

When God created the earth, he placed (modern) mankind in East Africa ≈200 000 years ago, as suggested by the phylogenetic and archeological studies.1,2 These hunter-gatherers populated the land and started to migrate, first to the west and southern tip of Africa and then north toward North Africa. Mankind gradually crossed the Arabian Peninsula to the Middle East and Eurasia. Then, over thousands of years of northerly movement, man crossed the Bering land bridge that connected Siberia to Alaska and migrated down to the Americas. This out-of-Africa migration took thousands of generations. It was interrupted by the Ice Age, which led to depopulation and isolation of the surviving population followed by the new waves of migrations, resulting in multiple founding populations.3
At the genetic level, the tree of life was produced through replication of the genome. For the genome to replicate, cells are equipped with DNA polymerases to copy the parental DNA, which they do at blazing speeds.4 However, “no protein is perfect,” and neither are the DNA polymerases. So, the DNA polymerases commit occasional errors in copying the template (parental) DNA and incorporate wrong nucleotides into the new DNA strands. To correct the errors, nature afforded the cells with DNA repair enzymes, which are assigned the task of maintaining the integrity of the nucleotide pools before replication.5 Nevertheless, the process of DNA replication and editing, although exceedingly precise, is imperfect. The error rate of the DNA replication and editing machinery is estimated to be at 10−6 to 10−10 nucleotides incorporated into the new strand of DNA.6,7 The inheritance is forward. Therefore, the misincorporated nucleotides are passed on to the next generation, and with each generation, new errors in DNA replication are introduced. The occasional “errors of nature” in DNA replication, which was probably necessary for the survival of the mankind, form the basis for variations in the genomic DNA nucleotide compositions among the individuals. Population genetic diversity is further expanded by the fact that each genome is a mixture of maternal and paternal DNA and the large number of recombination that occurs between the 2 homologous chromosomes during meiosis. Further compounding genetic diversity are the nature’s forces exercised Darwinian selection, the Baldwin effect, and geopolitical forces. Collectively, these elements have afforded significant diversity to the population genome, which by and large follows the geographic blueprint of out-of-Africa migration. Hence, there is a genetic gradient of shared haplotypes that decreases as the geographic distance increases. The point is best illustrated in the European population, where genetic variations mirror Europe geography.8 As such, the geographic origin of a European individual could be inferred with remarkable accuracy on the basis of the genetic structure of the individual.8
The majority of “nature’s errors” in DNA replication occur at the single-nucleotide level, and hence, these variations are referred to as single-nucleotide polymorphisms (SNPs). However, the errors are not only restricted to SNPs but also include small and large insertion/deletions (indels), duplications, and rearrangements, which are collectively referred to as structural variations (SVs). Approximately two thirds of the SVs in the genome involve <10 000 base pairs of DNA, ≈15% involve >100 000 base pairs, and a few involve several million base pairs.9,10 SVs that change the copy number of the genes (from the normal 2 copies) are referred to as copy number variants (CNVs). Although SNPs prevail in the number, indels/SVs/CNVs by involving large segments of DNA affect a much larger number of the nucleotides. Because indels/SVs/CNVs involve genes implicated in various cellular functions, they could impose a significant clinical impact.9,11
The genetic diversity of the population has been recognized since the dawn of DNA sequencing by Sanger technique ≈3 decades ago.12 Several million SNPs have already been deposited in the SNP database (dbSNP, build 130). However, the extent and diversity of DNA sequence variability in the diploid genome of a single individual only became evident on analysis of Venter’s genome sequence, which was sequenced by Sanger sequencing over 3 years.13 The findings were mesmerizing. Venter’s genome contained ≈4.1 million DNA sequence variants, including ≈3.5 million SNPs, of which approximately one third were novel. Remarkably, 44% of Venter’s annotated genes were heterozygous for one or more sequence variants. The abundance of SVs was quite notable. SVs were less common in number than the SNPs. However, because of the size of the involved DNA fragments, SVs comprised approximately three fourths of the variant nucleotides. The findings for the first time highlighted the unique features of an individual genome. The advent of next generation DNA sequencing techniques enhanced the pace and reduced the cost of DNA sequencing by several orders of magnitude. It led to sequencing of the genomes of Dr. Watson, A Yoruba African man, a Korean man, a Han Chinese man and several other individuals’ exomes. Some variants encompassed several million base pairs of DNA. A fascinating aspect of each genome is that ≈20% to 30% of the SNPs and the majority of indels/SVs/CNVs were novel (Figure). Moreover, each genome had 5000 to 10 000 non-synonymous SNPs, which would be expected to impose some level of biological functions and potentially cause diseases. These discoveries imply that the individual genome is by and large personal (personal genome).14
Figure.
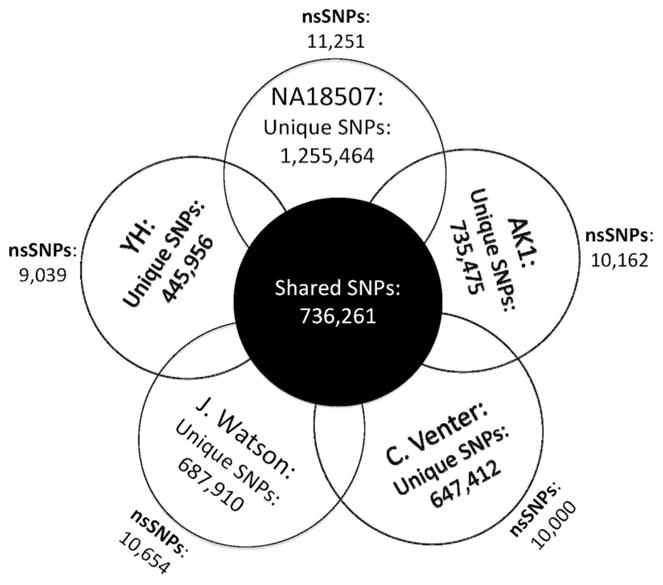
Venn diagram of personal genomes. The numbers of shared and unique SNPs and total number of nonsynonymous SNPs in 5 published genomes. NA18507 indicates Yoruba male; AK1, Korean male; HA, Han Chinese male.
Given the extent of DNA sequence variation among individuals, it is not surprising that phenotypic expression of diseases vary considerably. Sir William Osler, the father of modern medicine, astutely observed and advocated that no 2 individuals have identical diseases, more than a century ago. In fact, one may wonder why there is phenotypic similarity at all. Perhaps, it is the genetic gradient of the effect sizes that confer the phenotypic similarity. Accordingly, not all genetic variants impart the same level of phenotypic effect. There is a gradient of effects that ranges from miniscule to drastic. On one extreme are those variants that impart huge phenotypic effects and lead to single gene disorders. Such variant, when present, cause the phenotype even though the degree of its expression varies. On the opposite end of the spectrum are the variants with no or minimal effect sizes. Such variants contribute to the complex phenotypes, such as coronary atherosclerosis or the clinical phenotypes of single gene disorders, which are also complex phenotype. The prevalence of the DNA sequence variants mirrors the effect size gradient, albeit in the opposite orientation. Variants with major effect sizes are less common, partly because of the evolutionary pressure, and those with modest effects are more common. Accordingly, only a handful of DNA sequence variants are expected to exert major effects on a complex phenotype, whereas several hundred variants will exert modest and thousands minimal to barely discernible effects. The more complex the phenotype is, such as clinical phenotypes as opposed to biochemical phenotypes, the less impact of an individual variant and the greater the number of variants that contribute to the phenotype. This is best exemplified by the interindividual variability in height, which is primarily (≈80%) heritable. It is estimated that ≈93 000 SNPs explain 80% of the interindividual variation in height.15
With this background, 2 manuscripts in this issue of the Journal reports on phenotypes associated with the DNA sequence variants in genes known to cause Brugada and long-QT syndromes.16,17 The findings advocate the potential influence of genetic background on the clinical phenotype and illustrate dissociation of in vitro functional studies from the severity of the clinical phenotype. Both manuscripts raise several interesting points. First, the findings illustrate that not all nonsynonymous variants in genes known to cause monogenic disorders are causal mutations, even when rare. Many may function as modifier alleles. By definition, the causal mutation cannot be absent in family members with the phenotype. In contrast, modifier alleles are neither necessary nor sufficient to cause the phenotype, but when present, they affect phenotypic expression of the disease.18 The absence of the DNA sequence variant in the control population provides strong but not totally sufficient evidence of causality, as many nonsynonymous SNPs are rare. Likewise, near complete cosegregation of the DNA sequence variant with the phenotype in small-to-medium size families alone would not distinguish between causal and modifier variants. Nevertheless, the distinction between causal and modifier DNA sequence variants is somewhat diluted by nature’s genetic gradient and may be context dependent. A DNA sequence variant may function as a disease-causing mutation in certain backgrounds but as a modifier in another. Hence, not only there is a gradient of the effect sizes of various DNA sequence variants but also a phenotypic gradient of a single DNA variant.
Genetic studies are exposing the imperfection of the clinical phenotyping, which forms the essence of the current practice of medicine. Phenotypic plasticity of mutations in a single gene is well known. It is also the case for the SCN5A mutation and was also documented in the report by Probst et al.16 Likewise, phenotypic similarity of mutations in different genes is also well established. Expression of the Brugada phenotype in family members without the index SCN5A mutation, nonetheless, necessitates considering the possibility of another causal gene. The data also illustrate the point that biological and functional effects are not exclusive to the causal mutations. SNPs could also impart significant molecular, cellular, and clinical phenotypes. Given the rapid pace of advances in genetic sequencing techniques and the availability of individual genomes, researchers and clinicians are increasingly becoming aware that “no protein is perfect.” Thus, each protein could have common and rare nonsynonymous SNPs that exert biological functions but are not disease-causing variants. The complexity is expected to pose a significant challenge to the clinical utility of genetic screening.
The findings further illustrates the complexity of predicting the clinical phenotype based on in vitro modeling.17 Accordingly, despite the severity of the biophysical phenotype in the previously reported in vitro studies, the p.Y111C mutation in the KCNQ1, a known gene for long-QT syndrome, was associated with a low incidence of clinical events. The findings may be specific to the particular study population and not yet be generalized. The clinical phenotype of the p.Y111C mutation may vary in a different genetic background or environmental conditions. Nevertheless, the findings remind us that the results of an in vitro or in vivo study should be recognized in the context of the experimental conditions. An in vitro or in vivo study attempts to model the original, and no modeling is perfect. After all, “it’s only a model,” as Patsy, the loyal assistant to King Arthur, said when the Knights of the Round Table first got a glimpse of the Castle of Camelot (Monty Python and the Holy Grail).
The clinical phenotype is much more complex than commonly appreciated. The underlying complexity of the phenotype arises from intertwined nonlinear, dynamic, and often stochastic interactions among numerous genetic and nongenetic constituents that contribute to the phenotype. Likewise, the genome is much more complex than is discerned from the simple analysis of its sequence variants. The alphabets not only provide the codes for protein synthesis but also regulate noncoding RNAs, of which only microRNA are recognized for their influence on the phenotype.19 Similarly, the influence of epigenetic regulation of gene expression on the clinical phenotype remains to be understood.20 Moreover, 94% of the human genes undergo extensive alternative splicing.21 The influence of the alternative splicing on expression of the clinical phenotype is yet to be determined. Furthermore, a variety of posttranslational modifications, such as phosphorylation, acylation, glycosylation, lipoylation, ubiquitinylation, and disulfide bridges affect protein function and could influence the phenotype. The genome provides the stage on which various players choreograph the symphony. This is the case for every human phenotype. Yet, there is no phenotype that is solely genetics. Environmental factors directly contribute to expression of the phenotype or modulate the genetic determinants of the phenotype. Thus, to better understand the pluralism of causes and effects in any clinical phenotype, it is essential to analyze and incorporate all constituents of the phenotype into the modeling. Integration of signals from DNA sequence variants, mRNA splice variants, noncoding RNAs, proteome, metabolom, and the environment are essential in the modeling of a clinical phenotype. The late Dr Koshland’s “Cha-Cha-Cha” theory of scientific discoveries,22 however, necessitates delineating the fundamental biological mechanisms and applying these insights directly to the cure of disease. Only then, we could shift the current paradigm to individualized care, as Sir William Osler, the father of modern medicine, envisioned.
Acknowledgments
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute, a Clinical Scientist Award in Translational Research from the Burroughs Wellcome Fund, and the TexGen Fund from the Greater Houston Community Foundation.
Footnotes
The opinions expressed in this articles are not necessarily those of the editors or of the American Heart Association.
Disclosure
None.
References
- 1.McDougall I, Brown FH, Fleagle JG. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature. 2005;433:733–736. doi: 10.1038/nature03258. [DOI] [PubMed] [Google Scholar]
- 2.Mellars P. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science. 2006;313:796–800. doi: 10.1126/science.1128402. [DOI] [PubMed] [Google Scholar]
- 3.Liu H, Prugnolle F, Manica A, Balloux F. A geographically explicit genetic model of worldwide human-settlement history. Am J Hum Genet. 2006;79:230–237. doi: 10.1086/505436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 5.Hubscher U. DNA replication fork proteins. Methods Mol Biol. 2009;521:19–33. doi: 10.1007/978-1-60327-815-7_2. [DOI] [PubMed] [Google Scholar]
- 6.Osheroff WP, Jung HK, Beard WA, Wilson SH, Kunkel TA. The fidelity of DNA polymerase beta during distributive and processive DNA synthesis. J Biol Chem. 1999;274:3642–3650. doi: 10.1074/jbc.274.6.3642. [DOI] [PubMed] [Google Scholar]
- 7.Sutton MD, Walker GC. Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc Natl Acad Sci USA. 2001;98:8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, Stephens M, Bustamante CD. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders AC, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, Mac-Donald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marian AJ. Clinical implications of the “personal” genome. Curr Atheroscler Rep. 2008;10:361–363. doi: 10.1007/s11883-008-0055-6. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 16.Probst V, Wilde AAM, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scounarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson A, Meregalli PG, Le Marec H, Tan HL, Schott J-J. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009;2:552–557. doi: 10.1161/CIRCGENETICS.109.853374. [DOI] [PubMed] [Google Scholar]
- 17.Winbo A, Diamant U-B, Stattin E-L, Jensen SM, Rydberg A. Low incidence of sudden cardiac death in a Swedish Y111C-LQT1 population. Circ Cardiovasc Genet. 2009;2:558–564. doi: 10.1161/CIRCGENETICS.108.825547. [DOI] [PubMed] [Google Scholar]
- 18.Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol. 2002;17:242–252. doi: 10.1097/01.HCO.0000013803.40803.6A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol. 2009;21:461–469. doi: 10.1016/j.ceb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, Misawa Y, Isomura T, Shimada K, Mano H. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14:69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshland DE., Jr Philosophy Of Science. The Cha-Cha-Cha theory of scientific discovery. Science. 2007;317:761–762. doi: 10.1126/science.1147166. [DOI] [PubMed] [Google Scholar]