

Abstract
Background
Systolic (Sa) and diastolic (Ea) myocardial velocities measured by tissue Doppler (TD) imaging (TDI) recently were shown to be decreased in subjects who have mutations causing hypertrophic cardiomyopathy (HCM) but who do not have left ventricular (LV) hypertrophy. By studying these subjects at a later date, we sought to determine whether TDI predicts the subsequent evolution of the HCM phenotype.
Methods and Results
Serial 2D and Doppler echocardiography were performed in 12 subjects (age range, 17 to 51 years) with HCM-causing mutations on 2 occasions: before development of hypertrophy and 2 years later. Twelve age- and gender-matched family members without mutations were included as control subjects. In those with mutations, mean septal thickness and LV mass were 1.07±0.14 cm and 103.0±11 g at baseline, respectively, and increased to 1.30±0.36 cm and 193.0±78 g at follow-up (P<0.01), with 6 subjects satisfying HCM diagnostic criteria. Sa and Ea velocities in those with mutations were lower compared with control subjects at baseline and follow-up (lateral Sa, 15±1.2 versus 8.2±2.1 cm/s; lateral Ea, 16.5±2.8 versus 8.1±2.3 cm/s; P<0.01). At 2 years, left atrial volume and pulmonary venous flow indices of LV filling pressures increased, whereas TD early and late diastolic velocities decreased (all P<0.05) in those with the mutations. Control subjects had no significant interval changes of the above parameters.
Conclusions
Subsequent development of HCM in subjects with initially reduced TD velocities establishes TDI as a reliable method for early identification of HCM mutation carriers.
Keywords: cardiomyopathy, genetics, hypertrophy, screening
Elucidation of the molecular genetic basis of hypertrophic cardiomyopathy (HCM) has emphasized the need for early detection of mutation carriers. The ultimate goal is for early detection of mutation carriers and intervention to prevent evolution of the cardiac phenotype. Molecular genetic screening, although desirable, has been compounded by the diversity of known genes and mutations as well as the presence of yet-to-be-identified genes. Furthermore, penetrance is highly variable and unpredictable. In general, HCM seldom develops before puberty and may not develop until later in life.1
Recent observations in a transgenic rabbit model of HCM2,3 and in humans4–6 have shown reduced myocardial Doppler velocities in subjects with HCM mutations but without cardiac hypertrophy and have raised the possibility of using tissue Doppler (TD) imaging (TDI) for the early identification of individuals with such mutations. The clinical relevance of this finding remains uncertain, however, until it is determined whether this is an early stage of the disease with subsequent evolution of the HCM phenotype. Therefore, we carried out the present study to assess whether TDI can identify subjects with HCM-causing mutations who later develop left ventricular (LV) hypertrophy.
Methods
Study Population
Two age- and gender-matched adult groups from HCM families were evaluated. The control group included 12 asymptomatic members of the families who had not inherited the causal mutations. They had normal physical examinations and echocardiograms. The second group included 12 family members (age range, 17 to 51 years) with the causal mutations but without cardiac hypertrophy (wall thickness <13 mm) at the time of first examination. Subjects with the HCM mutation previously had been shown to have reduced myocardial systolic (Sa) and early diastolic (Ea) velocities in the absence of hypertrophy.4 None had coexisting diseases or were receiving medications. The causal mutations were detected by direct sequencing. Phenotypic characterization was repeated at the end of a 2-year follow-up period.
Echocardiographic Studies
Patients were imaged and data analyzed by a single observer without knowledge of the subjects’ names, genotypes, and previous clinical findings. LV septal, posterior, and maximal wall thicknesses; end-diastolic and end-systolic dimensions; ejection fraction; mass; and left atrial volumes were determined per established7 criteria. Mitral inflow was analyzed for peak E and A velocities, E/A ratio, acceleration time and deceleration time of E velocity, and isovolumic relaxation time. Pulmonary venous flow was analyzed for the systolic filling fraction (systolic time velocity integral/total forward time velocity integral), as well as peak velocity and duration of the atrial reversal (Ar) signal. The difference between Ar duration and that of transmitral A velocity was calculated (Ar-A duration). Pulse-Doppler was applied to allow for a spectral display of mitral annulus velocities at septal and lateral corners. Sa, Ea, and late diastolic (Aa) velocities were measured. The Ea/Aa and E/Ea ratios were computed at both corners of the mitral annulus.8
Statistical Analysis
Measurements at both time points in each group were compared by paired t tests. The changes over 2 years in the mutation-positive subjects were compared with those in control subjects by unpaired t tests. Regression analysis was used to correlate changes in LV mass to baseline TD velocities. Significance was set at P<0.05.
Results
The causal mutations in the 12 individuals without initial evidence of hypertrophy were: V606M (4 subjects) and R719W (1 subject) in β-myosin heavy chain, and InsG791 in myosin-binding protein C (7 subjects). One subject with an R92Q mutation in cardiac troponin T could not be reached for repeat echocardiography. Mean age (35±12 and 36±14 years) and gender (75% women) of the 2 groups were similar (P>0.3). All patients had satisfactory studies for analysis.
Evolution of Cardiac Hypertrophy
The mean values in the control subjects exhibited no significant change in LV dimensions, mass, or left atrial volume. In contrast, LV wall thickness (septal and maximal), mass, and left atrial volume all increased significantly during the 2-year follow-up period in those with HCM mutations (Table 1). In 6 of the 12 subjects with HCM mutations, septal thickness increased to >1.3 cm, fulfilling the diagnostic criteria for HCM, with 2 (of the 6) developing chordal systolic anterior motion of the mitral valve.
TABLE 1.
2D and Doppler Echocardiographic Measurements
| Control Subjects (n=12) |
Mutation-Positive Subjects Without Hypertrophy (n=12) |
|||
|---|---|---|---|---|
| Baseline | Follow-Up | Baseline | Follow-Up | |
| Heart rate, bpm | 74±8 | 72±8 | 75±9 | 74±10 |
| LV end-diastolic dimension, cm | 4.2±0.5 | 4.2±0.7 | 4.1±0.24 | 4.2±0.8 |
| LV end-systolic dimension, cm | 2.5±0.5 | 2.5±0.5 | 2.5±0.2 | 2.5±0.5 |
| Ejection fraction, % | 68±6 | 66±7 | 67±7 | 65±5 |
| Left atrial maximal volume, mL | 33±8 | 35±7 | 32±9 | 51±13* |
| Septal thickness, cm | 1±0.2 | 1±0.14 | 1.07±0.14 | 1.3±0.36* |
| Posterior wall thickness, cm | 0.96±0.1 | 1±0.11 | 1±0.1 | 1±0.12 |
| Maximal wall thickness, cm | 1±0.2 | 1±0.2 | 1±0.12 | 1.5±0.3* |
| LV mass, g | 102±10 | 111±11 | 103±11 | 193±78* |
| Peak E velocity, cm/s | 78±9.5 | 82±12 | 76±9.6 | 80±12 |
| E/A ratio | 1.4±0.3 | 1.4±0.4 | 1.4±0.4 | 1.4±0.4 |
| Isovolumic relaxation time, ms | 63±6 | 61±10 | 67±10 | 77±13 |
| Acceleration time, ms | 77±24 | 79±24 | 79±24 | 85±21 |
| Deceleration time, ms | 153±23 | 143±30 | 156±36 | 200±63 |
| Ar velocity, cm/s | 18±6 | 20±5 | 16±8 | 26±8* |
| Systolic filling fraction | 0.35±0.12 | 0.37±0.13 | 0.37±0.14 | 0.39±0.17 |
| Ar-A duration, ms | 0±10.5 | −7±12 | 0±16 | 19±15* |
P<0.01 vs baseline.
Changes in Ventricular and Atrial Filling Dynamics
No significant interval changes were noted in either group in the transmitral flow velocities. Only borderline significant changes were noted in deceleration time and isovolumic relaxation time (P<0.06 and 0.08, respectively) in those who had HCM mutations.
In contrast, a significant increase (both P<0.05) was noted in Ar velocity and Ar-A duration in the pulmonary venous flow pattern only in the subjects with HCM mutations. These 2 Doppler indices of LV end-diastolic pressure8 increased in 7 of the 12 patients.
Changes in TD Velocities
Sa and Ea velocities were significantly lower in subjects with HCM mutations in comparison with control subjects at follow-up (Table 2 and the Figure). Ea and Aa velocities were essentially unchanged in the control group but decreased significantly in the subjects with HCM mutations. The decrease in Sa velocity, however, was only borderline significant (P=0.07). The ratio of Ea/Aa remained significantly lower, and the ratio of E/Ea remained significantly higher at follow-up in those with mutations compared with control subjects (Table 2), indicating higher LV filling pressures. There was a significantly larger decrement in Ea (P=0.039) and Aa (P=0.014), along with a larger increment in E/Ea ratio (P=0.03) at follow-up in those with HCM mutations versus the control group. Significant inverse correlations were present between the increase in LV mass and baseline TD velocities (lateral Ea, −0.86; lateral Sa, −0.83; septal Ea, −0.8; septal Sa, −0.6; all P<0.05), with larger increments in LV mass noted in subjects with lower baseline velocities.
TABLE 2.
Tissue Doppler Velocities
| Control Subjects (n=12) |
Mutation-Positive Subjects Without Hypertrophy (n=12) |
|||
|---|---|---|---|---|
| Baseline | Follow-Up | Baseline | Follow-Up | |
| Lateral Sa, cm/s | 15.6±2 | 15±1.2 | 8.7±1.7* | 8.2±2.1* |
| Lateral Ea, cm/s | 16±2.5 | 16.5±2.8 | 9.5±2* | 8.1±2.3*† |
| Lateral Aa, cm/s | 9.4±1.1 | 9.1±1.5 | 9.8±2.2 | 8.3±1.4*† |
| Lateral Ea/Aa | 1.8±0.3 | 1.8±0.4 | 0.92±0.3* | 0.95±0.3* |
| Lateral E/Ea | 5.4±1.45 | 5.3±1.8 | 7.99±1.4* | 10±1.7*† |
| Septal Sa, cm/s | 14.5±1.4 | 13±1.2 | 7.96±1.6* | 7.4±1.9* |
| Septal Ea, cm/s | 15±2 | 14.7±2.5 | 8.5±2.27* | 7.8±2.5*† |
| Septal Aa, cm/s | 9.2±1.6 | 9.2±1.8 | 9.5±2.8 | 8.3±2.1*† |
| Septal Ea/Aa | 1.85±0.34 | 1.8±0.26 | 0.93±0.4* | 0.94±0.2* |
| Septal E/Ea | 5.8±0.92 | 5.6±0.92 | 8.65±1.5* | 10.26±2*† |
P<0.01 vs control subjects both at baseline and follow-up.
P≤0.01 vs mutation-positive subjects at baseline.
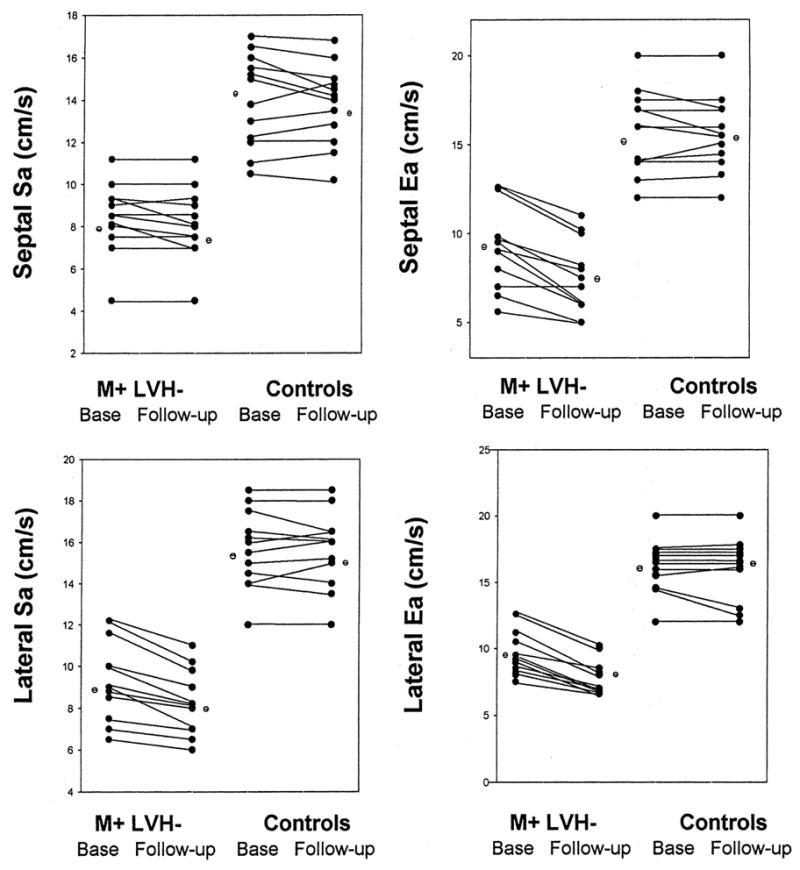
Individual data points showing septal and lateral Sa and Ea velocities at baseline and follow-up in both groups: the 12 subjects who had inherited the causal mutations for HCM and the 12 individuals in the control group. M+ LVH− indicates mutation-positive, LV hypertrophy–negative subjects; Base, baseline.
Discussion
Serial echocardiography was performed over a 2-year period in 12 subjects (age range, 17 to 51 years) who had inherited causal mutations for HCM. All had reduced TD velocities but no discernible hypertrophy at the time of initial evaluation. A significant increase in wall thickness occurred, and 6 developed septal hypertrophy to the extent that met the diagnostic criteria for HCM. Further impairment of myocardial function detected by TDI was also observed. In addition, several echocardiographic indices confirmed the occurrence of elevated LV filling pressures. These findings provide strong support for the clinical utility of TDI in early detection of preclinical HCM.
Although we4 and others5,6 have documented reduced TD velocities in humans with causal mutations for HCM and no hypertrophy, the present study is the first to document the evolution of HCM phenotype in such subjects. The strength and the significance of the present data reside in the use of TDI to identify mutation carriers who subsequently develop HCM. The rapidity whereby the TDI abnormalities progressed to LV hypertrophy, and the significant inverse correlation between the extent of hypertrophy and severity of baseline dysfunction by TDI, support both the specificity of the predictive role of this technique and the hypothesis that impaired sarcomeric function is a stimulus for hypertrophy.1 In addition, concomitant with the development of LV hypertrophy, myocardial relaxation deteriorated further (Ea velocity decreased). The findings with regard to hypertrophy (septal and maximal wall thickness and mass) and increased filling pressures (increased atrial volume, pulmonary Ar velocity and duration, and E/Ea; decreased Aa velocity) were concordant for several indices.
Although it is still unknown whether subjects with HCM mutations and reduced TD velocities but no hypertrophy are at an increased risk of sudden death, our data strongly indicate that members of HCM families with reduced TD velocities who have inherited the causal mutation are at risk of developing HCM. We propose that such individuals be monitored closely and assessed for the development of morphological and clinical features of HCM.
The results of the present study and previous studies4–6 support the utility of TDI in early detection of family members who have inherited the causal mutations. Early detection of mutation carriers could provide the opportunity for early intervention, including risk stratification and pharmacological therapies to prevent evolving cardiac phenotypes. This is particularly pertinent to recent studies in HCM animal models showing that losartan,9 simvastatin,10 and diltiazem11 reversed the evolving cardiac phenotype. Although genetic testing is of value in identifying those at risk for HCM, its clinical utility is compounded by variable penetrance and variable expression of phenotype. TDI would be invaluable in complementing the results of genetic testing—which remains significant—by identifying those likely to develop HCM soon, thus providing the opportunity for genetic counseling and implementation of early preventive measures.
Limitations
The observations in this study are limited to 12 subjects, and additional studies with larger samples are needed to further establish the role of TDI in the preclinical diagnosis of HCM. However, the present investigation, along with the published studies so far,4–6 supports the potential application of this technique in individuals without coexisting cardiac disorders (see references 4 and 5 for optimal cutoffs of TDI velocities). Because TD velocities decrease with age, one needs to apply age-specific values to achieve the highest accuracy of TDI. Furthermore, the specificity of TDI in an older population with coexisting cardiac disease (hypertension, coronary disease, pericardial or valvular disorders) is unknown and may represent a limitation for the proposed clinical application.
Acknowledgments
This work was supported by a grant from the National Heart, Lung, and Blood Institute, Specialized Centers of Research (P50-HL42267-01), and a Scientist Development grant (0030235N) from the American Heart Association National Center, Dallas, Tex.
References
- 1.Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marian AJ, Wu Y, Lim DS, et al. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest. 1999;104:1683–1692. doi: 10.1172/JCI7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagueh SF, Kopelen HA, Lim DS, et al. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102:1346–1350. doi: 10.1161/01.cir.102.12.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagueh SF, Bachinski LL, Meyer D, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho CY, Sweitzer NK, McDonough B, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992–2997. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 6.Cardim N, Perrot A, Ferreira T, et al. Usefulness of Doppler myocardial imaging for identification of mutation carriers of familial hypertrophic cardiomyopathy. Am J Cardiol. 2002;90:128–132. doi: 10.1016/s0002-9149(02)02434-7. [DOI] [PubMed] [Google Scholar]
- 7.Schiller NB, Shah PM, Crawford M, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 8.Nagueh SF, Lakkis NM, Middleton KJ, et al. Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation. 1999;99:254–261. doi: 10.1161/01.cir.99.2.254. [DOI] [PubMed] [Google Scholar]
- 9.Lim DS, Lutucuta S, Bachireddy P, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel R, Nagueh SF, Tsybouleva N, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semsarian C, Ahmad I, Giewat M, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]