

Abstract
Somatoform disorders are troubling to both patients and physicians. The diagnosis regrettably relies on the presence of subjective distress in the absence of objective findings. As a result, there is always the possibility that a diagnosis will be “missed.” There is a clear underlying physiology of distress, which implies that there is a two-way street—both psychosomatic and somatopsychic in terms of production and experience of somatoform symptoms. Studies on communication pathways from the immune system to the brain provide exciting new information on the pathophysiology of inflammation-associated symptoms.
Keywords: psychiatric diagnosis, somatoform disorder, inflammation, pain, fatigue
INTRODUCTION
At the heart of every clinical interchange is the doctor’s attempt to reconcile the patient’s subjective complaints with the objective findings, a 2 × 2 table, so to speak (Table 1). Medicine is typically most comfortable when these two areas are in agreement. When—for instance—objective findings and subjective complaints are present, one recognizes an “ideal disease.” Similarly, when neither objective findings nor subjective complaints are present, one happily recognizes “no disease.” Unfortunately, it is not uncommon for disparities between findings and complaints. The bulk of this paper will discuss the situation where objective findings are absent but subjective complaints are present. This situation may be viewed either as “undiagnosed disease” or alternatively as “somatoform disorder.”
TABLE 1.
Epistemology: Reconciling Objective Findings With Subjective Complaints
| Subjective Complaints | Objective Findings |
|
|---|---|---|
| Present | Absent | |
| Present | “Ideal” disease | Undiagnosed disease somatoform |
| Absent | Occult disease denial or stoicism | No disease |
“Somatoform disorders” represent a very heterogeneous group of patient presentations, ranging from conversion disorder to hypochondriasis to somatization disorder to body dysmorphic disorder to pain disorder, etc. The neologism was introduced in the Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition in the hopes of finding a neutral-sounding all-encompassing diagnostic label and in the recognition that many patients present with somatic distress that does not “fit” in the rubric of anxiety, mood, or psychotic disorders. Like Wilsonian efforts to redraw the map of Europe after World War I, this diagnostic category grouped together some uneasy companions. For the purpose of this paper, we use “somatoform” to refer to one segment of this disorder—psychiatric patient presentations associated with significant somatic distress (e.g., pain, fatigue).
It is the thesis of this paper that a substantial reservoir of undiagnosed disease is found in somatoform disorders. Before developing this premise, however, it is necessary to complete the 2 × 2 table. There is one further cell of the table that is at least as troubling as the somatoform disease category. Not infrequently, physicians encounter objective findings in the absence of subjective complaints. Such a situation is common in occult disease and is also common in denial or stoicism. A great deal could be written on this latter cell, but this paper focuses primarily on the cell wherein the diagnosis of somatoform disease is typically made.
Four Ways That Unrecognized Diseases Get Labeled as Somatoform Diseases
First, it is by no means unlikely that patients’ illnesses get labeled as somatoform diseases because the doctor has simply missed the diagnosis by insufficient attention to the history, physical examination, or adjunctive laboratory tests. This happens rather more often than one would like. Hong and Dimsdale recently carried out a pilot study of exercise as a treatment of fatigue in breast cancer patients (unpublished). They recruited a small number of breast cancer survivors who had devastating amounts of fatigue and who were willing to try an exercise regimen. As part of the baseline data, they obtained basic relevant laboratory studies and found that 40% of the patients were frankly hypothyroid. How did it come about that this eminently treatable cause of fatigue was not diagnosed? Breast cancer survivors are typically passed back and forth between oncologist and primary care provider. Neither of these physician pairs worked up the patients for their fatigue. Rather, they both assumed the fatigue was a result of chemotherapy or perhaps was a form fruste of depression in response to the stress of illness.
A second way that unrecognized diseases get labeled as somatoform diseases is by not relying on contemporary diagnostic techniques. There have been astonishing breakthroughs in clinical chemistry as well as anesthesia, imaging, and medical instrumentation. As a result, we are now recognizing a huge reservoir of unrecognized diseases because we previously lacked the techniques to study them. Obstructive sleep apnea (OSA), first discussed in a fascinating case report 50 years ago, is now recognized as an extremely prominent sleep disorder. Whereas the initial diagnosis of OSA was confined to rare “zebra” presentations marked by profound obesity and somnolence, contemporary diagnosis, made possible by ever smaller sleep monitoring systems, reveals that OSA is prevalent in 4% to 9% of the population, depending on population characteristics and definitional cut points (1). Similarly, celiac disease, once diagnosed as a rare malabsorption disorder in infancy, is now recognized as prevalent in 1% of the population. Accompanied by ill-defined findings and symptoms such as mild anemia and fatigue, the diagnosis can now be made thanks to progress in clinical chemistry, such as development of endomysial antibody tests (2) and pharmacological developments to make possible conscious sedation as well as biomedical instrument development to facilitate endoscopy. We now know that a huge population has an eminently treatable disease, if only we stop to think about the possibility of that diagnosis. We mention OSA and celiac disease in this context because both are characterized by fatigue and emotional distress and thus could readily be misdiagnosed as a somatoform disorder.
There is a third way that unrecognized diseases get labeled as somatoform diseases. Insights from basic science research can have profound ramifications for clinical symptoms. Symptoms, such as pain and fatigue, are core presentations in many patients with somatoform illness. However, those symptoms are tightly related to each other and have the potential to be worsened by iatrogenic factors. Fundamental research in sleep, for instance, has demonstrated that sleep disruption lowers pain threshold. Other work demonstrates that opioids—the mainstay for treatment of severe pain—disrupt sleep (3). Thus, one has the potential for a vicious circle in the treatment of pain. We treat the pain with opioids, thereby interrupting sleep and increasing daytime fatigue, thereby ensuring a need for more opioids, etc. How many of our pain patients who bear a label as “somatoform disorder” patients do so as a result of well-intended clinical interventions that have unexpected adverse outcomes?
Finally, it is important to acknowledge, as Pliny the Elder stated in 75 AD that “new diseases, unknown in past years, have come . . . .” Hepatitis C with its unpleasant cargo of fatigue and depression is perhaps one of the clearest instances of such new diseases. Unrecognized until sensitive laboratory tests were developed, this disease is currently found in ~4% of individuals aged 40 years in the United States population, (4) and again, the early symptoms are invariably ill-defined subjective complaints—a setup for (mis)diagnosing somatoform diseases.
Getting Beyond the Black Box of Symptom Reporting
The “mischief” comes from relying solely on self-report for symptom verification. Self-report is at the heart of all medical complaints, but in the absence of physical findings, understanding the meaning of self-report gets very complicated. Oddly enough, three developments in contemporary medicine allow a new “window” on self-report. One involves a refreshingly new way of looking at symptom reporting. Another involves contemporary neural imaging studies to shed light on self-report of symptoms. A third applies contemporary insights on neural immune properties as a way of understanding paradoxical self-report of symptoms. Together, the three approaches amount to a challenge of the “black box” that has historically characterized symptom reporting, a black box that has heretofore defied efforts at understanding the psychology and physiology of such reports.
Arthur Barsky and others asked, “What determines the ‘volume’ level of symptoms, how is it that some people amplify their symptoms and others de-amplify them?” (5) This is a felicitous approach in that it carries no implicit assumptions of underlying neuroticism, etc., but rather asks the question from a cognitive neural science perspective. Do some individuals “amplify” their symptoms characteristically or, perhaps, under unusual stressors? Is that “amplification” neurally driven? Does the amplification reflect the fact that patients have different explanatory models for understanding the significance of their symptoms?
Oddly enough, two brief case reports involving construction injuries with nails demonstrate the phenomenon beautifully. In one report (6), Fisher et al. described the case of a builder who jumped down onto a 7-inch nail, which pierced his boot at the toe level (Figure 1, left panel). The man was in pain and required intravenous sedation in the emergency room. However, when the boot was cut away, it turned out that the nail had fortunately passed between his toes as opposed to its apparent impaling of the foot. The man’s agonizing pain was elicited solely by his misperception—a case of somatic amplification. On the other hand, a report in USA Today described a construction worker who had unknowingly shot himself in the head with a nail gun (Figure 1, right panel) and who was unaware of the injury. He perceived a toothache and went to a dentist 6 days later, wherein the cause of the rogue toothache was discovered. In this case, one would conclude that somatic deamplification was at work. The patient was unaware of the injury and attributed the sensation to more familiar sources.
Figure 1.
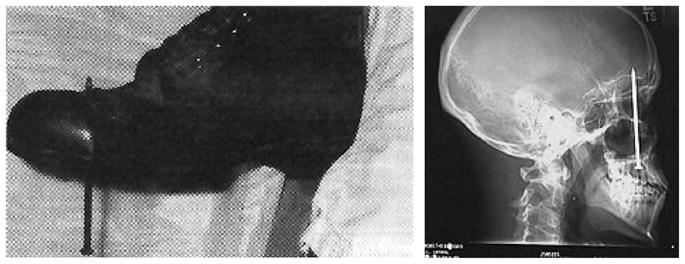
Symptom perceptions elicited by nails. The left panel exemplifies somatic amplification; reprinted from Fisher JP, Hassan DT, O’Connor N. Minerva. Br Med J 1995;310:70, with permission from BMJ Publishing Group Ltd. The right panel exemplifies somatic deamplification; reprinted with permission from Associated Press, Wide World Photos. 1/16/05.
Neural imaging studies have taken up the quest for understanding symptom perception. Raij et al. (7) used functional magnetic resonance imaging to examine how the brain responded to painful heat laser simulation to the hand versus to the hypnotic suggestion of laser stimulation. In healthy subjects tested repeatedly, they observed many common neural patterns of response to the two pain stimuli. The next step was made by Coghill et al., (8) who contrasted individuals’ neural imaging responses to thermal stimuli. In this case, the stimuli were identical but normal individuals were contrasted in terms of whether they reported high versus low self-report of pain sensitivity. Individuals with high sensitivity had increased activation of the anterior cingulate cortex, somatosensory cortex, and prefrontal cortex but there was no difference in the thalamic response between the high pain sensitivity and the low pain sensitivity subjects. Sadly, this sort of study has not been made of patients with somatoform disorders. Do these patients have a different pattern of response to painful stimuli? If so, what are the implications for etiology and treatment of the symptoms? Most astute clinicians do not question the authenticity of patients’ complaints of pain. Rather, they try to understand its origins and direct interventions that help manage the pain. Pain is inherently subjective. It can be treated with analgesic medication, distraction, hypnosis, etc., regardless of whether there is a known focal stimulus for the pain. The case reports of nail injuries, together with the neural imaging studies, suggest that a helpful focus for future clinical interventions in patients with somatoform illness must include an analysis of how their symptoms are perceived, interpreted, and amplified by the brain.
The focus on neutral terms such as somatic amplification as well as the employment of neural imaging probes may help shed light on the “black box” area of symptom reporting in somatoform disorders. Other developments in physiology may similarly help expose a somatopsychic as well as a psychosomatic underpinning of these disorders. One of the most dramatic areas of knowledge advances has come with our understanding of neural immune trafficking. Immune factors may underpin subtle perception of pain, fatigue, depression—what some have called “sickness behavior.”
Insights From the Psychoneuroimmune Perspective
Psychoneuroimmunology studies the interactions between the central nervous system and the immune system. The field has made major advances in understanding how brain functions can modulate the activity of the immune system and the discovery (particularly relevant for this paper) that mediators produced by cells of the immune system exert profound influences in the brain.
As a typical example, many breast cancer survivors experience persistent fatigue up to 5 to 10 years after diagnosis despite the termination of chemotherapy and radiotherapy. Fatigued breast cancer survivors display increased inflammatory biomarkers (9). Similar associations between self-reports of fatigue and inflammatory markers have been reported in patients with coronary heart disease (10). The co-occurrence of decreasing energy, general malaise, and minor depression in the weeks that precede a myocardial infection has been termed “vital exhaustion” (11). High levels of inflammatory biomarkers have been found in apparently healthy patients who scored high on vital exhaustion (12).
The mechanisms that are responsible for the association between subjective health complaints and inflammation have been elucidated over the last decade (13,14). Activation of the innate immune system by pathogen-associated molecular patterns induces the local production of proinflammatory cytokines. These molecules are responsible for the development of the local inflammatory response and the systemic response to inflammation. This acute-phase reaction includes the production of acute-phase proteins by hepatocytes and the occurrence of fever, which is a regulated metabolic response to pathogens. The fever is “coordinated” in the anterior preoptic area of the hypothalamus and is triggered by the action on the brain of proinflammatory cytokines that are released at the periphery.
Proinflammatory cytokines do not need to enter the brain to target the hypothalamus because the brain is able to form a cellular and molecular representation of the peripheral immune response (Figure 2). During the course of an inflammatory response, brain innate immune cells produce proinflammatory cytokines. These cytokines are produced in response either to blood-borne pathogen-associated molecular patterns or to circulating proinflammatory cytokines that are sensed by macrophage-like cells residing in circumventricular organs. Because circumventricular organs have a deficient blood-brain barrier, they are able to monitor changes in the composition of the internal milieu. The cytokines that are produced in the circumventricular organs gradually diffuse into the brain side of the blood-brain barrier and recruit microglial cells in the brain parenchyma.
Figure 2.

Immune-to-brain communication. Proinflammatory cytokines are produced at the periphery by innate immune cells in response to pathogen-associated molecular patterns or to danger signals, such as heat shock proteins released by dying cells. Peripheral proinflammatory cytokines induced the production of the same proinflammatory cytokines in the brain. The brain proinflammatory cytokines acting on various brain areas induce nonspecific symptoms of sickness, such as fatigue, depressed mood, and altered cognition. The production and action of proinflammatory cytokines are regulated both at the periphery and in the central nervous system by a number of opposing molecules including anti-inflammatory cytokines, steroid hormones such as glucocorticoids and neuropeptides such as α-melanotropin (α-MSH) and vasopressin (AVP).
Another important pathway of communication from the immune system to the brain is represented by the afferent nerves that innervate the bodily site in which the inflammation is taking place. Activation of these afferent nerves promotes the perception of the sensory components of inflammation (calor or heat and dolor or pain) and the expression of brain proinflammatory cytokines in response to peripheral inflammatory cytokines. A bilateral section of the vagus nerves blocks the immune-to-brain transmission of inflammation that takes place in the abdominal cavity (15,16), whereas the section of the trigeminal nerves does the same for an oral inflammation (17).
In addition to their role in the genesis of fever, brain proinflammatory cytokines are also responsible for the subjective and behavioral components of illness, which accounts for why one feels sick and behaves in a sick way when one is ill. Conversely, sickness symptoms that develop during the course of a peripheral activation of the innate immune system can be blocked by administration of various cytokine antagonists in the brain (18).
Studies of the brain effects of cytokines have shown that, although cytokine-induced sickness behavior is normally reversible on the resolution of the infectious episode, it persists when the innate immune system is chronically activated and can even culminate in major depression in vulnerable individuals (19,20). Psychological symptoms of depression (e.g., anhedonia, depressed mood, irritability) coexist with neurovegetative signs of depression (e.g., fatigue, reduced appetite) in vulnerable patients whose immune system is chronically activated. Patients with more depressive symptoms are more likely to become depressed in response to an activation of the immune system than those who have fewer such symptoms (21). The same applies to patients whose pituitary-adrenal axis is more responsive to the immune stimulation (22).
An important aspect of the pathophysiology of immune-to-brain communication is the existence of a cross-sensitization process between stressors and cytokines. Exposure to inescapable electric shocks, for instance, sensitized the peripheral and central cytokine response to lipopolysaccharide in rats (23) for a minimum of 4 days after stress. Reciprocally, a prior episode of interleukin (IL)-1-induced sickness sensitized the pituitary-adrenal response to inescapable electric shock up to 2 to 3 weeks after the cytokine treatment (24). Sensitization can also occur when the same cytokine is administered twice at an interval of several days or weeks, and it affects both cytokine-sensitive neurotransmitter metabolism and pituitary-adrenal responsiveness to cyokines (25).
These protracted effects of stressors and cytokines on brain functions likely play an important role in the pathophysiology of somatic amplification. The clearest demonstration of the clinical relevance of the sensitizing effects of cytokines is in the field of pain. The perception of pain is strongly amplified under the effect of proinflammatory mediators produced by activated glial cells in the spinal cord (26). It is important to note that glial activation is not restricted to the spinal cord but also occurs in the brain in situations of chronic inflammation associated, for instance, with progressive neurodegeneration (27), obesity (28), or aging (28). In these conditions, the brain cytokine system seems to be sensitized in that it responds to a greater extent to a further activation of the peripheral innate immune system, resulting in a more intense cytokine-induced sickness behavior and/or a delayed recovery from sickness.
The main medical implication of this view is that many somatization symptoms including depressed mood, fatigue, and pain may represent the expression of a previously sensitized brain cytokine system that is reactivated by infectious or noninfectious trauma. At the clinical level, there is not yet consensus as to which biomarkers best relate somatization symptoms to inflammation. Only peripheral markers of inflammation are currently available. Circulating levels of acute-phase proteins (e.g., C-reactive protein) provide a gross index of the inflammatory response that can be refined using plasma levels of cytokines, such as IL-6 together with its soluble receptor. Because most other cytokines act in a paracrine/autocrine manner rather than as hormones, the production of cytokines by stimulated monocytes in culture is a better index of activity of the innate immune system than circulating levels that only reflect overflow of local cytokines. Because cytokines are produced by many cell types other than innate immune cells, it can be useful to more precisely assess the involvement of macrophages in the inflammatory response by measuring circulating levels of neopterin. Last but not the least, the possible impact of immune activation on serotoninergic neurotransmission can be studied by measuring the circulating concentrations of kynurenine, the main metabolite of tryptophan.
At the therapeutic level, treatments that specifically target activation of the brain cytokine system are not yet available. However, there is already evidence that pharmacological (e.g., antidepressants) (29) and nonpharmacological (e.g., aerobic exercise) (30) therapies are able to attenuate some somatic symptoms by down-regulating inflammation.
CONCLUSION
Although medicine’s goal is always to allay suffering, there is no one universal remedy other than courtesy and respect and kindness. Specific remedies may be applied only when an accurate diagnosis has been made. Somatoform disorders are among the hardest disorders to diagnose and thus to treat. This paper suggests two rather different conclusions. First, somatoform disorder may be misdiagnosed due to complex factors that lead to underrecognition of another underlying disorder other than somatoform disorder. Second, one must study the underlying physiology of symptoms in somatoform disorder in terms of the cognitive processes involved in recognition of symptoms and the complex physiology of distress, increasingly recognized as immune in nature, which augments non-specific symptoms.
Acknowledgments
This research has been supported by Grants HL36005, HL44915, CA23100, R01 MH-71349, and MH-079829 from the National Institutes of Health.
References
- 1.Caples SM, Gami AS, Somers VK. Obstructive sleep apnea. Ann Intern Med. 2005;142:187–97. doi: 10.7326/0003-4819-142-3-200502010-00010. [DOI] [PubMed] [Google Scholar]
- 2.Reddick BK, Crowell K, Fu B. Clinical inquiries: what blood tests help diagnose celiac disease? J Fam Pract. 2006;55:1088, 1090–3. [PubMed] [Google Scholar]
- 3.Dimsdale J, Norman D, Dejardin D, Wallace MS. The effects of opioids on sleep architecture. J Clin Sleep Med. 2007;3:33–6. [PubMed] [Google Scholar]
- 4.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705–14. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 5.Barsky A, Wyshak G, Klerman G. The somatosensory amplification scale and its relationship to hypochondriasis. J Psychiatr Res. 1990;24:323–34. doi: 10.1016/0022-3956(90)90004-a. [DOI] [PubMed] [Google Scholar]
- 6.Fisher JP, Hassan DT, O’Connor N. Minerva. BMJ. 1995;310:70. [Google Scholar]
- 7.Raij TT, Numminem J, Narvanen S, Hiltunen J, Hari R. Brain correlates of subjective reality of physically and psychologically induced pain. Proc Nat Acad Sci. 2005;102:2147–51. doi: 10.1073/pnas.0409542102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coghill RC, McHaffie JG, Yen YF. Neural correlates of interindividual differences in the subjective experience of pain. Proc Nat Acad Sci. 2003;100:8538–42. doi: 10.1073/pnas.1430684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collado-Hidalgo A, Bower JE, Ganz PA, Cole SW, Irwin MR. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res. 2006;12:2759–66. doi: 10.1158/1078-0432.CCR-05-2398. [DOI] [PubMed] [Google Scholar]
- 10.Janszky I, Lekander M, Blom M, Georgiades A, Ahnve S. Self-rated health and vital exhaustion, but not depression, is related to inflammation in women with coronary heart disease. Brain Behav Immun. 2005;19:555–63. doi: 10.1016/j.bbi.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Appels A. Mental precursors of myocardial infarction. Br J Psychiatry. 1990;156:465–71. doi: 10.1192/bjp.156.4.465. [DOI] [PubMed] [Google Scholar]
- 12.Wirtz PH, von Kanel R, Schnorpfeil P, Ehlert U, Frey K, Fischer JE. Reduced glucocorticoid sensitivity of monocyte interleukin-6 production in male industrial employees who are vitally exhausted. Psychosom Med. 2003;65:672–8. doi: 10.1097/01.psy.0000062529.39901.c7. [DOI] [PubMed] [Google Scholar]
- 13.Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–9. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 14.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 200;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bluthe RM, Michaud B, Kelley KW, Dantzer R. Vagotomy blocks behavioural effects of interleukin-1 injected via the intraperitoneal route but not via other systemic routes. Neuroreport. 1996;7:2823–7. doi: 10.1097/00001756-199611040-00083. [DOI] [PubMed] [Google Scholar]
- 16.Laye S, Bluthe RM, Kent S, Combe C, Medina C, Parnet P, Kelley K, Dantzer R. Subdiaphragmatic vagotomy blocks induction of IL-1 beta mRNA in mice brain in response to peripheral LPS. Am J Physiol. 1995;268:R1327–31. doi: 10.1152/ajpregu.1995.268.5.R1327. [DOI] [PubMed] [Google Scholar]
- 17.Navarro VP, Iyomasa MM, Leite-Panissi CR, Almeida MC, Branco LG. New role of the trigeminal nerve as a neuronal pathway signaling brain in acute periodontitis: participation of local prostaglandins. Pflugers Arch. 2006;453:73–82. doi: 10.1007/s00424-006-0113-2. [DOI] [PubMed] [Google Scholar]
- 18.Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- 19.Capuron L, Ravaud A, Dantzer R. Early depressive symptoms in cancer patients receiving interleukin 2 and/or interferon alfa-2b therapy. J Clin Oncol. 2000;18:2143–51. doi: 10.1200/JCO.2000.18.10.2143. [DOI] [PubMed] [Google Scholar]
- 20.Constant A, Castera L, Dantzer R, Couzigou P, de Ledinghen V, Demotes-Mainard J, Henry C. Mood alterations during interferon-alfa therapy in patients with chronic hepatitis C: evidence for an overlap between manic/hypomanic and depressive symptoms. J Clin Psychiatry. 2005;66:1050–7. doi: 10.4088/jcp.v66n0814. [DOI] [PubMed] [Google Scholar]
- 21.Capuron L, Ravaud A. Prediction of the depressive effects of interferon alfa therapy by the patient’s initial affective state. N Engl J Med. 1999;340:1370. doi: 10.1056/NEJM199904293401716. [DOI] [PubMed] [Google Scholar]
- 22.Capuron L, Raison CL, Musselman DL, Lawson DH, Nemeroff CB, Miller AH. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160:1342–5. doi: 10.1176/appi.ajp.160.7.1342. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JD, O’Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun. 2002;16:461–76. doi: 10.1006/brbi.2001.0638. [DOI] [PubMed] [Google Scholar]
- 24.Tilders FJ, Schmidt ED, Hoogendijk WJ, Swaab DF. Delayed effects of stress and immune activation. Baillieres Best Pract Res Clin Endocrinol Metab. 1999;13:523–40. doi: 10.1053/beem.1999.0040. [DOI] [PubMed] [Google Scholar]
- 25.Anisman H, Merali Z, Hayley S. Sensitization associated with stressors and cytokine treatments. Brain Behav Immun. 2003;17:86–93. doi: 10.1016/s0889-1591(02)00100-9. [DOI] [PubMed] [Google Scholar]
- 26.Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–46. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bluthe RM, Michaud B, Delhaye-Bouchaud N, Mariani J, Dantzer R. Hypersensitivity of lurcher mutant mice to the depressing effects of lipopolysaccharide and interleukin-1 on behaviour. Neuroreport. 1997;8:1119–22. doi: 10.1097/00001756-199703240-00011. [DOI] [PubMed] [Google Scholar]
- 28.O’Connor JC, Satpathy A, Hartman ME, Horvath EM, Kelley KW, Dantzer R, Johnson RW, Freund GG. IL-1beta-mediated innate immunity is amplified in the db/db mouse model of type 2 diabetes. J Immunol. 2005;174:4991–7. doi: 10.4049/jimmunol.174.8.4991. [DOI] [PubMed] [Google Scholar]
- 29.Davidson KW, Kupfer DJ, Bigger JT, Califf RM, Carney RM, Coyne JC, Czajkowski SM, Frank E, Frasure-Smith N, Freedland KE, Froelicher ES, Glassman AH, Katon WJ, Kaufmann PG, Kessler RC, Kraemer HC, Krishnan KR, Lesperance F, Rieckmann N, Sheps DS, Suls JM National Heart, Lung, and Blood Institute Working Group. Assessment and treatment of depression in patients with cardiovascular disease: National Heart, Lung, and Blood Institute Working Group Report. Psychosom Med. 2006;68:645–50. doi: 10.1097/01.psy.0000233233.48738.22. [DOI] [PubMed] [Google Scholar]
- 30.Woods JA, Vieira VJ, Keylock KT. Exercise, inflammation, and innate immunity. Neurol Clin. 2006;24:585–99. doi: 10.1016/j.ncl.2006.03.008. [DOI] [PubMed] [Google Scholar]