

Abstract
Aims
To assess the relationship between genetic polymorphisms and indinavir pharmacokinetic variability and to study the link between concentrations and short-term response or metabolic safety.
Methods
Forty protease inhibitor naive-patients initiating indinavir/ritonavir containing HAART and enrolled in the COPHAR 2 - ANRS 111 trial were studied. At week 2, 4 blood samples were taken before and up to 6 hours following drug intake. A population pharmacokinetic analysis was performed using the Stochastic Approximation Expectation Maximization (SAEM) algorithm implemented in the MONOLIX software. Indinavir area under the concentration-time curve (AUC), maximal (Cmax) and trough concentrations (Ctrough) were derived from the population model and tested for correlation with short-term viral response and safety measurements, while ritonavir AUC, Cmax and Ctrough were tested for correlation with short-term biochemical safety.
Results
A one-compartment model with first-order absorption and elimination best described both indinavir and ritonavir concentrations. For indinavir, the estimated clearance and volume of distribution were 22.2 L/h and 97.3 L respectively. The eight patients *1B/*1B for CYP3A4 gene had an absorption decreased by 70% compared to *1A/*1B or *1A/*1A genotypes (0.5 versus 2.1, P=0.04, likelihood ratio test by permutation). Indinavir AUC and Ctrough were positively correlated with the HIV RNA decrease between week 0 and week 2 (r=−0.4, P = 0.03 and r=−0.4, P = 0.03, respectively). Patients with the *1B/*1B genotype had significantly lower indinavir Cmax (median 3.6 [range 2.1 – 5.2] ng/mL versus 4.4 [2.2 – 8.3] ng/mL, P=0.04) and a lower triglycerides increase during the first 4 weeks of treatment (0.1 [−0.7 – 1.4] versus 0.6 [−0.5 – 1.7] mmol/L, P = 0.02). For ritonavir, the estimated clearance and volume of distribution were 8.3 L/h and 60.7 L respectively and concentrations were not found to be correlated to biochemical safety. Indinavir and ritonavir absorption rate constants were found to be correlated, as well as their apparent volumes of distribution and clearances indicating correlated bioavailability of the two drugs.
Conclusion
CYP3A4*1B polymorphism was found to influence the pharmacokinetics of indinavir and in some extent the biochemical safety of indinavir.
Keywords: Adult; Anti-HIV Agents; adverse effects; pharmacokinetics; Antiretroviral Therapy, Highly Active; Area Under Curve; Clinical Trials as Topic; Cohort Studies; Cytochrome P-450 CYP3A; drug effects; Female; Genotype; HIV Infections; drug therapy; HIV Protease Inhibitors; adverse effects; pharmacokinetics; HIV-1; Humans; Indinavir; adverse effects; pharmacokinetics; Male; Metabolic Clearance Rate; Middle Aged; Models, Statistical; Multicenter Studies as Topic; Pharmacogenetics; Pilot Projects; Polymorphism, Genetic; Prospective Studies; Treatment Outcome
Keywords: Pharmacokinetics, Nonlinear Mixed Effects Modeling, Protease inhibitors, CYP3A4, Safety, Efficacy
1. Introduction
Indinavir has been one of the preferred protease inhibitor (PI) included in highly active antiretroviral therapy (HAART). Nowadays, even if not recommended as initial therapy, indinavir is still used in patients who initiated their therapy with this PI and kept a viral load below the limit of quantification with an acceptable safety profile. Further compared to others PI, indinavir exhibits a high penetration into viral reservoirs such as genital compartments and CNS [1]. It was found that the indinavir better distribution is leading to better outcomes in neurological complications related to HIV [2]. The pharmacokinetics (PK) of indinavir is characterised by high maximal concentrations leading to potential toxicity, notably nephrolithiasis [3] and low minimum concentrations with respect to the 95%inhibitory concentration of the virus. These low residual concentrations result from an extended oxidative metabolism by the cytochrome P450 (CYP3A) isoenzyme [4]. The co-administration of ritonavir, whose molecular structure leads to CYP3A inhibition, therefore enhances exposure to indinavir [5,6]. As a booster, ritonavir is given at lower dose than for therapeutic use, but was shown nevertheless to influence metabolic profiles especially with respect to lipids disorders [7,8].
The large inter-patient and intra-patient variability of indinavir pharmacokinetics is well referenced [9–11]. Genetic polymorphisms could partly explain this variability, as far as proteins involved in the metabolism and transport of PI are concerned. Few studies have investigated the impact on indinavir pharmacokinetics of ABCB1 polymorphisms, a gene coding for P-glycoprotein, and of CYP3A5 and CYP3A4*1B polymorphisms. Solas et al. [12] have shown that the ABCB1 C3435T genotype affects the absorption constant of indinavir whereas Verstuyft et al. [13] have reported an absence of association. Anderson et al. [14] have observed that CYP3A5 expressors (CYP3A5*1 carriers) have significantly faster oral clearance than non-expressors. To date no relationship was found between CYP3A4*1B polymorphism and alterations in CYP3A substrate metabolism, but clinical data have shown an association between CYP3A*1B polymorphism and disease risk/treatment toxicity [15].
Efficacy [16,17] as well as adverse events [18,3] has been related to indinavir plasma concentrations. Thus, therapeutic drug-monitoring appears to be a potent tool to achieve un-detectable HIV-RNA and prevent toxicity for this drug. The COPHAR 2 - ANRS 111 trial is a multi-centre non-comparative pilot trial of early therapeutic drug-monitoring in HIV-positive patients naive of protease inhibitor containing highly active antiretroviral treatment (HAART) [19]. We focused on the PK sub-study from the group of patients receiving indinavir boosted with ritonavir. The aims of this paper were to estimate the population PK parameters and variability of indinavir and ritonavir in HIV patients, to evaluate the impact of genetic polymorphisms on indinavir PK and to study the link between indinavir concentrations and short-term efficacy and metabolic safety.
2. Methods
2.1. Study
The COPHAR 2 - ANRS 111 study is a multi-centre non-comparative prospective pilot trial of early-dose adaptation in HIV-positive patients naive of protease inhibitors (PI) starting a PI containing HAART treatment. The trial started on July 2002 and was completed by the end of March 2005. The objective was to assess the benefit of pharmacological advice based on trough plasma concentrations of PI. The study involved three groups treated with indinavir, nelfinavir or lopinavir respectively. In the present study we analysed the data obtained during the first month of treatment in the indinavir group. A similar analysis of the data in the nelfinavir group was performed by Hirt et al [20,19], details of the study can be found in that paper.
Patients were required to have a baseline plasma viral load value > 1000 copies/mL, and to be naïve of treatment with protease inhibitor. Patients initiated a HAART treatment containing 400, 600 or 800 mg of indinavir b.i.d. associated with ritonavir booster (100 mg b.i.d.) and two nucleoside analogues. The first dose was left to physicians’ discretion and no dose adaptation was performed from week 0 (W0) to W4. A detailed pharmacokinetic study was performed at W2. Adherence was evaluated at W2 by means of a validated auto-questionnaire [21] and patients were classified as adherent when reporting no shift in their treatment schedule during the last 4 days and non-adherent otherwise.
Viral load and CD4 count were collected at baseline (D0) and at W2. Biochemical profiles of total cholesterol, high density lipoprotein cholesterol, triglyceride and glycaemia as well as creatinine clearance and clinical events (diarrhoea grade of 2) were collected 4 weeks before treatment initiation (W-4) and at W4.
The study was performed in accordance with the Declaration of Helsinki and its amendments. All subjects gave written informed consent and the protocol was approved by the Ethic Committee of the Bicêtre Hospital (France) as well as the amendment for the pharmacogenetic study.
2.2. Indinavir and ritonavir concentration measurements
During a visit to the hospital at W2, the patients were sampled on arrival to measure trough concentrations. Patients were asked to record the time at which the dose was taken on the previous evening. Then they were given their medications, and sampled again 1 h, 3 h and 6 h after drug administration. Plasma concentrations were assumed at steady state with trough concentrations considered as following the drug intake using the delay reported by the patient from previous dosing. Plasma concentrations were determined in the laboratories of the hospitals by specific high-performance liquid chromatography. The participant laboratories were cross-validated before starting the study. Results of the blind inter-laboratory quality control at three concentrations for indinavir and for ritonavir were within 15% of the target values for medium and high values and within 20% for low values. Lower limits of quantification (LOQ) were 0.02 mg/L for indinavir and 0.025 mg/L for ritonavir.
2.3. Genetic polymorphisms
All the genotyping analyses were performed in the same laboratory. Total deoxyribonucleic acid (DNA) was extracted from plasma samples by use of the QIAamp DNA Blood Mini Kit (Qiagen, Courtaboeuf, France). ABCB1 polymorphisms in exons 21 (GG, GT, TT) and 26 (CC, CT, TT) were determined by use of previously-published methods [22]. Genotyping of CYP3A5 (*1*1, *1*3, *3*3, *1*6, *6*6) was performed by real-time polymerase chain reaction by use of TaqMan MGB probe technology (Applied Biosystems). Genotyping for CYP3A4 (*1B*1B, *1B*1A, *1A*1A) was determined by polymerase chain reaction (PCR) followed by direct sequencing. PCR was performed by use of a GenAmp PCR System 9700 (Applied Biosystems, Courtaboeuf, France) according to a previously-published method [23]. Amplified DNA was purified by use of the QiaQuick DNA Purification System (Qiagen) and sequenced by use of BigDye Terminator chemistry and an ABI PRISM 3100 genetic analyser (Applied Biosystems). For each genotyping analysis, at least 2 positive controls were used: 1 homozygous for the wild-type allele and 1 heterozygous and, when available, 1 homozygous for the mutated allele. These controls were DNA that had already been sequenced.
Allele frequencies (p for the wild allele and q = 1 − p for the mutant allele) were estimated by gene counting. Departure from Hardy-Weinberg proportions (p2, 2pq, q2) was tested by a χ2 test with 1 degree of freedom within each ethnic group [24]. We used two approaches to define patients belonging to an ethnic group: i. classification on the patient town, birth area and nationality and ii. classification by means of their genotype information using the Structure software [25]. This software is based on a Bayesian approach and computes a posteriori probabilities for each individual of belonging to a given ethnic group. We assumed each locus to be at the Hardy-Weinberg equilibrium and patients to originate in one ethnic group (with its own characteristic set of allele frequencies).
2.4. Population pharmacokinetic analysis
We used a population approach to analyse the concentration-time data at W2 for indinavir and for ritonavir separately. Model fitting and estimation of the population model parameters were performed by use of the Stochastic Approximation Expectation Maximization algorithm (SAEM) for nonlinear mixed-effects models implemented in the MONOLIX software version 2.1 [26–28]. Both indinavir and ritonavir concentrations were fitted by a one-compartment model with first-order absorption and first-order elimination parameterised in absorption rate constant (ka), oral clearance (Cl/F) and oral volume of distribution (V/F). Each model was assumed at steady state with trough concentrations considered as following the drug intake.
An exponential model was used for inter-individual variability where random effects were assumed to follow a normal distribution with zero mean and diagonal variance matrix. Additive, proportional and combined error models were tested and model choice was based on the likelihood ratio test (LRT) and goodness-of-fit plots (observed vs. predicted population and individual concentrations, population and individual weighted residuals vs. predicted concentrations and vs. time). We performed a visual predictive check (VPC) with 1000 simulated data sets to evaluate the basic model [29].
Interaction between ritonavir and indinavir pharmacokinetics was evaluated with the individual parameters estimated from the basic model for each drug. All the different correlations were tested with the Spearman non-parametric correlation test.
2.5. Assessment of the effect of covariates
The effects of the following covariates were evaluated from the basic model: dose, concomitant use of the zidovudine lamivudine combination (AZT/3TC), co-infection by hepatitis C or B (VHC/VHB), adherence as previously defined, sex, ethnic group, the four studied genetic polymorphisms (ABCB1 exon 26, ABCB1 exon 21, CYP3A5 and CYP3A4) and the CDC classification for HIV infection as categorical variables, age, body mass index (BMI), body weight, creatinine clearance, albumin and orosomucoid levels as continous variables. The latter were centered to the median and log-transformed for model interpretation convenience.
Each of the four genetic polymorphisms was analysed by means of two binary categorisations: first, wild homozygotes versus heterozygotes or mutant homozygotes, second, heterozygotes or wild homozygotes versus mutant homozygotes. Categorisation in three classes was also tested: wild homozygotes versus heterozygotes versus mutant homozygotes. Missing continuous covariates were replaced with the median and patients with missing dicrete covariates were discarded for the corresponding analysis. The effects of covariates on the empirical Bayes estimates (EBE) of each individual pharmacokinetic parameters from the basic model were tested with the Wilcoxon non-parametric test for categorical variables and the Spearman non-parametric correlation test for continuous variables. The population covariate model was built with the covariates which were found to have an effect in this first step with a P-value < 0.1. When a genetic covariate was found to have an effect whatever the categorisation, the same categorisation as other genetic covariates also found to have an effect was chosen in model selection for consistency.
A forward selection of these covariates for the population model was performed by use of the likelihood ratio test (LRT) with a significance threshold at P < 0.05. From this ascending method, a backward elimination procedure was performed. In order to correct the inflation of the LRT type I error on small sample size [30], the backward selection was realized using permutation [31]. More specifically, 1000 data sets are generated by permuting the rows of the covariates matrix from the original data set. For each covariate, one likelihood ratio statistic LRTobs is estimated from the original data and one likelihood ratio statistic LRT perm is estimated from each of the 1000 data sets. Thus, we obtain j=1,…,1000 LRTpermj. The permutation P-value is the proportion: card(LRTpermj > LRTobs)/1000.
2.6. Short-term efficacy and safety and link with concentrations
As there was no change of dose before W4, we studied the link between concentration at W2 and efficacy or safety during the first two or four weeks of treatment. For short-term efficacy, the difference of log viral load between the day of treatment initiation and W2 (ΔlogVL) was studied. The significance of the viral load decrease was tested by a Wilcoxon non-parametric paired test.
Individual area under the concentration-time curve (AUC), maximal plasma concentrations (Cmax) and trough concentrations (Ctrough) of indinavir at steady-state were derived for each patient by use of the EBE of the individual parameters from the basic model and their corresponding dose of indinavir. The relationship between indinavir dose, indinavir AUC, Cmax, Ctrough and ΔlogVL was evaluated by use of the Spearman correlation test. A Wilcoxon non-parametric test was performed to compare ΔlogVL between patients with or without a Ctrough below the lower limit of the therapeutic range used in the COPHAR 2 - ANRS 111 trial: 150 ng/mL.
Safety was analysed through the difference between 4 weeks before and 4 weeks after treatment initiation in total cholesterol (ΔTC), high density lipoprotein cholesterol (ΔHDL), triglyceride (Δtrig) and glycaemia (Δgly) and also through appearance of diarrhoea (grade 2) between treatment initiation andW4. To our knowledge, no precocious biological markers exist for nephrolithiasis, however creatinine clearance has been found to relate to occurence of severe adverse events (including nephrolithiasis) in a multivariate analysis [3]. Thus, we also analysed the difference in creatinine clearance (ΔClCr), computed with the Cockcroft-Gault formula using body weigth and serum creatinine 4 weeks before and 4 weeks after treatment initiation. The significance of these differences was tested by use of a Wilcoxon non-parametric paired test.
We performed Spearman correlation tests between indinavir dose, indinavir AUC, Cmax, Ctrough and ΔTC, ΔHDL, Δtrig, Δgly and ΔClCr. We used Wilcoxon non-parametric tests to compare these differences between patients with or without an indinavir Ctrough over the upper limit defined in the therapeutic index (550 ng/mL). We studied the link between the appearance of grade 2 diarrhoea (yes/no) between treatment initiation and W4 and indinavir dose, indinavir AUC, Cmax, Ctrough using a Wilcoxon non-parametric test and we studied the association with or without an indinavir Ctrough > 550 ng/mL using a Fisher exact test.
We assessed the relation between the genetic polymorphisms remaining in the final population model and indinavir dose, indinavir AUC, Cmax, Ctrough and the relation between these genetic polymorphisms and the short-term efficacy and safety outcomes using Wilcoxon non-parametric tests.
We also derived AUC, Cmax, Ctrough for ritonavir and performed Spearman correlation tests with ΔTC, ΔHDL, Δtrig, Δgly and ΔClCr as well as Wilcoxon non-parametric tests on appearance of grade 2 diarrhoea.
3. Results
3.1. Patients
Forty-two patients were included in this treatment group of the COPHAR 2 ANRS - 111 trial, one patient withdrew from the study and one switched to another protease inhibitor during the first week of treatment. We therefore obtained pharmacokinetic data from 40 patients (27 men, 13 women) with a median age of 36.5 years (20.0 – 59.0). Table 1 describes the main characteristics of the 40 studied patients.
Table 1.
Characteristics of the 40 studied patients
| Median [Range] | |
|---|---|
| Age (years) | 36.5 [20.0 – 59.0] |
| BMI (kg/m2) | 22.6 [17.5 – 35.8] |
| Weight (kg) | 68.0 [45.0 – 103.0) |
| Creatinine clearance (mmol/L) | 95.4 [57.4 – 245.1] |
| Albumin (g/L) | 38.4 [25.5 – 47.4) |
| Orosomucoid (g/L) | 1.0 [0.5 – 2.9] |
| Number of patients (%) | |
| Dose (400/600/800mg) | 26 (65)/8 (20)/6 (15) |
| Coadministration of AZT/3TC (y/n) | 33 (83)/7 (17) |
| Coinfection VHB/VHC (y/n)* | 7 (18)/32 (82) |
| Good adherence (y/n) | 15 (38)/25 (62) |
| Sex (male/female) | 27 (68)/13 (32) |
| Ethnic group (African/Caucasian) | 20 (50)/20 (50) |
| CDC classification for HIV infection (A or B/C) | 30 (75)/10 (25) |
One patient had this information missing
Both approaches to allocate the ethnic group provided corroborating results. Using the civic information we allocated 20 patients to the African group and 20 to the Caucasian group. Given that one patient had missing information for all genotypes, the Structure software allocated 19 patients to the Caucasian group and 20 to the African group. In the resulting two ethnic groups, Hardy-Weinberg proportions were respected for all polymorphisms under study as shown in Table 2.
Table 2.
Distribution of the genetic polymorphisms within each ethnic group and P-values for the Hardy-Weinberg proportions test (H-W)
| Number of patients (%) | H-W P-value | |
|---|---|---|
| African | ||
| ABCB1 exon 26 (CC/CT/TT) | 11 (55)/9 (45)/0 (0) | 0.43 |
| ABCB1 exon 21 (GG/GT/TT) | 19 (95)/1 (5)/0 (0) | 0.99 |
| CYP3A5 (4 *1/3 *1/≤2 *1) | 0 (0)/8 (40)/12 (60) | 0.53 |
| CYP3A4*1B (*1A*1A/*1A*1B/*1B*1B) | 9 (45)/8 (40)/3 (15) | 0.86 |
| Caucasian | ||
| ABCB1 exon 26 (CC/CT/TT) | 2 (12)/12 (70)/3 (18) | 0.22 |
| ABCB1 exon 21 (GG/GT/TT) | 4 (21)/11 (58)/4 (21) | 0.79 |
| CYP3A5 (4 *1/3 *1/≤2 *1) | 18 (100)/0 (0)/0 (0) | 1 |
| CYP3A4*1B (*1A*1A/*1A*1B/*1B*1B) | 0 (0)/3 (16)/16 (84) | 0.93 |
Note: One patient had missing information for all genotypes, ABCB1 exon 26 and CYP3A4 genotypes were also both missing in another patient, in addition one patient had missing genotype for ABCB1 exon 26
3.2. Indinavir pharmacokinetics
For two patients, two samples were missing, the trough and the 6 h concentrations and only the trough concentration was available for another. Among the 155 samples, two indinavir plasma concentrations in one patient were below the limit of quantification (at 1 h and at trough) and were discarded from the analysis. Figure 1a displays indinavir plasma concentrations at W2 versus time. It shows that there is a high inter-individual variability.
Fig. 1.
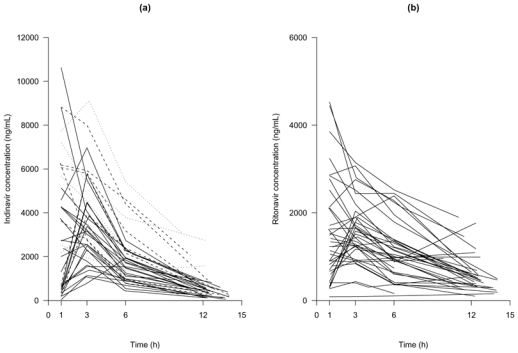
Observed plasma indinavir concentration (a) and plasma ritonavir concentration (b) versus time collected two weeks after treatment initiation, in 40 HIV naive-patients receiving indinavir plus 100 mg of ritonavir b.i.d. In the indinavir plot the plain lines correspond to an indinavir dose of 400 mg b.i.d., the dashed lines to 600 mg b.i.d. and the dotted lines to 800 mg b.i.d. Sampling times following drug administration were measured by the nurse. Concentrations were assumed at steady state, trough concentrations are displayed as following the drug intake at sampling times deduced from the patient record.
The best error model was a proportional error model. The population estimates are displayed in Table 3. All the relative standard errors (RSE) were below 25% with the exception of ka and ωV/F (around 30 and 60% respectively). The inter-individual variance of ka in this study was rather important (above 100%). The simulated median and the 90th interval are displayed in Figure 2a together with all the observed concentrations of indinavir. This graph shows good evidence of the adequacy of the model.
Table 3.
Population pharmacokinetic parameters of indinavir for the basic and the final model: estimates and relative standard error (RSE)
| Parameters | Basic model (N = 40) | Covariate model (N = 38) | |||
|---|---|---|---|---|---|
| Estimates | RSE (%) | Estimates | RSE (%) | ||
| ka(h−1) | 1.3 | 33.7 | 2.1 | 44.1 | |
| - | - | −1.3 | 42.0 | ||
| Cl/F (L/h) | 21.9 | 6.9 | 22.2 | 6.9 | |
| V/F (L) | 93.9 | 8.2 | 97.3 | 9.3 | |
| ωka (%) | 118.0 | 22.9 | 98.2 | 28.7 | |
| ωCl/F (%) | 34.4 | 15.0 | 34.9 | 15.0 | |
| ωV/F (%) | 19.3 | 66.8 | 21.6 | 57.8 | |
| σ (%) | 44.5 | 8.9 | 44.7 | 8.6 | |
RSE = relative standard error
Fig. 2.
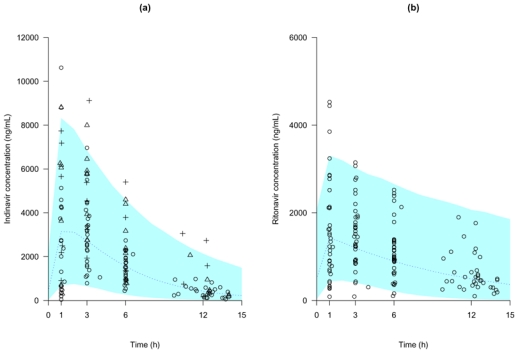
Visual predictive check of the basic population PK model: comparison between the median (continued line) and the 90th interval (colored area) predicted for 1000 simulated datasets and the observed concentrations of indinavir (a) and of ritonavir (b). In the indinavir plot the open circles correspond to an indinavir dose of 400 mg, the open triangles to 600 mg and the crosses to 800 mg
From that basic model, we first tested the effects of the covariates on the individual parameters estimates. Effects of age (P = 0.03) and ABCB1 exon 26 polymorphism (P=0.09) on Cl/F and of CDC classification (P = 0.09) and CYP3A4*1B polymorphism (P = 0.09) on ka were found. Both ABCB1 exon 26 and CYP3A4*1B polymorphism variables were dichotomised in mutant homozygotes versus other genotypes. After the forward selection based on LRT, the population model had CYP3A4 effect on ka (P = 0.02) and age effect on Cl/F (P = 0.03). The age effect on clearance was withdrawn from the model after the backward selection based on permutation test. In the final model, Absorption rate constant was decreased by 70% (P = 0.04, LRT by permutation) in patients *1B*1B for the CYP3A4 allele:
The population parameters of this final model and their RSE are given in Table 3 for the thirty-eight patients with available genotyping for CYP3A4*1B polymorphism. The inter-individual variability for ka decreased by 27% from the basic model with the incorporation of the covariate and residual variability was 44.7%.
3.3. Ritonavir pharmacokinetics
One patient who had indinavir concentrations had no concentration for ritonavir, so we only analysed ritonavir data for 39 patients. The same 5 samples for indinavir mentioned in the preceding paragraph were also missing. Among the 151 samples, two ritonavir plasma concentrations at 1 h and at trough in one patient and one concentration at 12 h in another patient were below the limit of quantification and were discarded. Observed plasma concentrations are displayed in Figure 1b, and it should be noticed that some patients exhibited high plasma concentrations (above 2000 ng/mL) for a dose of 100 mg b.i.d.
A proportional error model was selected. The population estimates are displayed in Table 4. All the RSE were below 25% with the exception of ka, partly attributable to the sparse design, and ωCl/F as observed for the indinavir data. The VPC obtained with the basic model parameters estimates is displayed in Figure 2b, together with the observed concentrations. The results of the basic model evaluation were very satisfactory.
Table 4.
Population pharmacokinetic parameters of ritonavir for the basic and the final model: estimates and relative standard error (RSE)
| Parameters | Basic model (N = 39) | Covariate model (N = 39) | |||
|---|---|---|---|---|---|
| Estimates | RSE (%) | Estimates | RSE (%) | ||
| ka(h−1) | 2.4 | 98.7 | 2.2 | 93.5 | |
| Cl/F (L/h) | 8.7 | 9.4 | 8.3 | 9.0 | |
| - | - | −0.8 | 46.5 | ||
| V/F (L) | 61.6 | 8.6 | 60.7 | 8.7 | |
| ωka (%) | 357.7 | 21.9 | 346.8 | 21.5 | |
| ωCl/F (%) | 55.9 | 12.2 | 52.4 | 12.4 | |
| ωV/F (%) | 22.8 | 53.9 | 23.3 | 51.5 | |
| σ (%) | 30.4 | 8.4 | 30.3 | 8.4 | |
RSE = relative standard error
Effects of orosomucoid (P=0.03), albumin levels (P=0.04) and CYP3A5 polymorphism (patients with 2 wild alleles at most vs other genotypes, P = 0.04) on Cl/F were found on the individual parameters by use of non-parametric tests as well as effects of HIV disease status (P = 0.05) on ka and creatinine clearance (P = 0.1) on V/F. In the final model, an increase of 0.5 g/L in orosomucoid from the median (1 g/L) was associated with a clearance decrease of 28% (P=0.03, LRT by permutation):
The population parameters of this model and their RSE are given in Table 4.
3.4. Link between indinavir and ritonavir PK parameters
Four positive correlations between individual parameters of ritonavir and indinavir were found significant. There was a relationship between indinavir and ritonavir absorption rate constant (r = 0.4, P = 0.005). Indinavir clearance was strongly correlated to ritonavir clearance (r = 0.6, P<0.0001) and to a smaller degree to ritonavir volume of distribution (r = 0.4, P < 0.01), while indinavir volume of distribution was highly correlated to ritonavir volume of distribution (r = 0.5, P < 0.002).
3.5. Concentrations link with short-term efficacy and safety
There was a significant decrease in viral load in the first two weeks of treatment and a significant increase in total cholesterol, glycaemia and triglycerides in the first four weeks of treatment, as shown in Table 5.
Table 5.
Median [range] of the studied short-term efficacy and safety measurements and of the change from baseline
| Baselinea | W2 or W4b | Difference from baseline | P-value | |
|---|---|---|---|---|
| Efficacy | ||||
| Log viral load (log copies/mL) | 4.9 [3.4 – 6.3] | 2.9 [1.8 – 4.1] | −1.8 [−2.8 – −0.5] | <0.001 |
| Safety | ||||
| Total cholesterol (mmol/L) | 4.3 [1.9 – 7.4] | 5.0 [2.9 – 7.5] | 0.8 [0.8 – 4.7] | <0.001 |
| HDL cholesterol (mmol/L) | 1.1 [0.5 – 1.8] | 1.1 [0.4 –2.1] | 0.1 [− 0.7 – 1.0] | 0.09 |
| Glycaemia (mmol/L) | 4.7 [3.4 – 6.0] | 4.9 [2.8 – 7.1] | 0.2 [−1.0 – 2.7] | 0.013 |
| Triglycerides (mmol/L) | 1.0 [0.4 – 3.0] | 1.4 [0.6 – 4.0] | 0.4 [−0.7 – 1.7] | <0.001 |
| Creatinine clearance (mL/min) | 98.4 [62.0 – 195.7] | 97.4 [62.8 – 252.0] | −1.0 [−38.0 – 56.4] | 0.5 |
Baseline = D0 for log viral load and W-4 for safety
W2 for log viral load and W4 for safety
The decrease in log viral load was significantly associated with higher indinavir AUC (r = −0.4, P = 0.03) and Ctrough (r = −0.4, P = 0.03), as shown in Figure 3. No significant difference in viral load decrease was found between the five patients with a Ctrough below the lower limit of the therapeutic range and the thirty five patients with a Ctrough above this value.
Fig. 3.
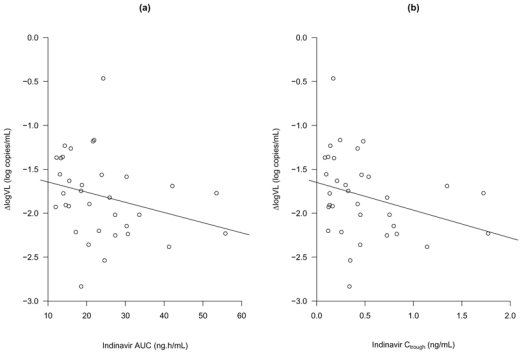
Differences in log viral load (ΔlogVL) observed between treatment initiation and week 2 versus area under the concentration-time curve (a) and trough plasma concentration of indinavir (b) predicted by the model
Further, no significant relationship was found between indinavir nor ritonavir concentrations and safety measurements or grade 2 diarrhoea. No nephrolithiasis has been reported in the COPHAR 2 - ANRS 111 trial, which has prevented us to analyze the link between concentrations and this adverse event associated with indinavir.
The genetic covariate kept in the final population pharmacokinetic model was the CYP3A4*1B polymorphism categorised in 2 classes: *1B*1B versus other genotypes. Both Cmax and triglycerides increase were found to be significantly associated with the CYP3A4*1B polymorphism, althought not significantly correlated. In patients’ homozygotes for the *1B allele, Cmax was significantly lower (median 3.6 [range 2.2 – 5.2] ng/mL compared with the other groups 4.4 [2.2 – 8.3] ng/mL, P=0.04) and the increase in triglycerides was also significantly smaller (0.1 [−0.7 – 1.4] mmol/L versus 0.6 [−0.5 – 1.7] mmol/L, P=0.02), as illustrated by Figure 4. Regarding the efficacy, no significant association was found between the CYP3A4*1B*1B genotype and the Ctrough or the log viral decrease.
Fig. 4.
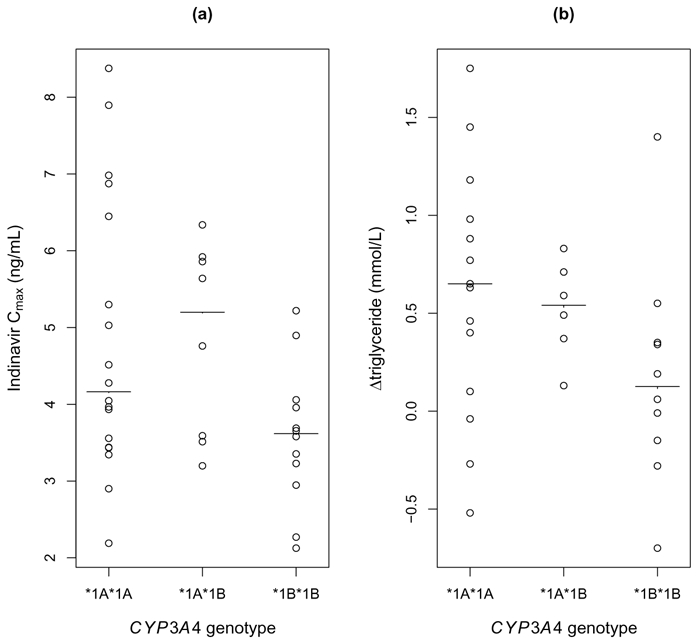
Peak indinavir concentrations predicted by the model (a) and differences in triglycerides (Δtriglyceride) 4 weeks before and after treatment initiation (b) versus CYP3A4 genotype. The solid-line represents the median in each group
The various doses of indinavir were not found to be associated with the CYP3A4*1B polymorphism, short-term efficacy or safety, which dismissed its potential confounding effect.
4. Discussion
The pharmacokinetics of indinavir was analysed by use of a one compartment model with first-order absorption and elimination at steady-state. The estimated clearance and volume of distribution were 22.2 L/h and 97.3 L respectively, both in the range of those obtained in previous studies [9,11,32]. In this study, the ABCB1 exon 26 and 21 and the CYP3A5*3 and *6 polymorphisms were not found to significantly influence the pharmacokinetics of indinavir. Whereas, the absorption rate was 0.6 h−1 for CYP3A4*1B*1B patients and 2.1 h−1 for CYP3A4*1A*1A or CYP3A4*1A*1B patients. The CYP3A enzymes are distributed in both hepatocytes and enterocytes [33] and their inhibition by ritonavir is well-documented [34–36]. In vivo, the genotype-phenotype correlation for CYP3A4*1B remains a subject of debate [37–40], however CYP3A4*1B has been related to increased transcription [41] in vitro. We hypothesised that in CYP3A4*1B*1B patients the ritonavir inhibition potency is lowered, which leads to higher first pass effect of indinavir although it does not impact on its clearance. The potential confounding effect of the ethnic group has been discarded, as this covariate was not significantly related to indinavir individual parameters in the sample. Still, this finding is more relevant clinically in an african population given the extremely low frequency of the CYP3A4*1B*1B genotype among caucasian. The primary objective of the COPHAR2 study was not to assess the influence of genetic polymorphisms on indinavir pharmacokinetics, and the use of modelling has helped to circumvent the limited sample size of forty patients. In addition, most of the tests in this study were performed at an exploratory step and final inclusion in the model was based on permutation to cope with departure from the asymptotic assumption [30]. No evidence for a gender effect was found as reported in some other studies on indinavir PK [9–11] but there were only thirteen women in the present study. The dose was not found to influence the pharmacokinetics of indinavir and using ritonavir as a boost hides dose non linearity of indinavir [42]. We did not assess the impact of the diet, as these data were not available, but patients were recommended to ingest the pills with food containing a sufficient amount of fats.
We also performed a population pharmacokinetic analysis of ritonavir concentrations. Ritonavir profiles were adequately described by a one compartment model with first order absorption and elimination processes, with estimates of the parameters in good agreement with previous studies [6,43,44]. The estimated inter-individual variance for the absorption constant was singularly large. We found a negative relationship between ritonavir clearance and the orosomucoid level in plasma. The affinity of ritonavir for orosomucoid protein as well as its impact on PI intracellular concentrations and efficacy has been described in both in vitro and in vivo studies [45–47]. In patients with high orosomucoid plasma level, the decrease in unbound fraction of ritonavir led to a lower clearance.
In the analysis of both protease inhibitors, the few concentrations (1.3% and 2% for indinavir and ritonavir, respectively) below the limit of quantification were discarded. This approach has been shown to provide using SAEM a less important bias than to set those to LOQ/2 [48] and in MONOLIX 2.1, no proper method to handle LOQ was implemented.
In the analysis of the link between indinavir and ritonavir concentrations, we chose not to include ritonavir as a covariate in the indinavir model, as performed in previous studies [10,11]. Indeed, such parameterisation assumes unidirectional influence of ritonavir on indinavir which is not true. Ritonavir concentrations when ritonavir is given with lopinavir are lower than when ritonavir is given with indinavir [44]. We have instead underlined the different levels of interaction between indinavir and ritonavir PK, especially at the absorption step, with the strong correlation between their absorption constant, but also in terms of bioavailability as the oral clearances and volumes of distribution were highly correlated. In order to properly model such an interaction between PI, a joint population analysis of concentrations of indinavir and ritonavir should be considered with correlated absorption constants and bioavailabilities.
We observed significant changes in viral load after two weeks of treatment and we confirmed the association between high indinavir trough and mean concentrations and a greater decrease of viral load, which has already been described in PI naive patients [49–51]. We did not find any relationship between CYP3A4*1B polymorphism and viral load decrease. We also observed a significant increase, after four weeks of treatment, of total cholesterol, glycaemia and triglycerides, as already reported [52], which was, however, not significantly related to indinavir concentrations at week 2. Ritonavir was found in singularly high levels in the study and is known to affect metabolic profiles, yet we have found no evidence of an association between ritonavir levels and safety measurements. In patients’ homozygotes for the CYP3A4*1B allele, the ritonavir-decreased inhibition on indinavir metabolism led to significantly lower indinavir Cmax and appeared to impact at a metabolic level trough a significantly lower increase in triglycerides in these patients.
5. Conclusion
We developed and validated models for indinavir and ritonavir PK with reduced sampling in indinavir HAART patients. Both the average and through concentrations were found to be predictor of the viral load decline. Only the CYP3A4*1B allele was found to influence indinavir absorption and biochemical safety, but no evidence was found of an impact of the five genetic polymorphisms under study on indinavir efficacy.
Acknowledgments
Steering committee of COPHAR2 ANRS-111 trial: principal investigators: D. Salmon-Céron, X. Duval, statistics: F. Mentré; other members: S. Auleley, M. Biour, M.J. Commoy, B. Diquet, C. Goujard, C. Katlama, C. Lascoux, M. Legrand, A. Métro, G. Peytavin, E. Rey, A.M. Taburet, J.M. Tréluyer.
Safety committee: S. Auleley, M. Biour, A. Métro, C. Lascoux, D. Salmon-Céron.
Pharmacological monitoring committee: X. Duval, E. Rey, J.M. Tréluyer.
Independent committee: C. Rouzioux, C. Piketti, P. Flandre, M. Zenut, P. Marquet.
Clinical centers: Dr. Bentata, Dr. Mansouri, Mme Touam, Pr. Sereni, Dr. Lascoux, Dr. Pintado, Dr. Goujard, MmeMole, Dr. Sellier, Dr. Bendenoun, Dr. Rami, Mme Parrinello, Dr. Jeantils, Mme Tassi, Pr. Vittecoq, Dr. Teicher, Mme Mallet, Pr. Dupont, Dr. Lahoulou, Soeur Azar, Pr. Rosembaum, Dr. Slama, Dr. Naï-Ighil Baakili, Dr. Courtial-Destembert, Pr. Vildé, Pr. Leport, Dr. Duval, Dr. Al Kaied, Pr. Salmon, Dr. Spiridon, Dr. Lesprit, Mme Chesnel, Pr. Katlama, Dr. Schneider, Mme Schoen, Pr. Molina, Dr. Ponscarme, Dr. Colin de verdière, Pr. Morlat, Dr. Bonarek, Dr. Joly, Dr. Ralaimazava, Mme Meridda, Mme Le Gac, P. Raffi, Dr. Allavena, Mme Hüe, Mme Sicot, Dr. Perré, Dr. Leautez, Dr. Aubry, Mme Suaud, Pr. Dellamonica, Dr. Rahelinirina, Pr. Michelet, Dr. Bouvier, Pr. Bazin, Dr. Goubin, Pr. May, Dr. Boyer, Pr. Rouveix, Dr. Dupont, Mme Berthé.
Pharmacological centers: Dr. Rey, Pr. Tréluyer, Dr. Abbara, Dr. Audoul, Dr. Tran, Dr. Sauvageon, Dr. Poirier, Dr. Taburet, Dr. Vincent, Dr. Aymard, Dr. Peytavin, Dr. Lamotte, Dr. Dailly, Dr. Garraffo, Dr. Lavrut, Dr. Mollimard, Dr. Titier, Dr. Tribut, Dr. Hulin, Dr. Huet, Dr. Delhotal, Dr. Hoizey.
Virological centers: Pr. Nicolas-Chanoine, Dr. Sousan, Pr. Dény, Dr. Baazia, Dr. Alloui, Pr. Brun-Vézinet, Dr. Chams, Pr. Fleury, Dr. Pellegrin, Dr. Garrigue, Pr. Fremut, Dr. Vabret, Pr. Lebon, Dr. Krivine, Pr. Calvez, Dr. Gourlain, Dr. Amellal, Pr. Bouvier-Alias, Pr. Norman, Dr. Idri, Pr. Chambreuil, Dr. Poirier, Pr. Mazeron, Pr. Le Faou, Dr. Vénard, Pr. Billaudel, Dr. Ferre, Pr. Rouzioux, Dr. Burgard, Pr. Lefevre, Dr. Cottalorda, Pr. Dussaix, Dr. Bensidhoum, Pr. Colimon, Dr. Ruffault, Dr. Maillard, Pr. Morinet, Dr. Palmer, Pr. Nicolas, Dr. Zalta.
Monitoring: S. Auleley, E. Marcault, F. Mentré.
Statistics: E. Bougen, F. Mentré, X. Panhard.
The authors thank the study participants and the Agence de Recherche Nationale sur le SIDA (ANRS, Essai 111) for financial support.
We also acknowledge Dr. Emmanuelle Comets, Pr. Marc Lavielle and Dr. Emmanuelle Génin for the precious advices they provided along the analysis.
During this analysis J. Bertrand was supported by a grant from Servier Research Group, France.
References
- 1.Solas C, Lafeuillade A, Halfon P, et al. Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003;47:238–43. doi: 10.1128/AAC.47.1.238-243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Letendre S, Marquie-Beck J, Capparelli E, et al. Validation of the CNS penetration-effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65:65–70. doi: 10.1001/archneurol.2007.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collin F, Chêne G, Retout S, et al. Indinavir trough concentration as a determinant of early nephrolithiasis in HIV-1-infected adults. Ther Drug Monit. 2007;29:164–70. doi: 10.1097/ftd.0b013e318030839e. [DOI] [PubMed] [Google Scholar]
- 4.Flexner C. HIV-protease inhibitors. N Engl J Med. 1998;338:1281–92. doi: 10.1056/NEJM199804303381808. [DOI] [PubMed] [Google Scholar]
- 5.Von Moltke LL, Greenblatt DJ, Grassi JM, et al. Protease inhibitors as inhibitors of human cytochromes P450: high risk associated with ritonavir. J Clin Pharmacol. 1998;38:106–11. doi: 10.1002/j.1552-4604.1998.tb04398.x. [DOI] [PubMed] [Google Scholar]
- 6.Hsu A, Granneman GR, Bertz RJ. Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet. 1998;35:275–91. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 7.Soriano V, Garcya-Gasco P, Vispo E, et al. Efficacy and safety of replacing lopinavir with atazanavir in HIV-infected patients with undetectable plasma viraemia: final results of the SLOAT trial. J Antimicrob Chemother. 2008;61:200–05. doi: 10.1093/jac/dkm413. [DOI] [PubMed] [Google Scholar]
- 8.Collot-Teixeira S, De Lorenzo F, Waters L, et al. Impact of different low-dose ritonavir regimens on lipids, CD36, and adipophilin expression. Clin Pharmacol Ther. 2009;375–8:85. doi: 10.1038/clpt.2008.243. [DOI] [PubMed] [Google Scholar]
- 9.Csajka C, Marzolini C, Fattinger K, et al. Population pharmacokinetics of indinavir in patients infected with human immunodeficiency virus. Antimicrob Agents Chemother. 48:3226–32. doi: 10.1128/AAC.48.9.3226-3232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brendel K, Legrand M, Taburet A, et al. Population pharmacokinetic analysis of indinavir in hiv-infected patient treated with a stable antiretroviral therapy. Fundam Clin Pharmacol. 2005;19:373–83. doi: 10.1111/j.1472-8206.2005.00315.x. [DOI] [PubMed] [Google Scholar]
- 11.Kappelhoff BS, Huitema ADR, Sankatsing SUC, et al. Population pharmacokinetics of indinavir alone and in combination with ritonavir in HIV-1-infected patients. Br J Clin Pharmacol. 2005;60:276–86. doi: 10.1111/j.1365-2125.2005.02436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solas C, Simon N, Drogoul MP, et al. Minimal effect of MDR1 and CYP3A5 genetic polymorphisms on the pharmacokinetics of indinavir in HIV-infected patients. Br J Clin Pharmacol. 2007;64:353–62. doi: 10.1111/j.1365-2125.2007.02903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verstuyft C, Marcellin F, Morand-Joubert L, et al. Absence of association between MDR1 genetic polymorphisms, indinavir pharmacokinetics and response to highly active antiretroviral therapy. AIDS. 2005;19:2127–31. doi: 10.1097/01.aids.0000196122.91633.04. [DOI] [PubMed] [Google Scholar]
- 14.Anderson PL, Lamba J, Aquilante CL, et al. Pharmacogenetic characteristics of indinavir, zidovudine, and lamivudine therapy in HIV-infected adults: a pilot study. J Acquir Immune Defic Syndr. 2006;42:441–49. doi: 10.1097/01.qai.0000225013.53568.69. [DOI] [PubMed] [Google Scholar]
- 15.Lamba JK, Lin YS, Schuetz EG, et al. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliver Rev. 2002;54:1271–94. doi: 10.1016/s0169-409x(02)00066-2. [DOI] [PubMed] [Google Scholar]
- 16.Lichterfeld M, Nischalke HD, Bergmann F, et al. Long-term efficacy and safety of ritonavir/indinavir at 400/400 mg twice a day in combination with two nucleoside reverse transcriptase inhibitors as first line antiretroviral therapy. HIV Med. 2002;3:37–43. doi: 10.1046/j.1464-2662.2001.00091.x. [DOI] [PubMed] [Google Scholar]
- 17.Duval X, Mentre F, Lamotte C, et al. Indinavir plasma concentration and adherence score are codeterminant of early virologic response in HIV-infected patients of the APROCO cohort. Ther Drug Monit. 2005;27:63–70. doi: 10.1097/00007691-200502000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Dieleman JP, Gyssens IC, van der Ende ME, et al. Urological complaints in relation to indinavir plasma concentrations in HIV-infected patients. AIDS. 1999;13:473–78. doi: 10.1097/00002030-199903110-00005. [DOI] [PubMed] [Google Scholar]
- 19.Duval X, Mentré F, Rey E, et al. Benefit of therapeutic drug monitoring of protease inhibitors in HIV-infected patients depends on PI used in HAART regimen - anrs 111 trial. Fundam Clin Pharmacol. 2008 doi: 10.1111/j.1472-8206.2009.00693.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirt D, Mentré F, Tran A, et al. Effect of CYP2C19 polymorphism on nelfinavir to M8 biotransformation in HIV patients. Br J Clin Pharmacol. 2008;65:548–57. doi: 10.1111/j.1365-2125.2007.03039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrieri P, Cailleton V, LeMoing V, et al. The dynamic of adherence to highly active antiretroviral therapy: results from the french national APROCO Cohort. J Acq Immun Def Synd. 2001;28:232–39. doi: 10.1097/00042560-200111010-00005. [DOI] [PubMed] [Google Scholar]
- 22.Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74. doi: 10.1067/mcp.2001.114164. [DOI] [PubMed] [Google Scholar]
- 23.Dally H, Edler L, Jager B, et al. The CYP3A4*1B allele increases risk for small cell lung cancer: effect of gender and smoking dose. Pharmacogenetics. 2003;13:607–18. doi: 10.1097/00008571-200310000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Crow J. Hardy, Weinberg and language impediments. Genetics. 1999;152:821–25. doi: 10.1093/genetics/152.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchard J, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal. 2005;49:1020–38. [Google Scholar]
- 27.Lavielle M. MONOLIX group; Orsay, France: 2008. MONOLIX (MOdèles NOn LInéaires à effets miXtes) http://software.monolix.org/index.php. [Google Scholar]
- 28.Lavielle M, Mentré F. Estimation of population pharmacokinetic parameters of saquinavir in HIV patients with the MONOLIX software. J Pharmacokinet Pharmacodyn. 2007;34:229–49. doi: 10.1007/s10928-006-9043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelman A, Carlin JB, Stern HS, et al. Bayesian data analysis. Chapman & Hall; London: 1995. [Google Scholar]
- 30.Bertrand J, Comets E, Mentré F. Comparison of model-based tests and selection strategies to detect genetic polymorphisms influencing pharmacokinetic parameters. J Biopharm Stat. 2008;18:1084–102. doi: 10.1080/10543400802369012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manly B. Randomization, bootstrap and monte carlo methods in biology. Chapman & Hall; London: 1998. [Google Scholar]
- 32.Goujard C, Legrand M, Panhard X, et al. High variability of indinavir and nelfinavir pharmacokinetics in HIV-infected patients with a sustained virological response on highly active antiretroviral therapy. Clin Pharmacokinet. 2005;44:1267–78. doi: 10.2165/00003088-200544120-00005. [DOI] [PubMed] [Google Scholar]
- 33.Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin Pharmacol Ther. 2007;81:270–83. doi: 10.1038/sj.clpt.6100050. [DOI] [PubMed] [Google Scholar]
- 34.Ikezoe T, Hisatake Y, Takeuchi T, et al. HIV-1 protease inhibitor, ritonavir: a potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004;64:7426–31. doi: 10.1158/0008-5472.CAN-03-2677. [DOI] [PubMed] [Google Scholar]
- 35.Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44:279–04. doi: 10.2165/00003088-200544030-00005. [DOI] [PubMed] [Google Scholar]
- 36.Rittweger M, Arastéh K. Clinical pharmacokinetics of darunavir. Clin Pharmacokinet. 2007;46:739–56. doi: 10.2165/00003088-200746090-00002. [DOI] [PubMed] [Google Scholar]
- 37.Spurdle A, Goodwin B, Hodgson E, et al. The CYP3A4*1B polymorphism has no functional significance and is not associated with risk of breast or ovarian cancer. Pharmacogenetics. 2002;12:355–66. doi: 10.1097/00008571-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 38.He P, Court MH, Greenblatt DJ, et al. Genotype-phenotype associations of cytochrome P450 3A4 and 3A5 polymorphism with midazolam clearance in vivo. Clin Pharmacol Ther. 2005;77:373–87. doi: 10.1016/j.clpt.2004.11.112. [DOI] [PubMed] [Google Scholar]
- 39.Hesselink DA, van Schaik RH, van der Heiden IP, et al. Genetic polymorphisms of the CYP3A4, CYP3A5, and MDR-1 genes and pharmacokinetics of the calcineurin inhibitors cyclosporine and tacrolimus. Clin Pharmacol Ther. 2003;74:245–54. doi: 10.1016/S0009-9236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- 40.Tran A, Jullien V, Alexandre J, et al. Pharmacokinetics and toxicity of docetaxel: role of CYP3A, MDR1, and GST polymorphisms. Clin Pharmacol Ther. 2006;79:570–80. doi: 10.1016/j.clpt.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Amirimani B, Ning B, Deitz AC, et al. Increased transcriptional activity of the CYP3A4*1B promoter variant. Environ Mol Mutagen. 2003;42:299–305. doi: 10.1002/em.10199. [DOI] [PubMed] [Google Scholar]
- 42.Yeh K, Stone J, Carides A, et al. Simultaneous investigation of indinavir nonlinear pharmacokinetics and bioavailability in healthy volunteers using stable isotope labeling technique: study design and model-independent data analysis. J Pharm Sci. 1999;88:568–73. doi: 10.1021/js9802392. [DOI] [PubMed] [Google Scholar]
- 43.Lu JF, Blaschke TF, Flexner C, et al. Model-based analysis of the pharmacokinetic interactions between ritonavir, nelfinavir and saquinavir after simultaneous and staggered oral administration. Drug Metab Dispos. 30:1455–61. doi: 10.1124/dmd.30.12.1455. [DOI] [PubMed] [Google Scholar]
- 44.Kappelhoff BS, Huitema ADR, Crommentuyn KML, et al. Development and validation of a population pharmacokinetic model for ritonavir used as a booster or as an antiviral agent in hiv-1-infected patients. Br J Clin Pharmacol. 2005;59:174–82. doi: 10.1111/j.1365-2125.2004.02241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schön A, del Mar Ingaramo M, Freire E. The binding of HIV-1 protease inhibitors to human serum proteins. Biophys Chem. 2003;105:221–30. doi: 10.1016/s0301-4622(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 46.Jones K, Hoggard PG, Khoo S, et al. Effect of alpha1-acid glycoprotein on the intracellular accumulation of the HIV protease inhibitors saquinavir, ritonavir and indinavir in vitro. Br J Clin Pharmacol. 2001;51:99–102. doi: 10.1046/j.0306-5251.2001.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Schooley R, Gerber J. The effect of increasing alpha1-acid glycoprotein concentration on the antiviral efficacy of human immunodeficiency virus protease inhibitors. J Infect Dis. 1999;180:1833–37. doi: 10.1086/315123. [DOI] [PubMed] [Google Scholar]
- 48.Samson A, Lavielle M, Mentré F. Extension of the SAEM algorithm to left-censored data in nonlinear mixed-effects model: Application to HIV dynamics model. Comput Stat Data Anal. 2006;51:1562–74. [Google Scholar]
- 49.Fletcher CV. Pharmacologic considerations for therapeutic success with antiretroviral agents. Ann Pharmacother. 1999;33:989–95. doi: 10.1345/aph.19075. [DOI] [PubMed] [Google Scholar]
- 50.Burger DM, Hugen PWH, Aarnoutse RE, et al. A retrospective, cohort-based survey of patients using twice-daily indinavir + ritonavir combinations: pharmacokinetics, safety and efficacy. J Acquir Immune Defic Syndr. 2001;26:218–24. doi: 10.1097/00042560-200103010-00003. [DOI] [PubMed] [Google Scholar]
- 51.Rayner CR, Galbraith KJ, Marriott JL, et al. A critical evaluation of the therapeutic range of indinavir. Ann Pharmacother. 2002;36:1230–37. doi: 10.1345/aph.1A467. [DOI] [PubMed] [Google Scholar]
- 52.Behrens G, Dejam A, Schmidt H, et al. Impaired glucose tolerance, beta cell function, and lipid metabolism: HIV patients under treatment with protease inhibitors. AIDS. 1999;13:F63–70. doi: 10.1097/00002030-199907090-00001. [DOI] [PubMed] [Google Scholar]