motif (ITAM) that is also found in many immunoglobulin receptors e.g. BCR. The K1 ITAM is comprised of two appropriately spaced Src-homology 2 (SH2) binding motifs and is required for BCR activation events (Lagunoff et al., 1999; Lee et al., 1998a; Lee et al., 1998b). We and others have shown that K1 is capable of activating B lymphocyte signal transduction and interacting with Syk (Damania et al., 2000; Lagunoff et al., 1999; Lee et al., 1998a). K1 signaling activity in B cells has been linked to K1 internalization (Tomlinson & Damania, 2008), and K1 also co-internalizes with the BCR (Tomlinson & Damania, 2008) resulting in the downregulation of BCR surface expression (Lee et al., 2000).
We previously reported that K1 activates the phosphatidyl-inositol-3’-OH-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway in both B cells and endothelial cells (Tomlinson & Damania, 2004; Wang et al., 2006). In addition, we first reported that K1 can prevent Fas-mediated apoptosis through activation of the PI3K/Akt pathway (Tomlinson & Damania, 2004). In epithelial and endothelial cells, K1 expression induced the secretion of angiogenic factors (Wang et al., 2004). Cumulatively, these data suggest a paracrine model in which K1-mediated secretion of cytokines is involved in the development of KSHV-associated tumorigenesis and angiogenesis (Wang et al., 2004).
Molecular chaperones are involved in various cellular processes including protein folding and cell signaling. For example, both heat shock protein (Hsp) 90 and the Hsp40/Hsp70 system act to enhance the Akt pathway which is important for cell survival (Gao & Newton, 2002; Sato et al., 2000). Hsp40, Hsp70, and Hsp90 are important for the assembly of signaling receptor complexes (Nathan & Lindquist, 1995; Picard et al., 1990; Smith et al., 1995). Hsp90 and the Hsp40/Hsp70 system have been shown to suppress tumor necrosis factor (TNF)- and Fas-induced apoptosis (Lewis et al., 2000; Pang et al., 2001). Additionally, Hsp90 overexpression has been detected in a variety of cancers (reviewed in (Schmitt et al., 2007)). The KSHV K1 protein targets similar signaling pathways as Hsp90 e.g. the PI3K/Akt pathway, and hinders apoptosis by modulating this pro-survival pathway. Hence the functions of Hsp90, Hsp40, and K1 appear to be concordant.
We identified Hsp90β and the ER-associated Hsp40 (Erdj3/DnajB11) as cellular binding partners of K1. Pharmacological inhibition of Hsp90 function reduced expression of the K1 protein. Similar results were obtained when siRNAs were used to knockdown Hsp90β or Hsp40/Erdj3. Knockdown of these chaperones also prevented K1 from promoting cell survival and inhibiting apoptosis. Finally, we report that the Hsp90 inhibitors were very effective in inhibiting the proliferation of KSHV-positive PEL cell lines.
RESULTS
Tandem affinity purification identifies Hsp90β and Hsp40 (Erdj3) as cellular binding partners of K1
In order to identify cellular binding partners of K1 in the context of mammalian cells, we performed tandem affinity purification (TAP) (Rigaut et al., 1999). We cloned the FLAG and HA epitopes in tandem right after the signal peptide sequence in the N-terminus of K1 using PCR. The double-tagged K1 gene was cloned into the pcDNA3 vector. Stable 293 cells expressing FLAG-HA-K1 (293-K1) or empty vector (293-Vec) were generated by transfection of pcDNA3-FLAG-HA-K1 or pcDNA3 vector into 293 cells, respectively. Cells were selected in 1 mg/ml G418 containing-media for two weeks.
For TAP, forty confluent T175 flasks per stable cell line were harvested and lysed in NP40 buffer. The cell lysate was centrifuged and the supernatant was subjected to TAP. The supernatant was first incubated with anti-FLAG resin and then transferred to a spin column. The resin was washed with NP40 buffer and protein complexes were eluted twice with 3× FLAG peptide and then pooled. Next, the eluates were incubated with anti-HA resin. This was followed by washes with NP40 buffer and bound proteins were then eluted and resolved by SDS-PAGE. The gel was Coomassie-stained; protein bands unique to K1 expressing cells were excised, trypsin-digested, and subjected to MALDI-TOF mass spectrometry analysis (Fig. 1A).
Figure 1. TAP identified Hsp90β and ER-associated Hsp40 as cellular binding partners of K1.
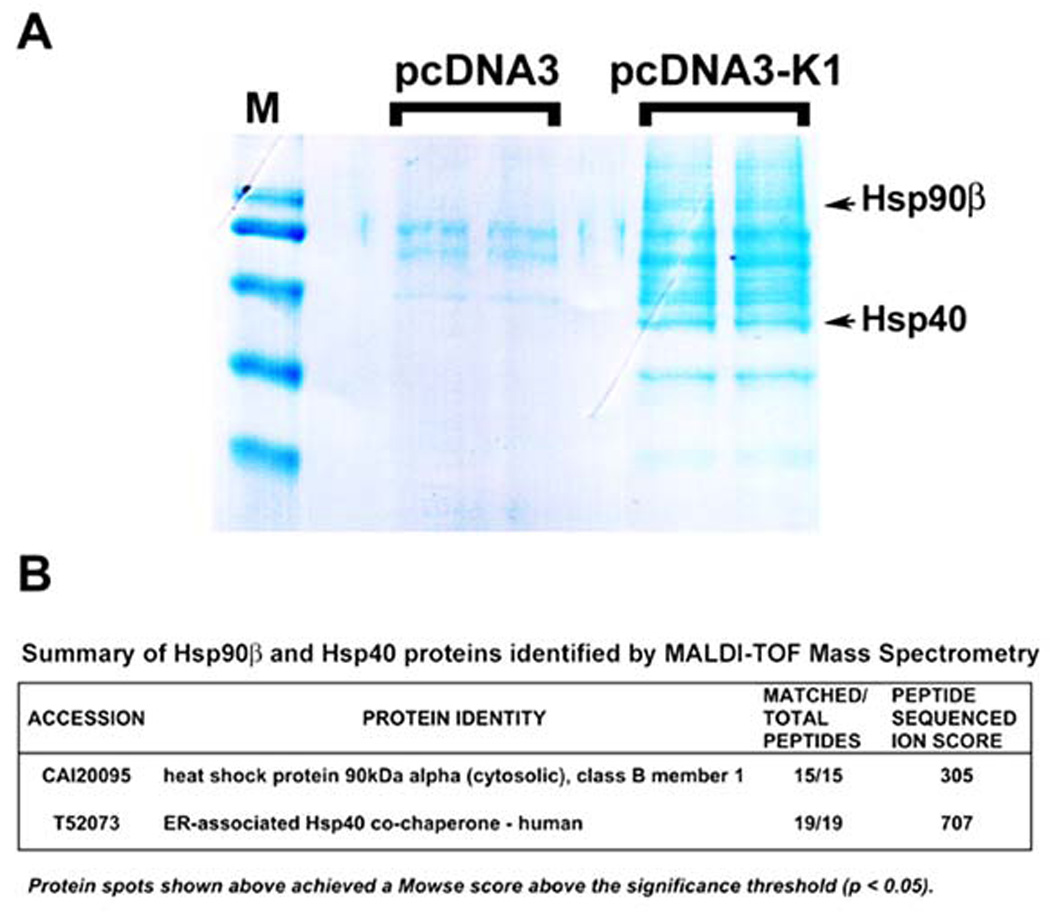
A. A Coomassie-stained gel of the K1 cellular partners identified by TAP is shown. After TAP, eluted proteins were resolved by SDS-PAGE and visualized by Coomassie staining. Protein bands unique to K1 expressing cells were excised and subjected to MALDI-TOF Mass Spectrometry (MS). M denotes marker. B. MS/MS data for the protein identities of Hsp90β and ER-associated Hsp40 are shown.
Mass spectrometry identified Hsp90β and the ER-associated Hsp40(Erdj3) as cellular binding partners of K1 (Fig. 1A). Importantly, the specific protein bands for both Hsp90β and Hsp40 were only visible in the K1 lanes, but not in the empty vector (pcDNA3) lanes (Fig. 1A). Furthermore, the digested peptides fully matched the peptide sequences of Hsp90β and Hsp40 (Erdj3) and achieved very high ion scores (Fig. 1B).
To confirm the TAP results, equivalent micrograms of pre-cleared 293-K1 and 293-Vec lysates were used to perform immunoprecipitation with anti-FLAG resin beads to immunoprecipitate FLAG-tagged K1 (Figs. 2A and 2C). The immunoprecipitates were subjected to Western blot analysis with anti-Hsp90β (Fig. 2A) or anti-Erdj3 antibody (Fig. 2C). Interactions of K1 with endogenous Hsp90β and Erdj3 were confirmed by reverse co-immunoprecipitation using anti-Hsp90β (Fig. 2B) or anti-Erdj3/Hsp40 (Fig. 2D), followed by immunoblotting with an anti-FLAG antibody to detect K1 expression. Immunoprecipitation assays performed in both directions strongly corroborated the TAP results, and demonstrated that K1 physically associates with Hsp90β and Erdj3/ER-associated Hsp40. To confirm specificity, we repeated the K1 immunoprecipitation assay with a normal mouse IgG antibody as a negative control. We found that immunoprecipitatons using an anti-Flag antibody to pull down K1 co-immunoprecipitated Hsp90β and Hsp40, while immunoprecipitation with the control normal mouse IgG antibody did not pull down K1, Hsp90β or Hsp40 (Supplemental Fig. 1).
Figure 2. K1 interacts with endogenous Hsp90β and ER-associated Hsp40.

A. Protein lysates from stable 293 cells expressing empty control vector (left lanes) or FLAG-K1 (right lanes) were immunoprecipitated with anti-FLAG antibody. Immunoprecipitation reactions were subjected to SDS-PAGE followed by Western blotting with an anti-Hsp90β antibody. Input lysates showed the presence of Hsp90β in all cell lysates, and K1 protein in the 293-K1 cell lysate. B. Reverse co-immunoprecipitations were also performed. Lysates from 293-Vec or 293-K1 stable cells were immunoprecipitated with an anti-Hsp90β antibody. Immunoprecipitations were subjected to SDS-PAGE and WB analysis with an anti-FLAG antibody to detect K1 protein expression. Input lysates showed the presence of Hsp90β in all cell lysates, and K1 protein in the 293-K1 cell lysate. These data are representative of at least three independent experiments. C. & D. Identical co-immunoprecipitations were performed as indicated in panels A and B, respectively, except that anti-Hsp40 antibody was used instead of Hsp90β antibody.
Since Hsp90α and Hsp90β show >80% identity, and each isoform possesses chaperoning activity (Picard, 2004), we also investigated whether K1 could interact with Hsp90α. Hsp90β is the constitutive form, whereas Hsp90α is more inducible and has been found in the medium and on the cell surface (Picard, 2004). We immunoprecipitated K1 using anti-FLAG resin beads from pre-cleared 293-K1 and 293-Vec lysates, and performed Western blot analysis using anti-Hsp90α antibody. We found that K1 associated with Hsp90α (Supplemental Fig. 2), indicating that K1 can interact with both α and β isoforms of Hsp90.
We next investigated whether K1 interacts with heat shock proteins other than Hsp40 and Hsp90. We immunoprecipitated K1 from 293-K1 stable cells with an anti-FLAG antibody. The immunoprecipitates were subjected to Western blot analysis with an anti-Hsp70 antibody (Fig. 3A). We found that K1 failed to interact with Hsp70 suggesting that K1 specifically interacts with Hsp90 and Erdj3/Hsp40.
Figure 3. K1 interacts with Hsp90β and Erdj3/Hsp40 in both 293 and BJAB cells.

A. Protein lysates from stable 293 cells expressing empty pcDNA3.1 control vector or FLAG-K1 were immunoprecipitated with anti-FLAG or control anti-normal mouse Ig antibody. Immunoprecipitation reactions were subjected to SDS-PAGE followed by Western blotting with an anti-Hsp70 antibody. Input lanes show the presence of Hsp70 in all protein lysates and K1 protein in the 293-K1 lysate only. B. Protein lysates from stable 293-Vec or 293-K1 cells after treatment with vehicle or apyrase were immunoprecipitated with anti-FLAG antibody. Immunoprecipitation reactions were subjected to SDS-PAGE followed by Western blotting with an anti-Hsp90β antibody. C. Identical co-immunoprecipitations were performed as described in Fig. 2A, except that protein lysates from BJAB B cells transfected with pcDNA3.1 vector or K1 expression plasmid were immunoprecipitated with anti-FLAG antibody. Immunoprecipitates were subjected to SDS-PAGE followed by Western blotting with an anti-Hsp90β antibody. Input lysates showed the presence of Hsp90β in all cell lysates, and K1 protein in the 293-K1 cell lysate. Immunoprecipitation was also performed with species-matched normal IgG antibody as a negative control.
Hsp90 possesses intrinsic ATPase activity critical for its chaperoning function (Pearl & Prodromou, 2006). Apyrase catalyzes the breakdown of ATP and ADP into AMP and inorganic phosphate thereby depleting the ATP content in cells (Pedersen et al., 2003; Zhang & Guy, 2005). We investigated whether K1’s interaction with Hsp90β was dependent on the presence of ATP. We incubated 293-K1 or 293-Vec stable cell lysates with 20 units/ml of apyrase for 30 min, in order to deplete ATP from the samples (Pedersen et al., 2003). The ATP-depleted extracts were used to immunoprecipitate K1 and the immunoprecipitates were subjected to SDS-PAGE and immunoblotting with an anti-Hsp90β (Fig. 3B) or anti-Erdj3/Hsp40 antibody. Notably, ATP depletion by apyrase resulted in less Hsp90β protein being immunoprecipitated with K1 (Fig. 3B), suggesting that ATP levels modulate K1’s interaction with Hsp90β. Interestingly, apyrase treatment did not alter the interaction of K1 with Erdj3/Hsp40, which lacks ATPase activity (data not shown).
Given that K1 expression can be detected in B cells and that K1 perturbs B cell signaling and function, we examined if K1 could interact with Hsp90β and Erdj3/Hsp40 in B cells. Lysates from BJAB cells transfected with FLAG-tagged K1 or pcDNA3 vector plasmids were subjected to immunoprecipitation using an anti-FLAG antibody to pull down K1 or normal mouse IgG as a negative control (Fig. 3C). The K1 immunoprecipitates as well as the input lysates were subjected to Western blot analysis to detect K1, Hsp90β, or Erdj3/Hsp40 (Fig. 3C). Similar to the situation in 293 epithelial cells, we detected specific interactions of Hsp90β with K1 and Erdj3 with K1 in B cells which suggests that these interactions might be relevant in KSHV-associated B lymphoproliferative diseases such as PEL and MCD.
To corroborate the above findings, we also performed co-localization assays for K1 and Hsp90 or Erdj3 in KSHV-positive BCBL-1 cells. Briefly, BCBL-1 were fixed, permeabilized, and then stained with anti-K1 antibody and anti-Hsp90/anti-Erdj3 antibodyand examined by confocal microscopy. We found that K1 colocalizes with Hsp90 and Erdj3/Hsp40 (Supplemental Fig. 3). Additionally, we also performed immunohistochemistry on PEL tumor sections (Supplemental Fig. 4). We found that both K1 and Hsp90 proteins were detected by immunohistochemical staining of tumor sections (Supplemental Fig. 4).
Hsp90β and Erdj3/Hsp40 associate with the amino-terminal domain of K1
To gain insight into the function and topology of the interaction, we used a panel of FLAG-tagged K1 domain deletion constructs ((ΔC: C-terminal deletion; ΔTM: transmembrane deletion; ΔN: N-terminal deletion)) to determine the K1 domain that interacted with Hsp90β and the ER-associated Hsp40. Expression plasmids for the K1 deletion mutants, the FLAG-tagged wild-type K1, and the control pcDNA3 vector, were transiently transfected into 293 cells. Protein lysates were harvested 48 h post-transfection. Co-immunoprecipitations for K1 with Hsp90β and Hsp40 were performed as described above for Fig. 2. FLAG-tagged K1 was immunoprecipitated with anti-FLAG beads, followed by immunoblotting with anti-Hsp90β (Fig. 4A) or anti-Hsp40/Erdj3 antibody (Fig. 4C). Hsp90β and Hsp40 were detected in all of the samples expressing K1 except for the sample expressing the K1ΔN mutant. Additionally, the reverse immunoprecipitation of Hsp90β (Fig. 4B) or Hsp40 (Fig. 4D) followed by Western blot analysis with anti-FLAG antibody also suggested that the K1ΔN mutant protein does not interact with Hsp90β or Hsp40. Collectively, our data suggests that the N-terminal domain of K1 interacts with the Hsp90β and Hsp40/Erdj3 molecular chaperones.
Figure 4. K1 interacts with endogenous Hsp90β and ER-associated Hsp40 through its Nterminus.
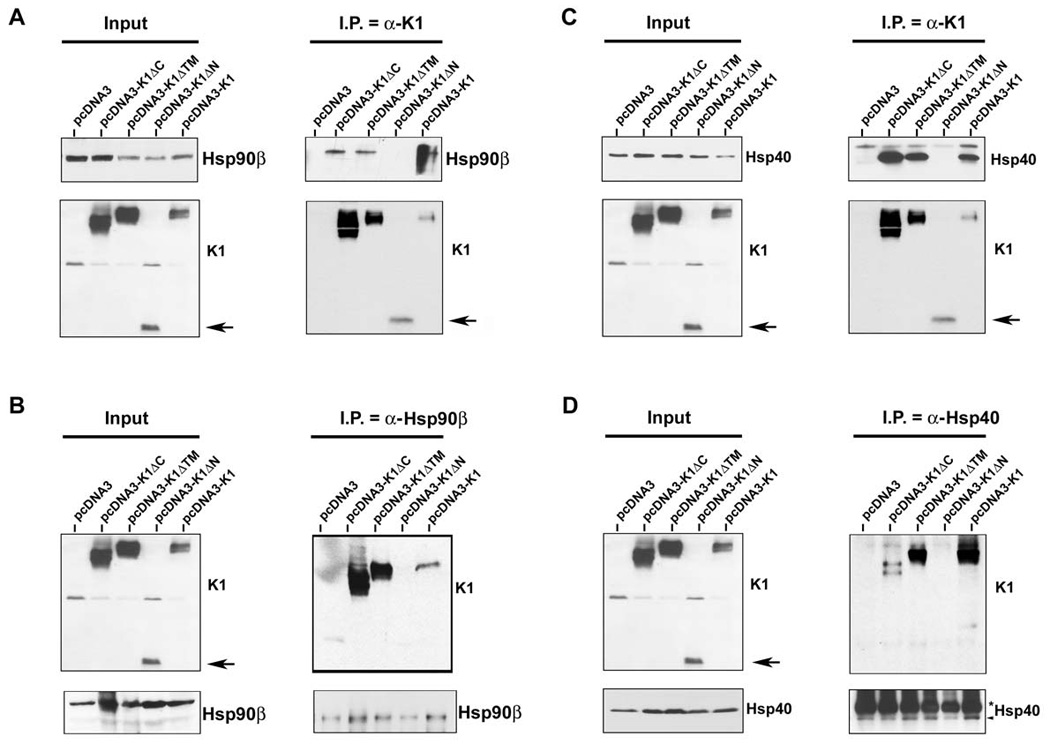
A. Co-immunoprecipitation of a panel of K1 constructs with endogenous Hsp90β. 293 cells were transfected with the empty vector or a panel of FLAG-K1 domain deletion mutants (ΔC: C-terminal deletion; ΔTM: transmembrane deletion; ΔN: N-terminal deletion), or the full-length FLAG-K1. Lysates were immunoprecipitated with anti-FLAG resin. Immunoprecipitation reactions were subjected to SDS-PAGE and Western blotting with an anti-Hsp90β antibody. Input lysates showed the presence of Hsp90β in all cell lysates and the presence of K1 mutants in the K1 mutant transfected cells. B. Reverse co-immunoprecipitations were also performed. Lysates from cells transfected with the vector control or the K1 panel of mutants were immunoprecipitated with an anti-Hsp90β antibody. Immunoprecipitation reactions were subjected to Western blot analysis with an anti-FLAG antibody to detect K1 protein expression. Input lysates showed the presence of Hsp90β in all cell lysates and the presence of K1 mutants in the K1 mutant transfected cells. These data are representative of at least three independent experiments. C. & D. Identical co-immunoprecipitations were performed as indicated in panels A and B, respectively, except that anti-Hsp40 antibody was used instead of Hsp90β antibody. In panels A–D, arrows indicate the N-terminal deletion mutant of K1.
K1 protein expression is dependent on Hsp90 activity
As heat shock proteins are molecular chaperones that confer protein stability to their client proteins, we next tested whether the chaperoning activity of Hsp90 affected K1 protein expression. The ansamycin antibiotic, geldanamycin (GA), was used to inhibit the ATPase activity of Hsp90, an activity that is essential for its chaperoning function. Thus, GA prevents the transfer of client proteins to Hsp90. GA inhibits all isoforms of Hsp90, but not Hsp40. After GA treatment, client proteins of Hsp90 are degraded by the proteasomal machinery (Whitesell et al., 1997; Xu et al., 2003). We treated stable 293-K1 or 293-Vec cells with 1µM GA, or vehicle (DMSO), for 6 h. Lysates were harvested and subjected to immunoblotting. We found that pharmacological inhibition of Hsp90 with 1µM GA in 293-K1 cells dramatically decreased K1 protein level when compared to DMSO treatment (Fig. 5A). Expectedly, the protein level of phosphorylated Akt (S473), a known client protein of Hsp90 (Xu et al., 2003), was also diminished in the presence of GA.
Figure 5. K1 protein expression is dependent on Hsp90 activity and the endogenous levels of Hsp90β and ER-associated Hsp40.
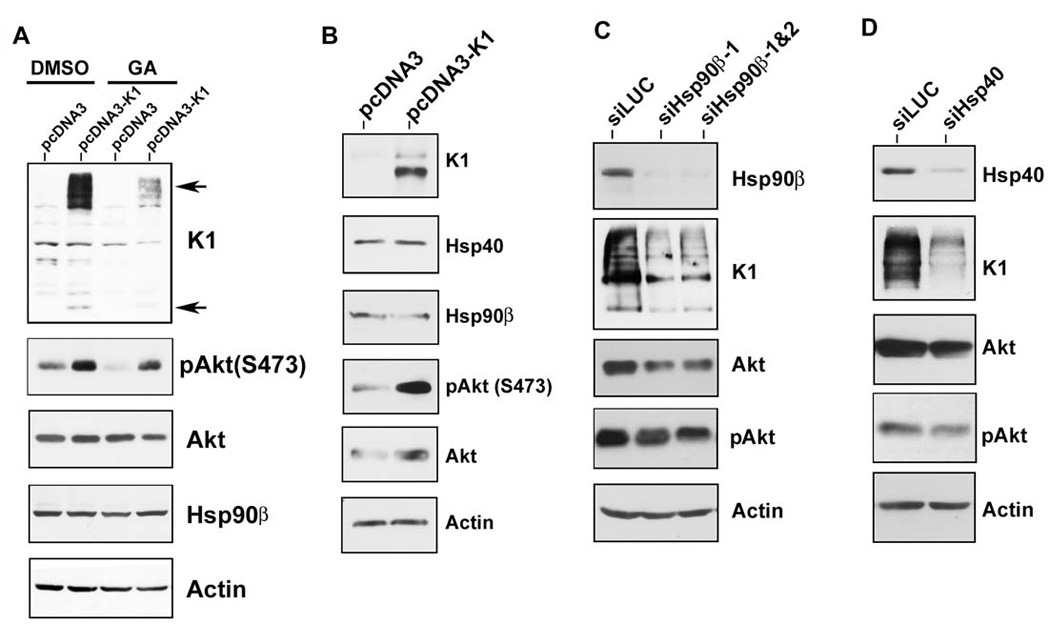
A. Stable 293-Vec cells expressing empty control vector (pcDNA3) or 293-K1 cells expressing FLAG-K1 were treated with 1 µM of the Hsp90 inhibitor, geldanamycin (GA), or DMSO (vehicle), for 6 h in the absence of serum. Cell lysates were harvested and resolved by SDS-PAGE and immunoblotting. Western blots were probed with the indicated antibodies. Arrows indicate monomeric and multimeric forms of the K1 protein. B. K1 does not affect the endogenous levels of Hsp90β and Hsp40. Stable 293-K1 or 293-Vec cell lysates were subjected to SDS-PAGE and Western blot analysis using the indicated antibodies. Each panel is representative of at least three independent experiments. C. 293-K1 cells were subjected to siRNA targeting luciferase (siLUC; control), or Hsp90β (siHsp90β-1 or siHsp90β-1&2) for 48 h. Cell lysates were harvested and subjected to SDS-PAGE and Western blot analysis. D. 293-K1 cells were subjected to siRNA targeting luciferase (siLUC; control) or a pool of siRNAs targeting ER-associated Hsp40 (siHsp40) for 48 h. Cell lysates were harvested and subjected to SDS-PAGE and Western blot analysis.
We previously reported that K1 can activate and phosphorylate Akt in B and endothelial cells (Tomlinson & Damania, 2004; Wang et al., 2006). Here we observed the same phenomenon in epithelial cells, where 293-K1 cells showed increased p-Akt (S473) levels compared to 293-Vec samples (Fig. 5B). We next tested whether K1 had any effect on the endogenous levels of Hsp90β and ER-associated Hsp40. Western blot analysis of 293-K1 and 293-Vec cell lysates did not show an appreciable difference in Hsp90β and Hsp40 protein abundance, albeit the total Akt and p-Akt (S473) levels were increased in K1-expressing 293 cells (Fig. 5B).
siRNA-mediated depletion of Hsp90β and ER-associated Hsp40/Erdj3 reduces K1 protein expression
To confirm the results obtained with pharmacological inhibition of Hsp90, and to rule out any off-target effects of GA that may lead to decreased K1 protein levels, we performed genetic knockdown of Hsp90β and Erdj3/ER-associated Hsp40 using siRNAs. Briefly, 293-K1 cells were transfected with 200pmol of pooled Hsp90β-targeting siRNAs or a control luciferase-targeting siRNA (siLUC). Cells were harvested 48 h post-transfection and the protein lysates were subjected to Western blot analysis. The Hsp90β siRNAs robustly depleted the endogenous level of Hsp90β without affecting endogenous actin levels (Fig. 5C). Similar to GA treatment, Hsp90β siRNA expression resulted in a reduction of K1 protein expression compared to control luciferase siRNA (Fig. 5C). Hsp90β siRNA knockdown also led to reduced total Akt and p-Akt (S473) levels.
We also tested if K1 protein level is dependent upon ER-associated Hsp40/Erdj3 expression. 293-K1 cells were transfected with a pool of three different siRNAs directed against Hsp40/Erdj3 (siHsp40 in Fig. 5D) or the irrelevant siLUC for 48 h. The protein lysates were then subjected to immunoblotting. As shown in Fig. 5D, the pooled siHsp40 siRNAs effectively reduced the endogenous level of Hsp40/Erdj3 compared to the siLUC transfected sample, but did not alter endogenous actin levels. We found that siRNA knockdown of Hsp40/Erdj3 also inhibited K1 protein expression and was accompanied by a slight reduction of the total Akt and p-Akt (S473) levels (Fig. 5D). Taken together, these data suggest that Hsp90 and Hsp40/Erdj3 are required for the protein stability of K1, and that K1 is a client protein of Hsp90 and the Hsp40/Hsp70 chaperone system.
Interaction with Hsp90β and Erdj3 regulates K1 anti-apoptotic function
We next interrogated whether Hsp90β and Erdj3’s association with K1 can modulate its function. In order to examine the impact of Hsp90β or Erdj3 knockdown on K1 function, we tested the ability of K1 to prevent Fas-mediated apoptosis (Berkova et al., 2009; Tomlinson & Damania, 2004; Wang et al., 2007) in the presence of Hsp90β and/or Erdj3 siRNAs described above. 293-K1 or 293-Vec cells were transfected with 200pmol of siRNAs targeting Erdj3 and/or Hsp90β, or K1. To control for any undesirable effect of siRNA transfection on apoptosis, non-targeting siRNA, as well as a GFP-targeting siRNA were used for comparison. Thirty-six hours post-siRNA transfection, the cells were subjected to Fas-receptor antibody stimulation for 12 h to stimulate Fas-receptor dependent apoptosis. Equivalent number of cells were harvested, lysed, and subjected to a fluorescence-based assay that measures caspase-3 activity as a readout of apoptosis (Tomlinson & Damania, 2004). Caspase-3 activity in the antibody stimulated 293-K1 cells was calculated as a percent of the caspase-3 activity in the antibody stimulated 293-Vec control cells. Fig. 6A shows the percent of activated caspase-3 detected in the Fas antibody-stimulated 293-K1 cells compared to the stimulated 293-Vec cells with the same siRNA transfection. Overall, the 293-K1 cells displayed a fifty percent decrease in caspase-3 activation compared to the 293-Vec cells, after Fas-receptor stimulation. This is in concordance with what we and others have previously reported (Berkova et al., 2009; Tomlinson & Damania, 2004; Wang et al., 2007). Transfection of either Hsp90β siRNA, Hsp40/Erdj3 siRNA, GFP siRNA, or non-targeting siRNA into the 293-K1 cells, followed by anti-Fas-receptor antibody stimulation, yielded similar levels of caspase-3 activity in these cells (Fig. 6A) indicating that single knockdown of either the Hsp90β or Hsp40 chaperone did not alter K1’s ability to prevent Fas-mediated apoptosis. However, when both Hsp90β siRNAs and Hsp40/Erdj3 siRNAs were co-transfected into 293-K1 and 293-Vec cells, the 293-K1 cells now displayed similar levels of caspase-3 activity as the 293-Vec cells (Fig. 6A), suggesting that both Hsp90β and Erdj3 are required for K1 to exert its anti-apoptotic function, and that knockdown of both chaperones increases the sensitivity of K1-expressing cells to Fas-mediated apoptosis. The absolute data for the caspase-3 assays are provided in Supplemental Fig. 5A.
Figure 6. K1 anti-apoptotic function is dependent on the endogenous levels of Hsp90β and ER-associated Hsp40.

A. 293-K1 and 293-Vec stable cells were transfected with the indicated panel of siRNAs. At 36 h post-transfection, anti-Fas antibody was added for 12 h to simulate Fas-receptor-dependent apoptosis. Cells were harvested and subjected to a fluorescence-based caspase-3 assay. Caspase-3 activity was measured using the caspase-3 fluorescent substrate, DEVD-AMC. Percent caspase-3 activity in 293-K1 cells compared to 293-Vec cells is plotted on the y-axis, and the siRNA transfected samples are depicted on the x-axis. Error bars represent standard deviations from the mean. The graphs are representative of three independent experiments. B. The experimental setup was identical to that in panel A, except Fas-antibody induction time was increased to 18 h. Cells were then stained with Trypan blue and counted using a hemocytometer. Each sample was performed in quadruplicate. Cell death in 293-K1 cells was plotted as a percentage of cell death in 293-Vec cells. Error bars represent standard deviations from the mean. C. 293-K1 cells were transfected with K1 siRNA or control siLUC for 48h. Protein lysates were subjected to SDS-PAGE and Western blot analysis using an anti-FLAG or actin antibody.
In order to look at the role of Hsp90β and Hsp40/Erdj3 on K1’s ability to prevent cell death, we repeated the experiment with Fas antibody stimulation but extended the incubation time with the antibody to 18 h (instead of 12 h) to analyze cell death of 293-K1 versus 293-Vec cells transfected with the indicated siRNAs (Fig .6B). Cells were stained with trypan blue and the numbers of viable and dead cells were counted under microscopy in four different fields. Cell death in the Fas antibody stimulated 293-K1 cells was calculated as a percentage of cell death seen in 293-Vec cells stimulated with the same antibody. Overall, the trend in Fig. 6B recapitulates that in Fig. 6A. Supplemental Fig. 5B shows the absolute number of dead cells seen in Fas antibody stimulated 293-K1 cells compared to 293-Vec cells. As a control, a Western blot of K1 expression in 293-K1 cells transfected with siLUC versus K1 siRNA is depicted in Fig. 6C to demonstrate effective knockdown of K1 protein levels in the K1 siRNA transfected cells.
Hsp90 inhibitors decrease the proliferation of KSHV-positive PEL-derived B cell lines in vitro
Since Hsp90 and Hsp40 regulate the protein expression and anti-apoptotic function of K1, we wanted to determine if Hsp90 inhibitors can block proliferation of KSHV-positive PEL cells. PEL cells have previously been shown to express K1 transcripts and protein at low levels (Bowser et al., 2002; Lee et al., 2003). We treated PEL cells with DMSO (vehicle control) or 50nM of the GA-derived Hsp90 inhibitors, 17-AAG and 17-DMAG, that are currently in clinical trials (Goetz et al., 2005; Ivy & Schoenfeldt, 2004) for 0, 24, 48, 72, and 96 hours. We found that both inhibitors suppressed the growth of KSHV-harboring PEL cell lines, BCP-1, BCBL-1, and JSC-1, in a MTS-based cell proliferation assay (Fig. 7A). We also determined the IC50 of 17-AAG and 17-DMAG in BCP-1, BCBL-1, and JSC-1 cell lines 72h post-treatment. The IC50 of 17-AAG and 17-DMAG were ~50nM and ~10nM, respectively, in all three cell lines (Fig. 7B and 7C). These IC50 values are consistent with other tumor cell lines that are dependent on Hsp90 (Hollingshead et al., 2005). Hence, 17-AAG and 17-DMAG can effectively inhibit KSHV-infected primary effusion lymphoma cell lines in vitro. We also investigated the effects of 17-DMAG on 293-K1 cells compared to 293-Vec cells. 1×106 293-K1 and 293-Vec cells were incubated with 17-DMAG and a Trypan blue exclusion assay was performed at 24, 48 and 72 hours post-treatment. We found that at all three timepoints tested, there were more live cells present in the 293-K1 cells compared to the 293-Vec cells. This suggests that K1 confers a survival advantage to 293 cells upon Hsp90 inhibition (Supplemental Fig. 6) and that higher concentrations of drug are required to inhibit K1-expressing 293 cells compared to 293 cells that do not express the K1 protein.
Figure 7. The Hsp90 inhibitors, 17-AAG and 17-DMAG, inhibit the proliferation of KSHV-positive PEL cell lines.

A. BCP-1, JSC-1, and BCBL-1 were incubated with 50 nM of 17-AAG or 17-DMAG and subjected to a MTS assay over a period of four days. The absorption at 490 nm in the presence of 50 nM of 17-AAG or 17-DMAG, or equivalent volume of vehicle control (DMSO) is shown on the vertical axis, and time in days after addition of the drugs is shown on the horizontal axis. Error bars represent standard deviation from the mean. B. IC50 values of 17-AAG on BCP-1, JSC-1, and BCBL-1 were determined after 72 h incubation with 17-AAG. A range of concentrations of 17-AAG was used: 0, 1, 10, 100, 1000, or 10000 nM. A nonlinear fit, sigmoidal curve was generated by plotting percent growth inhibition against the log concentration of 17-AAG. Error bars represent SEM. C. IC50 values of 17-DMAG on BCP-1, JSC-1, and BCBL-1 were determined by MTS assays after 72 h incubation with 17-DMAG.
To address whether the decrease in proliferation of 50nM 17-AAG and 17-DMAG is due to the induction of cell death, we treated the same three PEL lines above with 50nM or 500nM of each inhibitor, or DMSO, for 0, 24, 48, 72, and 96 h and assessed cell viability by a Trypan blue exclusion assay. We found that 500nM of 17-AAG and 17-DMAG induced an increase in cell death of PEL cells over time (Fig. 8A). In agreement with this observation, we have found that the LC50 values of 17-AAG and 17-DMAG in BCP-1 are 464.4nM and 523.5nM, respectively (Fig. 8B & 8C) at 72 hours post-treatment. We also confirmed that similar to GA treatment (Fig. 5A), treatment of 293-K1 cells with 500nM of 17-DMAG inhibited K1 protein expression compared to 293-K1 cells treated with vehicle (DMSO) (Fig. 8D).
Figure 8. The Hsp90 inhibitors, 17-AAG and 17-DMAG, induce cell death of KSHV-positive PEL cell lines.
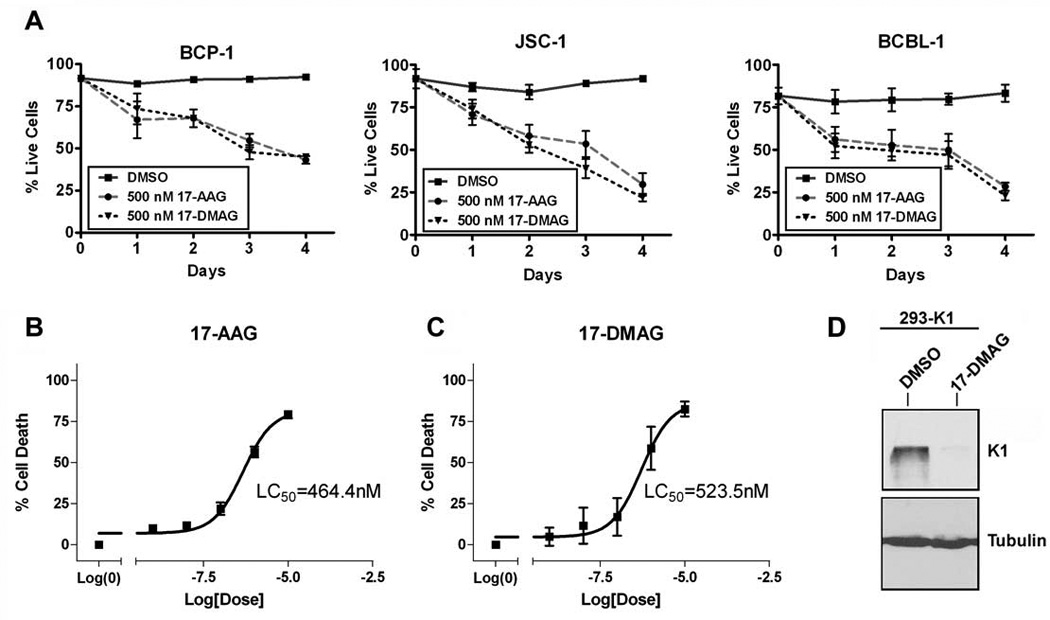
A. BCP-1, JSC-1, and BCBL-1 were incubated with 500 nM of 17-AAG, 17-DMAG, or the equivalent volume of DMSO, and subjected to a Trypan blue exclusion assay over a period of four days. The cells were stained with Trypan blue and counted at the indicated timepoints. Live cells were plotted as a percentage of total cells on the y-axis and the time periods of incubation with 17-AAG or 17-DMAG is depicted on the x-axis. Error bars represent standard deviation from the mean. B. LC50 value of 17-AAG on BCP-1was determined after 72 h incubation with 17-AAG. A range of concentrations of 17-AAG was used: 0, 1, 10, 100, 1000, or 10000 nM. A nonlinear fit, sigmoidal curve was generated by plotting percent growth inhibition against the log concentration of 17-AAG. Error bars represent SEM. C. LC50 value of 17-DMAG on BCP-1 was determined in the same manner as in panel B, except that 17-DMAG was used instead of 17-AAG. D. 293-K1 cells were treated with 500 nM of 17-DMAG or DMSO vehicle for 72 h. Protein lysates were subjected to SDS-PAGE and Western blot analysis using an anti-FLAG antibody to detect K1 protein levels. An anti-tubulin blot is shown as a control.
Based on the inhibitory effect of 17-AAG/17-DMAG on PEL cell proliferation, we speculated that both Hsp90 inhibitors may alter the cell cycle of PEL. We performed cell cycle analysis to address this possibility. JSC-1 and BCBL-1 were treated with 50nM 17-AAG and 17-DMAG for 48 h, and then subjected to propidium staining and flow cytometry. Both PEL cell lines displayed significant G0/G1 arrest and suppressed DNA synthesis upon Hsp90 inhibition (Fig.9). 17-DMAG was a stronger inhibitor of the cell cycle than 17-AAG (Fig. 9). Thus, the cell cycle arrest phenotype seen in the presence of 17-AAG and 17-DMAG corroborates the inhibition of cell proliferation we observed in the MTS assay for the different PEL cell lines (Fig. 7).
Figure 9. 17-AAG and 17-DMAG affect induce G0/G1 arrest of KSHV-positive PEL cell lines.
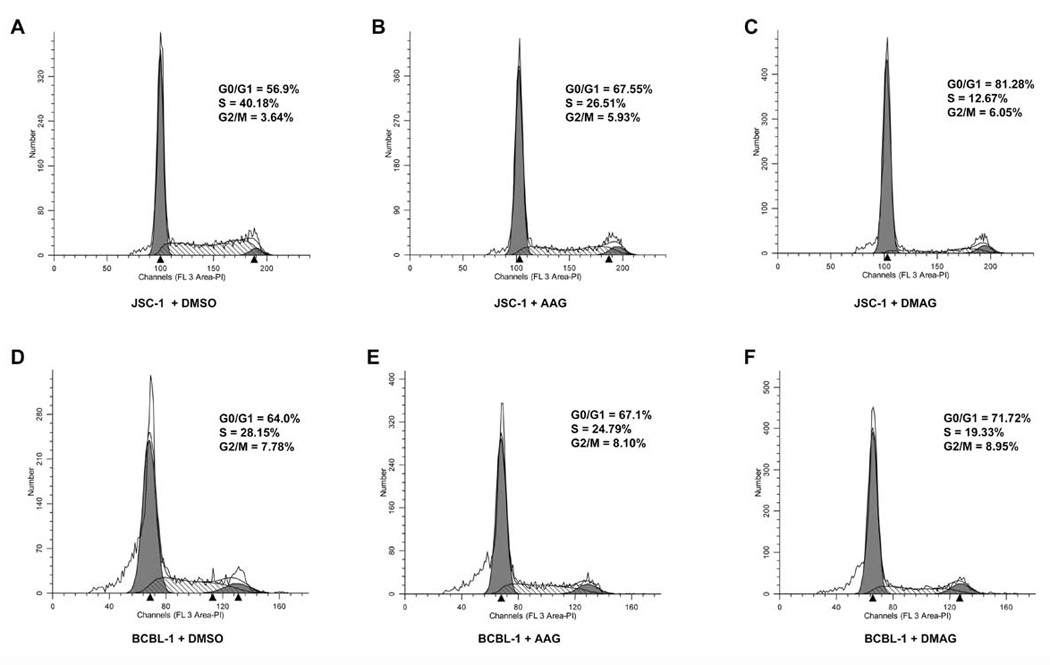
1×106 live JSC-1 (A–C) and BCBL-1 (D–F) were incubated with 50 nM of 17-AAG (B & E), 17-DMAG, (C & F) or equivalent volume of DMSO (A & D) for 48 h. The cells were washed with 1× PBS, fixed in 70% ethanol, and stained with propidium iodide and subjected to cell cycle analysis using flow cytometry. The percentages of cells at different stages in the cell cycle (G0/G1, S, G2/M) are shown.
DISCUSSION
KSHV K1 is a viral oncoprotein that has been shown to transform cells, induce tumor formation (Damania et al., 1999; Prakash et al., 2002), as well as inhibit apoptosis (Berkova et al., 2009; Tomlinson & Damania, 2004; Wang et al., 2007). Furthermore, the K1 protein activates the PI3K/Akt signaling pathway in endothelial cells and B cells (Tomlinson & Damania, 2004; Wang et al., 2006). However, relatively little is known about how K1 protein expression and function are modulated. Using TAP, we identified Hsp90β and the ER-associated Hsp40/Erdj3 protein as cellular partners of K1. This was corroborated by co-immunoprecipitations that confirmed these protein-protein interactions. Importantly, we have shown that K1’s interaction with Hsp90β (but not Hsp40/Erdj3) is dependent on the presence of ATP. We also found that K1 can interact with the Hsp90α isoform. Interestingly, both Hsp90 and K1 have individually been shown to modulate the phosphorylation or function of several important signaling molecules such as VEGF receptor (Le Boeuf et al., 2004; Wang et al., 2006; Wang et al., 2004), Akt (Sato et al., 2000; Tomlinson & Damania, 2004; Wang et al., 2006), Lyn (Lee et al., 2005; Prakash et al., 2005; Prakash et al., 2002; Trentin et al., 2008), PDK1 (Fujita et al., 2002; Wang et al., 2006), BCR (Lee et al., 2000; Shinozaki et al., 2006; Tomlinson & Damania, 2008), and mTOR (Ohji et al., 2006; Wang et al., 2006).
In KSHV, as well as other herpesviruses, Hsp90 also regulates subcellular trafficking and function of viral proteins (Burch & Weller, 2005; Field et al., 2003; Livingston et al., 2008). A novel finding of this report is that K1 appears to be a viral client protein of Hsp90 and the ER-associated Hsp40/Hsp70 system. This is particularly intriguing as it suggests that the protein expression of the K1 viral oncogene can be manipulated with existing Hsp90 inhibitors as well as siRNA/small hairpin RNA targeting against Hsp90 and/or Hsp40/Erdj3 chaperones. Additionally, we have shown that K1 interacts and colocalizes with Hsp90 and Hsp40/Erdj3 in B cells. K1 and Hsp90 expression can also be detected in PEL tumors. Thus, our data validate Hsp90 as an important therapeutic target for treating KSHV-associated lymphomas.
Using the Hsp90 inhibitor GA, we showed that Hsp90 ATPase activity and chaperoning function is required for optimal expression of K1. GA inhibits all the known Hsp90 homologues, including cytosolic/extracellular Hsp90α and Hsp90β, as well as Grp94/Gp96 (ER-associated Hsp90), and Trap1 (mitochondria-associated Hsp90). Thus, it is possible that Hsp90 homologues other than Hsp90α and Hsp90β may also associate with K1 and modulate its expression and function. Similar to GA treatment, genetic knockdown of either Hsp90β or Hsp40/Erdj3 using siRNA also dramatically reduced K1 protein expression.
We found that depletion of both endogenous Hsp90β and ER-associated Hsp40 negated the ability of K1 to prevent Fas-mediated apoptosis. However, depletion of only Hsp90β or Hsp40 by itself had no effect. One explanation is that knockdown of both Hsp90β and Hsp40 reduced K1 expression levels more robustly than single knockdowns of these proteins. A second possibility is that there are two different pools of K1 whose expression and/or function are regulated by Hsp90β and Hsp40. K1 has been shown to be expressed on the plasma membrane as well as in the ER (Lee et al., 2000). Since Hsp90β is predominantly cytosolic and Hsp40/Erdj3 is ER-associated, one may speculate that these different pools of K1 are topologically distinct. Hence, knockdown of the ER-restricted chaperone may reduce K1 levels in the ER, but not affect cytosolic or cell surface levels of K1, and vice versa. Thus, single knockdowns of either Hsp90β̣ or Hsp40/Erdj3 may not have reduced K1 protein levels in all cellular compartments to the same extent as double knockdowns of both these chaperones.
We found that K1 could interact with both Hsp90β and Hsp90α isoforms. Besides being cytosolic, Hsp90α (Eustace et al., 2004; Li et al., 2007) and Hsp90β (Sidera et al., 2004) have been found on the cell surface i.e. extracellularly. Thus, the N-terminal domain of K1 may co-internalize with extracellular Hsp90. Since K1 internalization has been linked to its signaling function (Tomlinson & Damania, 2008), Hsp90 likely plays a role in chaperoning K1 as it is endocytosed. Additionally, since the N-terminal domain of K1 interacts with Hsp90β, it is also possible that Hsp90β interacts with the K1 N-terminus during de novo biogenesis of K1 protein when the growing peptide is transiting from the cytoplasm to the ER. Indeed, Hsp90β has been shown to be involved in the protein translation of the BCR (Shinozaki et al., 2006). Furthermore, since the ER-associated Hsp40/Erdj3 functions as a co-chaperone with Hsp70/BiP for unfolded/nascent proteins, including the unassembled immunoglobulin heavy chain (Shen & Hendershot, 2005), Hsp40/Erdj3 may also participate in the folding of newly synthesized/unfolded or misfolded K1 within the ER.
Our data demonstrate that Hsp90 inhibition by 17-AAG and 17-DMAG at low concentrations results in decreased cell proliferation and G0/G1 arrest, albeit at higher concentrations, 17-AAG and 17-DMAG can also induce cell death of KSHV-positive PEL cells. A potential mechanism for these observations is that Hsp90 inhibition leads to a decrease in K1 protein expression, and this has a two pronged effect on the PI3K/Akt/mTOR pathway. This is because Hsp90 inhibition suppresses activation of the PI3K/Akt/mTOR pathway which is normally activated by the K1 viral oncoprotein, and indirectly by Hsp90 through stabilization of and maintenance of Akt kinase activity. Since PI3K, Akt, and mTOR are cell survival kinases, inhibition of Hsp90 destabilizes K1 protein and suppresses its ability to enhance PEL cell proliferation and cell survival through this pathway. This model would predict that Hsp90 inhibition would lead to decreased proliferation of cells that do not express K1 compared to proliferation of cells that do express K1. Indeed, we observed that more 293-K1 cells survived in the presence of Hsp90 inhibitor compared to 293-Vec cells (Supplemental Figure 6).
We also speculate that there are several other KSHV proteins that utilize molecular chaperones to modulate their expression and function. Field et al. previously reported that the KSHV latent viral FLICE inhibitory protein (vFLIP) requires Hsp90 to complex with IκB kinase (IKK) and activate the NF-κB pathway (Field et al., 2003). Here we report that both Hsp90 and Hsp40 chaperones were needed for K1 protein expression and its anti-apoptotic function. Taken together, our studies provide additional rationale for using Hsp90 inhibitors to treat PEL and other KSHV-related malignancies.
MATERIALS AND METHODS
Cell culture
293-K1 and 293-Vec stable cells were established and maintained in 1mg/ml G418 selection in DMEM medium supplemented with 10% FBS in 5% CO2. BCP-1, JSC-1, and BCBL-1 cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS, 2mM L-glutamate, 0.05mM 2-mercaptoethanol, and 0.075% sodium bicarbonate in 5% CO2.
Antibodies
Rabbit anti-K1 antibody was a kind gift from Dr. Jae Jung. Anti-Hsp90 (ab1429) and anti-Hsp70 antibodies (ab2787) were purchased from Abcam. Anti-Hsp90α and Hsp90β antibodies were purchased from Stressgen (SPS-771 and SPA-843). Anti-Hsp40 (DNAJB11) antibody was obtained from Sigma (HPA010814). Anti-Akt and anti-phospho-Akt (S473) were purchased from Cell Signaling, while anti-actin antibody was purchased from Santa Cruz (C16). Anti-FLAG M2 resin was obtained from Sigma for immunoprecipitation of K1. Normal mouse IgG (sc-2025), normal rabbit IgG (sc-2027), and protein A/G PLUS-Agarose (sc-2003) were purchased from Santa Cruz. HRP-conjugated anti-ECS antibody used for FLAG immunoblotting was purchased from Bethyl (A190-101P).
Inhibitors and small-interfering RNAs (siRNAs)
Geldanamycin, 17-(Allylamino)-17-demethoxygeldanamycin (17-AAG) and 17-Dimethylamino-ethylamino-17-demethoxygeldanamycin (17-DMAG) were purchased from Invivogen. Stealth siRNAs targeting Hsp90β and Erdj3 were purchased from Invitrogen. Anti-Luc siRNA-1, Accell non-targeting siRNA pool, and GFP siRNA duplex were purchased from Thermo Scientific. The siRNAs directed against K1 (CCACAACAATTGCAGGATT-UU and CCATGCAACCACACATAAA-UU) were designed by Dharmacon siDESIGN® Center Custom siRNA Design Tool and purchased from Dharmacon.
Tandem affinity purification
We generated the FLAG HA tandem tagged K1 construct (TAP-K1) by QuikChange site-directed mutagenesis. The oligonucleotides 5'-ACGACGACAAGGGTACCTACCCATACGACGTCCCAGACTACGCTCTTTATGTGCTAT CGTC-3' and 5'-GACGATAGCACATAAAGAGCGTAGTCTGGGACGTCGTATGGGTAGGTACCCTTGTC GTCGT-3' were used to introduce the HA epitope sequence between FLAG and K1 at the N-terminus. pcDNA3-K1 and empty pcDNA3 vector were transfected into 293 cells using FuGENE 6 reagent (Roche) and selected with 1mg/ml G418 to establish 293-K1 and 293-Vec stable cells, respectively. Forty confluent T175-flasks of each cell line were harvested and washed twice in cold PBS. Cells were pelleted and stored at −80°C. 293-K1 and 293-Vec stable cell pellets were thawed and lysed in NP40 buffer supplemented with PMSF, protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktails 1 & 2 (Sigma). The lysates were pelleted and the supernatants (20mg/ml) were subjected to TAP using the FLAG HA Tandem Affinity Purification Kit (Sigma). After TAP, the samples were eluted in 2× Laemmli sample buffer (LSB) and resolved on a 12% NuPAGE Novex Bis-Tris Mini Gel (Invitrogen). The gel was Coomassie-stained and submitted to the UNC Proteomics Center for mass spectroscopy. Protein bands unique to K1 expressing cells were excised, trypsin-digested, and subjected to MALDI-TOF mass spectroscopy (MS) analysis.
Immunoprecipitation
For each reaction, confluent cells in a 10-cm dish were harvested, washed twice in ice cold PBS, and lysed in RIPA buffer supplemented with PMSF, and protease and phosphatase inhibitors. Extract was incubated with 1µg of primary antibody at 4°C for 4 h and Protein A/G PLUS-Agarose (or FLAG resin) was added to the lysate and incubated at 4°C overnight. After binding, the resins were washed 4× with RIPA buffer. Immunoprecipitated protein complexes were eluted in 2× LSB (or 3× FLAG peptide).
MTS assay
The MTS assay was performed according to the manufacturer’s protocols (Promega). Cells were incubated for 4 days with indicated amounts of drug at a seeding density of 1×105 cells/ml in triplicate. Absorbance was read daily at a wavelength of 490 nm.
Caspase-3 assay
Cells were transfected with siRNAs using Superfect (Qiagen). At 36 h posttransfection, the transfected cells were stimulated with anti-Fas cross-linking antibody (clone CH11, Millipore) at 0.2µg/ml in 1% FBS, and cells were harvested 12 h later (Tomlinson & Damania, 2004). Apoptosis was analyzed by using an ApoAlert caspase-3 fluorimetric assay kit from Clontech. Absorbance was read on a FLUOstar Optima fluorometric plate reader with a 400-nm excitation and 505-nm emission filter.
Cell-cycle analysis
Cells fixed in 70% ethanol were resuspended in PBS with 20µg/mL propidium iodide and 200µg/mL RNaseA. Flow cytometric analysis was performed with a Becton Dickinson FACScan and ModFitLT V3.2.1 program.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prasanna Bhende for help with immunohistochemistry and cell cycle analysis and Stuart Krall for technical assistance. We thank Dr. Jae Jung for providing us with the anti-K1 antibody and members of the Damania and Dittmer lab for informative discussions. We also thank the UNC Proteomics Center for processing of the protein samples and subsequent mass spectroscopy analysis. BD is a Leukemia & Lymphoma Society Scholar, American Heart Association established investigator, and a Burroughs Wellcome Fund Investigator in Infectious Disease. This work was supported by grants CA096500 and CA019014 from NIH, a University of Pennsylvania CFAR pilot project grant (P30-AI045008) to BD. KWW was supported in part by NIAID training grant T32-AI007001 and MSTP grant T32-GM008719. BD is a Leukemia & Lymphoma Society Scholar and Burroughs Welcome Fund Investigator in Infectious Disease.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary information is available from Oncogene’s website.
REFERENCES
- Berkova Z, Wang S, Wise JF, Maeng H, Ji Y, Samaniego F. J Natl Cancer Inst. 2009;101:399–411. doi: 10.1093/jnci/djn516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser BS, DeWire SM, Damania B. J Virol. 2002;76:12574–12583. doi: 10.1128/JVI.76.24.12574-12583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch AD, Weller SK. J Virol. 2005;79:10740–10749. doi: 10.1128/JVI.79.16.10740-10749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chandriani S, Ganem D. J Virol. 2010 doi: 10.1128/JVI.02723-09. J Virol. Mar 10. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Damania B, DeMaria M, Jung JU, Desrosiers RC. J Virol. 2000;74:2721–2730. doi: 10.1128/jvi.74.6.2721-2730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damania B, Li M, Choi JK, Alexander L, Jung JU, Desrosiers RC. J Virol. 1999;73:5123–5131. doi: 10.1128/jvi.73.6.5123-5131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL, Jay DG. Nat Cell Biol. 2004;6:507–514. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. J Cell Sci. 2003;116:3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Ishida A, Tsuruo T. J Biol Chem. 2002;277:10346–10353. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- Gao T, Newton AC. J Biol Chem. 2002;277:31585–31592. doi: 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- Gessain A, Sudaka A, Briere J, Fouchard N, Nicola MA, Rio B, Arborio M, Troussard X, Audouin J, Diebold J, de The G. Blood. 1996;87:414–416. [PubMed] [Google Scholar]
- Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, Adjei AA, Sloan J, Atherton P, Vasile V, Salazaar S, Adjei A, Croghan G, Erlichman C. J Clin Oncol. 2005;23:1078–1087. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- Hollingshead M, Alley M, Burger AM, Borgel S, Pacula-Cox C, Fiebig HH, Sausville EA. Cancer Chemother Pharmacol. 2005;56:115–125. doi: 10.1007/s00280-004-0939-2. [DOI] [PubMed] [Google Scholar]
- Ivy PS, Schoenfeldt M. Oncology (Williston Park) 2004;18:610, 615, 619–620. [PubMed] [Google Scholar]
- Lagunoff M, Ganem D. Virology. 1997;236:147–154. doi: 10.1006/viro.1997.8713. [DOI] [PubMed] [Google Scholar]
- Lagunoff M, Majeti R, Weiss A, Ganem D. Proc Natl Acad Sci U S A. 1999;96:5704–5709. doi: 10.1073/pnas.96.10.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Boeuf F, Houle F, Huot J. J Biol Chem. 2004;279:39175–39185. doi: 10.1074/jbc.M405493200. [DOI] [PubMed] [Google Scholar]
- Lee BS, Alvarez X, Ishido S, Lackner AA, Jung JU. J Exp Med. 2000;192:11–21. doi: 10.1084/jem.192.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Connole M, Tang Z, Harris NL, Jung JU. J Virol. 2003;77:8072–8086. doi: 10.1128/JVI.77.14.8072-8086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Lee SH, Feng P, Chang H, Cho NH, Jung JU. J Virol. 2005;79:12173–12184. doi: 10.1128/JVI.79.19.12173-12184.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. Mol Cell Biol. 1998a;18:5219–5228. doi: 10.1128/mcb.18.9.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. Nat Med. 1998b;4:435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- Lewis J, Devin A, Miller A, Lin Y, Rodriguez Y, Neckers L, Liu ZG. J Biol Chem. 2000;275:10519–10526. doi: 10.1074/jbc.275.14.10519. [DOI] [PubMed] [Google Scholar]
- Li W, Li Y, Guan S, Fan J, Cheng CF, Bright AM, Chinn C, Chen M, Woodley DT. Embo J. 2007;26:1221–1233. doi: 10.1038/sj.emboj.7601579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. J Virol. 2008;82:6324–6336. doi: 10.1128/JVI.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohji G, Hidayat S, Nakashima A, Tokunaga C, Oshiro N, Yoshino K, Yokono K, Kikkawa U, Yonezawa K. J Biochem. 2006;139:129–135. doi: 10.1093/jb/mvj008. [DOI] [PubMed] [Google Scholar]
- Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. Embo J. 2001;20:4478–4489. doi: 10.1093/emboj/20.16.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N. J Biol Chem. 2003;278:47449–47458. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- Picard D. Nat Cell Biol. 2004;6:479–480. doi: 10.1038/ncb0604-479. [DOI] [PubMed] [Google Scholar]
- Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Prakash O, Swamy OR, Peng X, Tang ZY, Li L, Larson JE, Cohen JC, Gill J, Farr G, Wang S, Samaniego F. Blood. 2005;105:3987–3994. doi: 10.1182/blood-2004-07-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G, Samaniego F. J Natl Cancer Inst. 2002;94:926–935. doi: 10.1093/jnci/94.12.926. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Samaniego F, Pati S, Karp JE, Prakash O, Bose D. J Natl Cancer Inst Monogr. 2001:15–23. doi: 10.1093/oxfordjournals.jncimonographs.a024252. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. J Leukoc Biol. 2007;81:15–27. doi: 10.1189/jlb.0306167. [DOI] [PubMed] [Google Scholar]
- Shen Y, Hendershot LM. Mol Biol Cell. 2005;16:40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki F, Minami M, Chiba T, Suzuki M, Yoshimatsu K, Ichikawa Y, Terasawa K, Emori Y, Matsumoto K, Kurosaki T, Nakai A, Tanaka K, Minami Y. J Biol Chem. 2006;281:16361–16369. doi: 10.1074/jbc.M600891200. [DOI] [PubMed] [Google Scholar]
- Sidera K, Samiotaki M, Yfanti E, Panayotou G, Patsavoudi E. J Biol Chem. 2004;279:45379–45388. doi: 10.1074/jbc.M405486200. [DOI] [PubMed] [Google Scholar]
- Smith DF, Whitesell L, Nair SC, Chen S, Prapapanich V, Rimerman RA. Mol Cell Biol. 1995;15:6804–6812. doi: 10.1128/mcb.15.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- Tomlinson CC, Damania B. J Virol. 2004;78:1918–1927. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson CC, Damania B. J Virol. 2008;82:6514–6523. doi: 10.1128/JVI.02637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentin L, Frasson M, Donella-Deana A, Frezzato F, Pagano MA, Tibaldi E, Gattazzo C, Zambello R, Semenzato G, Brunati AM. Blood. 2008;112:4665–4674. doi: 10.1182/blood-2008-02-139139. [DOI] [PubMed] [Google Scholar]
- Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Cancer Res. 2006;66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- Wang L, Wakisaka N, Tomlinson CC, DeWire SM, Krall S, Pagano JS, Damania B. Cancer Res. 2004;64:2774–2781. doi: 10.1158/0008-5472.can-03-3653. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang S, Maeng H, Young DP, Prakash O, Fayad LE, Younes A, Samaniego F. Blood. 2007;109:2174–2182. doi: 10.1182/blood-2006-02-003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Sutphin P, An WG, Schulte T, Blagosklonny MV, Neckers L. Oncogene. 1997;14:2809–2816. doi: 10.1038/sj.onc.1201120. [DOI] [PubMed] [Google Scholar]
- Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. Cancer Res. 2003;63:7777–7784. [PubMed] [Google Scholar]
- Zhang C, Guy CL. Plant Physiol Biochem. 2005;43:13–18. doi: 10.1016/j.plaphy.2004.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.