

Abstract
In contrast to many years of important research and clinical attention to the pathological effects of alcohol (ethanol) abuse, the past several decades have seen the publication of a number of peer-reviewed studies indicating beneficial effects of light-moderate, non-binge consumption of varied alcoholic beverages, as well as experimental demonstrations that moderate alcohol exposure can initiate typically cytoprotective mechanisms. A considerable body of epidemiology associates moderate alcohol consumption with significantly reduced risks of coronary heart disease and, albeit currently a less robust relationship, cerebrovascular (ischemic) stroke. Experimental studies with experimental rodent models and cultures (cardiac myocytes, endothelial cells) indicate that moderate alcohol exposure can promote anti-inflammatory processes involving adenosine receptors, protein kinase C (PKC), nitric oxide synthase, heat shock proteins, and others which could underlie cardioprotection. Also, brain functional comparisons between older moderate alcohol consumers and non-drinkers have received more recent epidemiological study. In over half of nearly 45 reports since the early 1990’s, significantly reduced risks of cognitive loss or dementia in moderate, non-binge consumers of alcohol (wine, beer, liquor) have been observed, whereas increased risk has been seen in only a few studies. Physiological explanations for the apparent CNS benefits of moderate consumption have invoked alcohol’s cardiovascular and/or hematological effects, but there is also experimental evidence that moderate alcohol levels can exert direct “neuroprotective” actions—pertinent are several studies in vivo and rat brain organotypic cultures, in which antecedent or preconditioning exposure to moderate alcohol neuroprotects against ischemia, endotoxin, β-amyloid, a toxic protein intimately associated with Alzheimer’s, or gp120, the neuroinflammatory HIV-1 envelope protein. The alcohol-dependent neuroprotected state appears linked to activation of signal transduction processes potentially involving reactive oxygen species, several key protein kinases, and increased heat shock proteins. Thus to a certain extent, moderate alcohol exposure appears to trigger analogous mild stress-associated, anti-inflammatory mechanisms in the heart, vasculature and brain that tend to promote cellular survival pathways.
Keywords: ethanol, coronary disease, ischemia-reperfusion, cytoprotection, heat shock protein, brain, preconditioning, neurodegeneration, nitric oxide, protein kinase C, focal adhesion kinase
Introduction
This minireview derived from an RSA roundtable coalesces emerging information about the epidemiology of heart disease, stroke and dementia in moderate, non-binge alcohol (ethanol) consumers with relatively recent results of laboratory studies of biochemical and physiological mechanisms that potentially underlie observed cytoprotective actions of moderate alcohol concentrations. The pathological damage and vast social havoc from addiction to and abuse of alcohol are well-known, and of necessity should continue to receive primary attention by doctors, scientific researchers and health professionals. However, results from human moderate consumption studies indicate that light-to-moderate, responsible alcohol consumption appears to carry certain health benefits.
Most of the epidemiological evidence for cytoprotection relates to the heart and vasculature. Experimentally, ischemia-reperfusion studies with cardiac myocytes and chronic alcohol-consuming mice have shown that moderate alcohol-dependent cardioprotection is linked to selective activation of protein kinase C (esp. PKCε isoform) and preservation of mitochondrial function. Furthermore, low-dose alcohol consumption in ischemic rats improves ventricular function while inducing cardioprotective proteins, e.g., heat shock proteins, superoxide dismutase, and nitric oxide synthase (NOS). When present in alcoholic beverages, the flavonoid resveratrol may play an important augmenting or independent role. Further studies also confirm that the endothelium is critical in moderate alcohol-dependent cardiovascular protection through changes in NOS as well as enhanced pro-fibrinolytic enzymes. Additionally, antecedent alcohol ingestion in an ischemia-reperfusion rat model has been shown to trigger entry of the microvasculature into a preconditioned, protected state that involves adenosine receptors, NOS, calcitonin gene-related peptide release and at least one heat shock protein, heme oxygenase.
Turning to the CNS, in the past decade or so a number of peer-reviewed cohort studies have reported significantly reduced risks of either cognitive decline or dementia, including Alzheimer’s, in light-moderate alcoholic beverage consumers in comparison to nondrinkers or never-drinkers. Although some of the vascular and hematological mechanisms triggered by moderate alcohol could favorably impact the aging brain, other more central brain molecular pathways possibly triggered by episodic low/moderate levels of alcohol that could exert anti-neuroinflammatory effects are possible contributors to neuroprotection.
MODERATE ALCOHOL CONSUMPTION AND RISK OF CORONARY DISEASE, STROKE AND DEMENTIA: AN EPIDEMIOLOGICAL OVERVIEW
The observed associations of alcohol consumed within recommended guidelines with a variety of chronic diseases at the population level continue to interest physicians and patients alike. Among the most important and complex of these are the associations of alcohol use with cardiovascular and neurovascular diseases, such as coronary heart disease (CHD) and stroke.
Two large prospective cohort studies have recently examined alcohol intake and these diseases: the Health Professionals Follow-up Study (HPFS) (Mukamal et al., 2003a) and the Cardiovascular Health Study (CHS) (Mukamal et al., 2006). Both are carefully-administered longitudinal studies with repeated measures of alcohol intake and validated assessment of cardiovascular disease incidence over 10–20 years. The populations differ to a substantial degree—HPFS enrolled over 50,000 middle-aged male health professionals from around the U.S., while CHS is a population-based study of almost 6,000 older adults from four U.S. communities—but they have produced remarkably similar findings on the relation of alcohol intake with cardiovascular disease.
The association of alcohol use with CHD has undoubtedly been the focus of the most observational studies. Several meta-analyses have come to generally similar conclusions about the relationship of average alcohol consumption with risk, suggesting ~25% lower risk throughout a range of intake from about ½ drink per day through two drinks per day, but potentially higher risk at or above six drinks per day (Cleophas, 1999; Corrao et al., 2000; Maclure, 1993). This association has been observed for over three decades (Klatsky et al., 1974) and in populations worldwide consuming a variety of alcoholic beverages (Brenner et al., 2001; Yuan et al., 1997). At the same time, concern continues to be raised about whether the lower risk associated with alcohol intake is related to bias by inclusion of former drinkers with pre-existing illness or confounding by lifestyle factors associated with moderate drinking (Freiberg and Samet, 2005).
In both HPFS and CHS (Figure 1), strong, graded, inverse associations of alcohol intake with risk of CHD were found (Mukamal et al., 2006; Mukamal et al., 2003a). Former drinkers were separated from longer-term abstainers in both cases, and the results demonstrated little difference in risk between abstainers and very light drinkers, suggesting that the choice of reference group did not bias the findings. It was possible to account for a large number of sociodemographic and clinical characteristics, and findings were similar for a variety of CHD-related outcomes. In both studies, risk was approximately 25–30% lower among consumers of 1 drink per day than among abstainers. No clear advantage was observed for any one beverage type in either study, suggesting that alcohol per se is most likely to be the active constituent, at least at the doses of alcoholic beverages normally consumed.
Figure 1. The relationships of alcohol intake with risks of coronary heart disease, ischemic stroke, and dementia among participants in the Cardiovascular Health Study.

Long-term abstainers were the reference category. The Y-axis indicates hazard ratios for coronary heart disease and ischemic stroke, and odds ratios for dementia. See text for appropriate references.
Several plausible mechanisms for the association of alcohol intake with lower risk of CHD exist. Randomized crossover feeding studies clearly show that alcohol intake raises high-density lipoprotein cholesterol (HDL-C) in an approximately dose-dependent manner and to a greater degree than currently available pharmacological options (Rimm et al., 1999). In the CHS and HPFS, alcohol intake correlates well with HDL-C levels, with coefficients of ~0.25–0.35. The effect of alcohol on HDL-C alone appears to explain 50–70% of the observed inverse association of alcohol with risk of CHD (Mukamal et al., 2005c; Rimm et al., 1999). Other feeding studies, including the longest randomized trial conducted to date (Marfella et al., 2006), also support a benefit of alcohol intake on insulin sensitivity (Davies et al., 2002) and on inflammatory markers (Sierksma et al., 2002b). Whether alcohol also provides direct cardioprotection from myocardial ischemia has not been determined in humans, but analyses of the Determinants of Myocardial Infarction Onset Study (which included nearly 4,000 patients with acute myocardial infarction) raise the possibility that alcohol consumed 6–12 hours prior to onset of ischemia could attenuate infarct size (Mukamal et al., 1999).
Despite similarities between CHD and stroke, their associations with alcohol intake appear quite different. In both HPFS (Mukamal et al., 2005a) and CHS (Figure 1) (Mukamal et al., 2005b), lower risk of ischemic stroke was limited to alcohol consumption weekly but less than daily and with a substantially weaker magnitude than was seen for CHD. In HPFS, alcohol intake above two drinks per day was clearly associated with higher ischemic stroke risk. Meta-analyses of alcohol and stroke support this fully (Reynolds et al., 2003), suggesting ~20% lower risk of ischemic stroke among less-than-daily drinkers in cohort studies, but increased risk with heavier drinking, and a dose-dependent increase in risk for hemorrhagic stroke. Consumption of three or more drinks in one day also appears to acutely trigger ischemic stroke (Hillbom et al., 1995).
Interest in the association of alcohol intake with dementia has been fueled by the recognition that dementia shares several risk factors with cardiovascular disease. In the CHS, a U-shaped relationship of alcohol intake with risk of dementia (see Figure 1) was observed, with similar findings for vascular dementia and Alzheimer disease. This U-shape exactly parallels findings on alcohol intake and MRI-identified cerebral white matter lesions (Mukamal et al., 2001), which are strong predictors of dementia in CHS (Kuller et al., 2003). Although dementia was not assessed in HPFS, findings from the Nurses’ Health Study, a parallel cohort of women, also support better cognitive function among women who consume alcohol within recommended ranges (Stampfer et al., 2005). However, data from other studies appear somewhat mixed (Dufouil et al., 2000; Larrieu et al., 2004; Luchsinger et al., 2004; Ruitenberg et al., 2002; Truelsen et al., 2002), and this remains an important area for future investigation.
Thus alcohol intake has a complex associations with cardiovascular and neurovascular diseases. These include dose-dependent associations of alcohol with lower risk of CHD but higher risk of hemorrhagic stroke throughout a recommended range of drinking, an increased risk of ischemic stroke with heavier drinking, and an emerging but somewhat variable association with lower risk of dementia.
MODERATE ALCOHOL AND PROTECTION AGAINST CARDIAC ISCHEMIA-REPERFUSION (I/R) INJURY: INVOLVEMENT OF PKCepsilon
Moderate alcohol consumption, one drink or less per day for women and two drinks or less per day for men, has been found to reduce the incidence and adverse consequences of heart disease in a series of epidemiological studies. For example, Mukamal and coworkers (Mukamal et al., 2005d) showed that alcohol consumption at least three to four days per week was associated with a lower risk of myocardial infarction among men and women. Importantly, this association was attributed to physiologically relevant trends in HDL cholesterol, fibrinogen, and hemoglobin A1C. Clinical trials of short-term alcohol intake are scarce but suggest that moderate drinking enhances insulin sensitivity (Davies et al., 2002) and decreases plasma levels of C-reactive protein and fibrinogen (Sierksma et al., 2002a). Long-term clinical trials to examine effects of moderate drinking on coronary artery disease outcomes are highly controversial and not likely to be conducted within the United States.
With a focus on the identification of intracellular signaling pathways activated by moderate alcohol consumption that preserve viability and contractile function of cardiac tissue during stress, adult rodent protocols have been carried out that simulate human drinking patterns to test the hypothesis that moderate alcohol concentrations trigger protective signaling in ventricular heart cells, thereby increasing resistance to acute ischemia-reperfusion injury. In such studies, moderate concentrations in rats produced sustained cardioprotection requiring activation of mitochondrial KATP channels (Zhu et al., 2000), consistent with experiments published by other laboratories using canine models (Pagel et al., 2002). Also, acute (Chen et al., 1999) and chronic (Zhou et al., 2002) cardioprotection stimulated by physiological concentrations of alcohol were associated with the selective activation of protein kinase Cepsilon (PKCε). This isoenzyme is recognized as a powerful cytoprotectant in myocytes, through interactions with substrates and anchoring molecules in numerous compartments (Ping et al., 2001).
In a typical experiment, adult male C57BL/6 mice ingest 10% ethanol (vol/vol) as the sole source of drinking water for 12 weeks (Zhou et al., 2004). The protocol produces blood alcohol concentrations of approximately 10 mg/dL or 4 mM at the time of sacrifice (and maximal night-time concentrations in comparable alcohol consumption studies of approximately 13 mM or 67 mg/dL remain in a moderate range) (Chen et al., 2003). Alcohol-fed animals appear normal in the cage environment and maintain predicted body weights. However, the hearts of alcohol-fed mice are much more resistant to stress than control hearts, with decreased infarction size and improved contractile recovery after simulated ischemia-reperfusion. Importantly, cardiac myocytes isolated from alcohol-fed mice are resistant to in vitro stress. As shown in Figure 2, control cells rapidly lose their capacity to exclude trypan blue dye during incubation with H2O2-supplemented culture medium. In contrast, cell survival under the same oxidative conditions is significantly greater in myocyte cultures prepared from the ventricles of alcohol-fed animals. These results confirm that moderate alcohol concentrations are cardioprotective, at least in part, through direct and sustained effects on cellular mechanisms intrinsic to ventricular myocytes.
Figure 2. Moderate drinking and mitochondrial protein kinase Cε localization.

Adult male C57BL/6 mice were fed 10% alcohol (vol/vol) in drinking water for 12 weeks as described (Zhou et al., 2004). Mitochondrial fractions were prepared from isolated hearts by differential centrifugation. (A) Western blot analysis of mitochondrial fractions from normoxic hearts. Moderate alcohol intake had no significant effects on mitochondrial protein kinase Cε expression. (B) Alcohol consumption produced a 3-fold increase in mitochondrial protein kinase Cε in hearts following 25 min ischemia and 30 min reperfusion on a Langendorff apparatus.
Mitochondria produce most of the ATP required for cardiac contractile activity. In addition, these organelles generate reactive oxygen species that contribute to both physiological heart function and pathological tissue damage. Previous experience with the effects of alcohol on mitochondrial KATP channel activity and PKCε expression prompted investigations of PKCε localization in heart mitochondrial fractions before and after chronic alcohol feeding and acute ischemia-reperfusion. As indicated in Figure 3A, chronic moderate alcohol consumption does not produce significant differences in mitochondrial PKCε expression under baseline conditions. However, localization of PKCε to mitochondrial fractions is 3-fold greater after ischemia-reperfusion in the hearts of alcohol-fed mice compared to controls. These data suggest that alcohol pretreatment promotes rapid recruitment of PKCε during stress to heart mitochondria, where PKCε has been shown by other groups to preserve organelle integrity (Baines et al., 2003).
Figure 3. Protein kinase Cε is a key factor in alcohol-mediated cardioprotection.

Submitochondrial particles were prepared as described (Zhou et al., 2004) from hearts of alcohol-fed (black bars) and control (white bars) mice and used to measure respiratory chain complex activities after ischemia-reperfusion. (A) Moderate alcohol intake significantly improved mitochondrial NADH-oxidase activity after stress. In contrast, sustained cardioprotection was blocked by a protein kinase Cε antagonist peptide and in knockout mice. (B) Chronic protection against NADH-Q1 reductase (Complex I) injury was also abolished by protein kinase C inhibition. n = 6 hearts per condition. *P<0.05 vs. control ischemia-reperfusion hearts.
Accordingly, microassays to measure the activity of NADH-ubiquinone (Q1) reductase, or Complex I, in submitochondrial particles prepared from individual mouse hearts have been developed (Zhou et al., 2004). As shown in Figure 3B, moderate alcohol concentrations did not produce significant differences in mitochondrial Complex I activity under baseline conditions. However, alcohol ingestion doubled Complex I activity after ischemia-reperfusion compared to controls. Pretreatment of isolated hearts with a specific PKCε antagonist peptide (Gray et al., 2004) prior to ischemia-reperfusion blocked alcohol-induced cardioprotection. In the above report, hearts from PKCε null mice fed 10% alcohol for 12 weeks did not exhibit protection against Complex I injury compared to controls. These results signify that moderate alcohol concentrations decrease damage to the mitochondrial respiratory chain complex most sensitive to the stress of ischemia-reperfusion and support the key role of PKCε activation in sustained alcohol-mediated cardioprotection.
Overall, a mouse model of consumption that realizes moderate but not high alcohol concentrations points to sustained PKCε activation in ventricular myocytes as mediator of resistance to ischemia-reperfusion injury. The experimental system is highly stable and may be more relevant to human cardiac physiology than genetically engineered mice that express a constitutively active form of PKCε (Ping et al., 2001). Hearts from alcohol-treated animals are being used to explore downstream mechanisms of chronic cardioprotection through conventional protein methods and unbiased approaches such as microarray analysis, with one principal objective of this research to better understand the relationship between alcohol consumption and heart disease, particularly for individuals who drink in moderation. Alternatively, this line of investigation may reveal cardioprotective targets that do not require alcohol ingestion, thus circumventing toxic effects on other organ systems.
ALCOHOL AND CARDIOVASCULAR PROTECTION via NITRIC OXIDE PATHWAYS
Coronary heart disease (CHD) remains the leading cause of death in both men and women in the United States (Klatsky, 1996). In patients with cardiovascular risk factors such as hypercholesterolemia, hypertension or aging (Zeiher et al., 1993) endothelial dysfunction predisposes to the development of structural vascular changes (Ross, 1993) and may play a critical role in acute myocardial infarction and sudden death. Heavy alcohol consumption has long been associated with vascular as well as myocardial complications (Klatsky, 1996), whereas, as discussed earlier in this summary, several epidemiological studies suggest an inverse association between long-term moderate alcohol consumption and the risk of CHD and myocardial infarction (Thun et al., 1997) as well as a reduction in mortality (Doll, 1997).
This cardioprotection has been attributed to the direct effects of moderate alcohol and/or polyphenols in increasing high-density lipoproteins (Gaziano et al., 1999; McConnell et al., 1997), decreasing platelet aggregation (Renaud and Ruf, 1996), enhancing fibrinolytic activity, decreasing fibrinogen (Aikens et al., 1998; Ridker et al., 1994) and decreasing ischemia-reperfusion injury (Miyamae et al., 1997). There is accumulating evidence that cardioprotection associated with moderate alcohol consumption involve cellular and molecular mechanisms related to nitric oxide (NO). Nitric oxide is generated by three isoforms of nitric oxide synthase (NOS). Endothelial NOS (eNOS) is a key regulator of vascular homeostasis, including basal vascular tone (blood flow) and blood pressure (Patel et al., 1999; Radomski and Moncada, 1993). Cardiac myocytes also constitutively express eNOS, which contributes to the regulation of myocardial contractility, heart rate (Kojda and Kottenberg, 1999) and cardiac oxygen consumption (Loke et al., 1999). Upregulation of eNOS has been postulated to play a protective role in both congestive heart failure (Khadour et al., 1998) and myocardial ischemia-reperfusion (Kanno et al., 2000; Sumeray et al., 2000). NO also inhibits smooth muscle proliferation, platelet aggregation and monocyte adhesion making it an anti-thrombogenic agent (Lamas et al., 1998).
In a paired feeding paradigm, male Sprague-Dawley rats consumed an alcohol (ethanol)-containing or equicaloric liquid diet in which alcohol comprised 0, 9 or 18% of the total calories for 8 weeks. At 8 weeks, animals were anesthetized and myocardial function accessed after 20 min ischemia and 30 min reperfusion using a modified Langerdorf preparation. Moderate alcohol consumption enhanced post-ischemic myocardial systolic and diastolic function as well as attenuated the ischemia-induced increase in coronary vascular resistance. In a separate series of experiments, vascular function was assessed in isolated thoracic aorta ring segments. Acetylcholine was added to stimulate endothelial cell NO formation via the mobilization of intracellular Ca++, which activates eNOS and invokes endothelium-dependent relaxation. Moderate alcohol consumption for 8 weeks significantly increased maximal amplitude for vascular relaxation, consistent with upregulation of eNOS. There was a paradoxical impairment in vessel relaxation with higher doses of alcohol consumption. Immunolocalization of eNOS protein confirmed moderate alcohol consumption increased eNOS protein expression in the vascular endothelium of the vascular endothelium. Finally, total nitrates and nitrites were increased in the blood of rats after 8 weeks consumption of moderate alcohol, suggesting a functional eNOS protein increase (Abou-Agag et al., 2005).
The characteristics of the cardioprotection associated with consumption of alcoholic beverages are indicative of mechanisms resulting in improved functional and metabolic performance in both the myocardium and vasculature. There are a number of actions of NO that would be predicted to be cardioprotective, including decreased cardiac contractility (Kitakaze et al., 1996; Lamas et al., 1998), decreased coronary resistance (Kitakaze et al., 1996), diminished myocardial oxygen consumption (Okubo et al., 1999), and improved metabolic function (Kanai et al., 1997). Increased NO bioavailability may play an important role in alcohol-dependent cardioprotective mechanisms through the regulation of antioxidant and NO producing enzymes.
ALCOHOL CARDIOPROTECTION MECHANISMS WITH ADDITIONAL CONSIDERATION OF THE CONTRIBUTION OF RESVERATROL
Epidemiological evidence strongly suggests that light-to-moderate alcohol drinking, usually defined as consumption of 1 to 2 drinks per day [less than 30 gm/day], is associated with a reduced risk of CHD including ischemic heart disease, cardiomyopathy, diabetes, and stroke (Zakhari, 1999). Heavy drinking (more than 3 drinks per day), on the other hand, adds further to the complications in CHD. A J- or U-shaped relationship exists between the amount of alcohol drinking and CHD (Djousse et al., 2002), indicating that risk of death due to CHD is the lowest among the moderate drinkers as compared to abstainers or heavy drinkers. Cardioprotective effects of moderate alcohol drinking include reduction of platelet aggregation and fibrinogen levels leading to increased fibrinolysis, coronary blood flow and reduced blood pressure; increased HDL and reduced LDL resulting in lowering of risk due to cholesterol; reduction of insulin resistance and increase in insulin sensitivity; and reduction of blood homocysteine levels (Figure 4A).
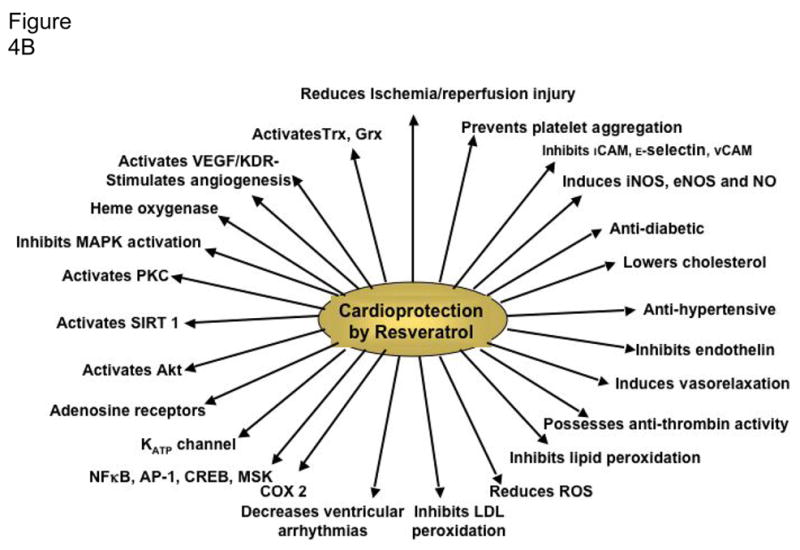
Figure 4.
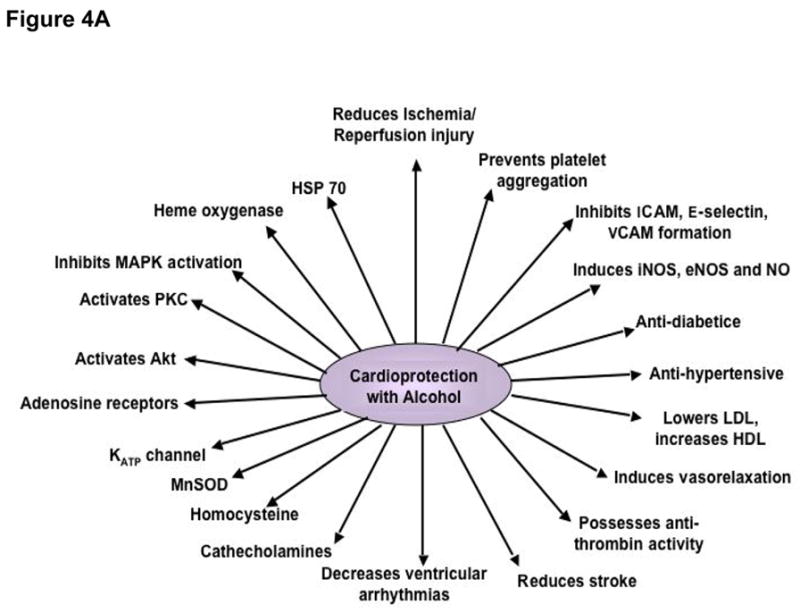
A. Cardioprotective and molecular targets of alcohol.Cardioprotective targets are shown on the right whereas the molecular targets are shown on the left.
B. Cardioprotective and molecular targets of resveratrol. Cardioprotective targets are shown on the right whereas the molecular targets are shown on the left.
Precise mechanisms for the cardioprotective effects are not known; however, several experimental studies have shed some light on the mechanisms of action of light to moderate drinking. In one of such studies, guinea pigs were given alcohol in their drinking water for 3–12 weeks and at the end, the hearts were isolated and subjected to ischemia reperfusion injury (Miyamae et al., 1997). Hearts from animals drinking alcohol showed improved functional recovery and decreased myocyte damage when compared with controls. Adenosine A1 receptor blockade abolished the protection, suggesting a role of adenosine A1 receptor in cardioprotection. In another study, the rats were made diabetic with streptozotocin, and then given alcohol for a month. The diabetic rats fed an alcohol diet showed a 52% reduction in advanced glycation end product when compared with diabetic controls (Al-Abed et al., 1999). In rabbit heart alcohol was found to exert a preconditioning-like effect mediated through adenosine receptors, PKC, and mitochondrial KATP channels (Krenz et al., 2004). In in vitro studies mentioned earlier, rat heart cardiomyocytes were protected from ischemic injury by alcohol through the upregulation of PKCε (Chen et al., 1999), and also, alcohol-mediated cardioprotection was achieved through the upregulation of eNOS expression (Abou-Agag et al., 2005). In rat heart, direct alcohol feeding led to a reduction of apoptotic cardiomyocyte death by reducing apoptotic signaling cascades, JNK-1 and c-Jun, and activating heat shock protein (HSP) 70, heme oxygenase and superoxide dismutase (Sato et al., 2004). Another study, also cited earlier in this summary, demonstrated that alcohol triggered anti-death signals by activating Akt (Zhou et al., 2002). The mechanisms of action of alcohol-mediated cardioprotection known to date are summarized in Figure 4A.
There is no direct clinical study demonstrating the mechanisms of cardioprotection afforded by light-to-moderate alcohol consumption. Nevertheless, clinical studies exist showing that moderate alcohol consumption leads to a reduction of serum lipids, lipoproteins, platelets, LDL and inflammatory cytokines, and increased HDL (Lucas et al., 2005). Overwhelming epidemiological evidence, however, exists in support of alcohol-mediated cardioprotection. All the risk factors of CHD including diabetes, hypercholesterolemia, smoking and stroke are modulated by low-to-moderate alcohol consumption (Djousse et al., 2002).
A last point is that most of the experimental studies related to alcohol-mediated cardioprotection that originate in Europe use wine, especially red wine, while American studies typically use alcohol itself. Although part of the wine-mediated cardioprotection may be attributed to the alcohol (wines typically contain 10–12% alcohol), cardioprotective properties of red wine can be correlated with the presence of polyphenolic antioxidants, especially resveratrol, which is concentrated in red wine (Das and Maulik, 2006), but also in other alcoholic beverages (albeit in lower amounts) such as beers (Callemien et al., 2005). Whether resveratrol is the essential component that is responsible for much of the cardioprotective effects of wine or the alcohol that is present in wines and is responsible for cardioprotection remains under considerable debate, but there is no doubt that resveratrol may protect the heart in many ways (Das et al., 2008; Juric et al., 2007; Lekli et al., 2008; Mokni et al., 2007), not to mention the brain with regard to stroke (Tsai et al., 2007). At any rate, it is interesting to note that there are many similarities in the cardioprotective mechanisms of action and molecular targets between resveratrol and alcohol (Figure 4A and Figure 4B) that may force the rethinking of the relative significance of alcohol vs. resveratrol in health and disease.
ANTECEDENT ALCOHOL PREVENTS MICROVASCULAR DYSFUNCTION IN I/R THROUGH ANTI-INFLAMMATORY AND ANTI-ADHESIVE MECHANISMS
The possibility that ingestion of alcoholic beverages might be associated with cardioprotective effects was first appreciated by consideration of epidemiologic data indicating that long-term, regular consumption of red wine at low to moderate levels (1 to 3 drinks per day for months to years) correlated with decreases in the incidence of coronary artery disease as well as improved survival in patients suffering myocardial infarctions (Das and Ursini, 2002; Korthuis, 2004). Although the antioxidant properties of red wine constituents were originally thought to largely explain its cardioprotective effects, subsequent epidemiologic evidence has indicated that consumption of white wine, beer, or spirits is also beneficial (Das and Ursini, 2002; Korthuis, 2004; Shigematsu et al., 2003). The latter observations pointed to the importance of alcohol per se in the beneficial actions of alcohol intake on cardiovascular mortality. While subsequent studies established that the cardioprotective effects of moderate alcohol consumption could be partially explained by effects on plasma lipids, platelet function, and fibrinolytic activity, alcohol intake maintains a significant association with reduced cardiovascular mortality even after controlling for lipoprotein and hemostatic factors. More recent work indicates that alcohol administration 24 hrs prior to I/R (referred to as antecedent alcohol, equivalent to alcohol preconditioning, but with trace residual alcohol levels during I/R) induces the development of an anti-inflammatory phenotype in postcapillary venules such that these vessels fail to express P-selectin or support leukocyte adhesion and emigration in postischemic tissues (Dayton et al., 2005; Gaskin et al., 2007; Kamada et al., 2004; Wang et al., 2007; Yamaguchi et al., 2007). In addition, alcohol consumption at low to moderate levels is associated with reduced C-reactive protein levels, a plasma marker of inflammation, and decreased production of inflammatory cytokines such as interleukin-6 (Albert et al., 2003; Das and Ursini, 2002; Korthuis, 2004; Korthuis, 2006). Given the critical importance of infiltrating leukocytes in the pathogenesis of atherosclerosis and I/R injury, these new observations provide novel insight regarding the mechanisms whereby alcohol ingestion reduces the likelihood and extent of I/R injury in individuals at risk for cardiovascular disease.
The anti-adhesive effects of antecedent alcohol intake on postischemic leukosequestration has been extensively studied using intravital microscopic approaches, which allow direct quantification of leukocyte rolling, stationary adhesion, and emigration in single postcapillary venules in real time. Yamaguchi and coworkers (Yamaguchi et al., 2007) were the first to demonstrate that acute intubation of a low-moderate dose of alcohol in mice (>0.5 g/kg; ~300 μl vol.) completely prevents adhesive interactions between circulating leukocytes and the endothelium from developing in tissues exposed to subsequent I/R. The temporal expression of this protected or preconditioned state induced by alcohol ingestion was biphasic. The initial phase (acute alcohol preconditioning) develops rapidly, involves activation of pre-existing effector molecules, and is short-lived, with peak anti-inflammatory effects (50% reduction in leukocyte adhesion) occurring 2 to 3 hours after intake, and then disappearing by 4 hours after administration. The second or delayed phase of ischemic tolerance induced by alcohol emerges 12 to 24 hours after alcohol intake, is longer-lived (24 hrs or more), requires the expression of new gene products, and is notable for its magnitude of protection, completely preventing postischemic leukocyte rolling and adhesion (Kamada et al., 2004; Korthuis, 2006; Yamaguchi et al., 2007). In these studies, plasma alcohol levels peaked at 45mg/dL within 30 minutes of gastric instillation by gavage and returned to control levels within 60 minutes of ingestion. This observation indicates that neither the acute phase of protection that arises within 1 to 3 hours of alcohol ingestion nor the late phase that reemerges 12 to 24 hours later are due to direct effects of alcohol. Indeed, alcohol must be largely absent before the cardioprotective effects of the alcohol become apparent.
Because late phase alcohol preconditioning exerted much more powerful and long-lived effects, most subsequent studies have focused on elucidation of the mechanistic underpinnings for this latter phase. As a result of this work, it has become apparent that the factors that contribute to the development of tolerance to ischemia can be conceptually compartmentalized into: 1) triggers that inaugurate entrance into the anti-inflammatory phenotype, 2) downstream signal transducers that are activated by the initiating factors and induce an increase in activity or expression of 3) end-effectors that mediate the anti-adhesive effects noted during I/R 24 hrs after ingestion (Kamada et al., 2004; Korthuis, 2006).
Early work in this area focused primarily on uncovering the initiators of alcohol preconditioning, as this may provide important clues towards the development of rational therapeutic interventions that could be used in lieu of alcohol to induce similar cardioprotective effects, but avoid potential untoward physiologic and social effects associated with alcohol consumption. The well-known effect of alcohol to inhibit nucleoside transporters in cell membranes (thereby limiting adenosine reuptake into cells) led to the postulate that adenosine might serve as a trigger for entrance into the anti-inflammatory state induced by adenosine. Pharmacologic inhibitor studies support this concept and indicated that adenosine A2 receptor occupancy is required for this process (Yamaguchi et al., 2007). Uncovering the role of adenosine A2 receptors was an important mechanistic distinction as the downstream signaling elements activated by adenosine A1/A3 versus A2 receptors are very different. Indeed, adenylyl cyclase or protein protein kinase A blockade, but not PI3 kinase inhibitors, prevents the late phase of alcohol preconditioning (Kamada et al., 2004; Korthuis, 2006). On the other hand, administration of adenosine A2 receptor agonists, cell-permeant cyclic AMP analogs, or adenylyl cyclase activators (e.g., isoproterenol, forskolin) mimics the postischemic anti-inflammatory effects of late-phase alcohol preconditioning.
Because alcohol enhances both basal and flow-stimulated NOS activity and NO production in vivo and in cultured endothelial cells, it has been suggested that production of this gaseous monoxide during the period of alcohol exposure may also serve as an important triggering element for the late phase of alcohol preconditioning (Wallerath et al., 2003; Yamaguchi et al., 2002; Yamaguchi et al., 2007). Support for this concept is derived from four lines of evidence. First, administration of NOS antagonists just prior to, but not 1 hour after, alcohol administration on day 1 abolishes the anti-adhesive effects of late alcohol preconditioning on day 2 (Yamaguchi et al., 2002; Yamaguchi et al., 2003; Yamaguchi et al., 2007). The latter observation supports the concept that NO plays an important role in promoting the development of the anti-inflammatory phenotype that becomes apparent 18 to 24 hours later. Second, plasma levels of nitrite/nitrate, a marker for NO production, are increased during the period of alcohol exposure (Yamaguchi et al., 2007). Third, tissues pretreated with NO donors in lieu of alcohol develop an antiinflammatory phenotype 24 hours after administration (Yamaguchi et al., 2002; Yamaguchi et al., 2007). Finally, the anti-inflammatory phenotype induced by alcohol does not appear in mice that are genetically deficient in eNOS (Yamaguchi et al., 2007). This last finding not only provides the fourth line of evidence supporting a role for NO as a triggering element in alcohol preconditioning, but also indicates that the eNOS isoform is essential for the development of the anti-inflammatory phenotype in response to alcohol. Interestingly, NO donors remain effective as preconditioning stimuli in eNOS-deficient mice, indicating that downstream mediators of preconditioning remain effective in these mice.
As mentioned in an earlier section of this workshop summary, the factors responsible for increasing eNOS activity in late alcohol preconditioning (or any form of preconditioning for that matter) are largely unknown; however, it now appears that this effect is triggered by an adenosine A2 receptor dependent mechanism (Yamaguchi et al., 2002; Yamaguchi et al., 2007). This notion is supported by the observations that ligation of adenosine A2 receptors increases the activity of cAMP-dependent kinase (PKA), which in turn activates eNOS by phosphorylating Ser-1177. Moreover, adenosine stimulates l-arginine transport and NO biosynthesis by activation of A2 receptors on human umbilical vein endothelial cells. More recent work has established that AMP kinase activation may also play a critical role as an upstream signaling step in eNOS-dependent preconditioning by alcohol (Gaskin et al., 2007).
Another well-known effect of alcohol is to increase the generation of reactive oxygen species, including superoxide and the hydroxyethyl radical (Das and Vasudevan, 2007). Although they are generally considered to exert deleterious effects in biologic systems, it is becoming increasingly apparent that reactive oxygen species may participate in a number of normal physiologic phenomena by serving as second messengers in transmembrane signaling processes. Indeed, administration of a cell-permeant superoxide dismutase (SOD) mimetic, the porphyrin MnTBAP, coincident with alcohol prevents the postischemic antiadhesive effects that become apparent 24 hours after ingestion of the alcohol (Yamaguchi et al., 2003). Moreover, exposing postcapillary venules to a superoxide generating system (hypoxanthine/xanthine oxidase) 24 hours prior to I/R mimicked the antiadhesive effects produced by antecedent alcohol exposure. Additional support for the concept that oxidants may participate in triggering the development of the anti-inflammatory phenotype in response to antecedent alcohol ingestion is derived from studies directed at elucidating their source of production. Inhibition of either xanthine oxidase or NADPH oxidase alone attenuated the anti-adhesive effects of alcohol preconditioning by 50 percent, whereas concomitant inhibition of both oxidant-producing enzymes effectively prevented the development of the protected phenotype (Yamaguchi et al., 2003). The latter studies indicate that xanthine oxidase and NADPH oxidase are important enzymatic sources of the reactive oxygen species that trigger entrance into the anti-inflammatory phenotype displayed by postcapillary venules exposed to alcohol 24 hours prior to I/R. These observations have been extended to stroke models where antecedent alcohol has been shown to reduce I/R-induced delayed neuronal death, neuronal and dendritic degeneration, oxidative DNA damage, glial cell activation and neutrophil infiltration. These beneficial actions of alcohol preconditioning were prevented by treatment with a SOD mimetic or an NADPH oxidase inhibitor (Wallerath et al., 2003).
As noted earlier, there is evidence implicating NO, formed secondary to adenosine A2-receptor-dependent activation of endothelial NOS, in the beneficial actions of antecedent alcohol ingestion. This raises the possibility that NO produced during the period of alcohol preconditioning initiates the protective effects of late alcohol-PC by a mechanism that involves its interaction with xanthine oxidase- and/or NAD(P)H oxidase-derived oxidants. This is an important issue because isoform-selective PKC translocation and activation appears to play a critical role as an obligatory downstream signaling element in late alcohol preconditioning (Dayton et al., 2005). However, NO and NO-releasing agents reversibly inactivate PKC. On the other hand, peroxynitrite, which is formed by the interaction of NO with superoxide, not only induces PKC activation, but has been implicated as a trigger for the beneficial actions of other forms of preconditioning including that induced by antecedent exposure to brief ischemia. Taken together, these observations suggest that reactive NO species formed secondary NO/superoxide interactions may be required to induce the development of alcohol preconditioning (Dayton et al., 2005; Yamaguchi et al., 2003).
Although the mediators of late phase alcohol preconditioning are unclear, the time course required for its development suggests that the appearance of the protected phenotype 18 to 24 hours after alcohol ingestion requires the formation of new gene products capable of producing anti-inflammatory agents. In this regard, it is tempting to speculate that alcohol might enhance the expression of cyclooxygenase-2, inducible NOS (iNOS), or heme oxygenase-1 (HO-1), which in turn generate prostacyclin and other eicosanoids, NO, and carbon monoxide, respectively, all of which produce robust antiadhesive effects in postcapillary venules. In addition to carbon monoxide, HO-1 also generates the powerful antioxidants bilirubin and biliverdin, which may act to prevent the formation of oxidant-dependent chemoattractants during I/R. It is also possible that alcohol may exploit the anti-adhesive properties of adenosine as a mediator of the preconditioned state secondary to enhanced production or decreased salvaging (via activation of 5′-nucleotidase or inhibition of adenosine kinase, respectively) of the nucleoside in postischemic tissues. The fact that an anti-inflammatory phenotype does not become apparent in mice treated with a pharmacologic HO inhibitor during reperfusion and fails to develop in HO-1 knockout mice suggests that this protective protein may serve as a major effector.
NEUROPROTECTIVE MECHANISMS EVOKED IN BRAIN BY PRECONDITIONING WITH MODERATE ALCOHOL CONCENTRATIONS
As indicated in Figure 1, moderate alcohol consumption has sometimes been associated in case-control studies with a lower risk of dementia (Mukamal et al., 2003b). Additionally, the epidemiological literature contains about 19 longitudinal cohort studies between 1997 and 2008 (Mehlig et al., 2008; Orgogozo et al., 1997) that compare relative risk factors for either cognitive decline or dementia—both Alzheimer’s and cerebrovascular—in older moderate alcohol consumers versus abstainers/never-drinkers. In these studies, over half (~54%) of the risk factors were significantly lower in moderate consumers. The remaining studies’ risk factors did not differ between the two groups, with the exception of 2 studies with risk factors (~5% of the total) that were increased by moderate drinking. Also, in 25 other cross-sectional or case-control reports of cognitive decline or dementia, a similar majority of the risk factors were significantly decreased in association with moderate consumption. The authors of the studies finding lower cognitive risk factors viewed the apparent age-dependent neuroprotection as the outcome of alcohol’s beneficial hematologic, cardiovascular and possibly cerebrovascular effects (see Figure 4A), as well as perhaps related to beverage anti-oxidant polyphenols (e.g., resveratrol—Figure 4B) (Pinder and Sandler, 2004) that can act within the nervous system. However, alcohol in moderate levels conceivably exerts its own “direct” neuroprotective effects that are under current study with antecedent exposure, co-exposure, and preconditioning models.
The phenomenon of preconditioning tissues or cells to generate a cytoprotective state or “phenotype” has been appreciated for some time (Gidday, 2006). Brief ischemia that can generate ischemic tolerance has been studied extensively in the heart and, more recently, the brain. Ischemic or hyperthermic preconditioning and brain neuroprotection, first described only about two decades ago (Chopp et al., 1989; Kitagawa et al., 1990), has spawned studies with traditionally toxic agents at exposures that mildly stress and thus precondition the system under study (Best et al., 1996; Leak et al., 2006; Nvue et al., 2004). Moreover, preconditioning-dependent protection by alcohol against ischemic insult of the heart is attested to in many studies, as related earlier in this review (Agarwal, 2002; Sato et al., 2004), and the process is considered a likely component of cardioprotection in moderate alcohol drinkers (Miyamae et al., 1997).
With the exceptions of several in vivo reports and a series of in vitro studies, neuroprotection due to either antecedent alcohol or alcohol preconditioning has been relatively overlooked. In an in vivo report mainly concerned with alcohol and the blood/brain barrier, neurons in brain cortices of adult male “alcohol-preferring” rats chronically consuming 15% alcohol/water were found to be protected from apoptosis due to peripherally injected pro-inflammatory lipopolysaccharide (Singh et al., 2007); however, a mechanistic interpretation was not provided. In ischemia studies mentioned above, antecedent moderate alcohol triggered significant neuroprotection against I/R hippocampal neuronal injury in an adult gerbil model (Wang et al., 2007), and reactive oxygen species (ROS) induction secondary to increased NADPH oxidase activity was implicated in alcohol’s protective effect. In adult rats, icv alcohol (~0.7 umoles over 4 hr) pre- (or post-) I/R was neuroprotective (Liao et al., 2003), and the mechanism was ascribed to ROS trapping by alcohol (Cohen et al., 1976).
In brain cultures, non-neurotoxic alcohol co-exposure can block excitotoxic (N-methyl-D-aspartate [NMDA] receptor-mediated) neurodegeneration (Belmadani et al., 2003; Cebere and Liljequist, 2003; Chandler et al., 1993; Wegelius and Korpi, 1995), consistent with NMDA receptor antagonism by alcohol. However, alcohol preconditioning (at least in vitro) circumvents the issue of direct blockade of NMDA receptor activity by alcohol during the neurotoxic insult in question. Alcohol preconditioning effects on inflammatory protein neurotoxicity were explored with organotypic slices of the maturing rat hippocampus-entorhinal cortex (HEC) complex—which incorporates two brain regions significantly impacted in Alzheimer’s dementia and other dementias—as well as with mixed cerebellar cultures (Collins et al., 2000). Several neurotoxic HIV-1 proteins have been studied, and notably, glycoprotein 120 (gp120), the most potent in the laboratory, which can cause neuronal degeneration and apoptosis at low picomolar (10−12 molar) concentrations in hippocampal slice cultures (Aggoun-Zouaoui et al., 1996) and other brain cultures (Brenneman et al., 1988). Of importance, moderate (15–30 mM) subchronic alcohol preconditioning of HEC slice and cerebellar cultures prevented gp120IIIB-induced neurodegeneration (Belmadani et al., 2001; Collins et al., 2000).
Neuroinflammatory processes originating from gp120IIIB interactions with glial chemokine receptors generate neurodegenerative cascades (Kaul and Lipton, 2006). The alcohol preconditioning, which was delayed in that it required >4d of alcohol exposure for neuroprotection, effectively blunted or suppressed several neuroinflammatory processes or events soon after gp120 addition—e.g., increases in [Ca+2]i, arachidonic acid release, tissue superoxide, and extracellular glutamate (Belmadani et al., 2001). Considerable research indicates that heat shock proteins (hsp) can be putative neuroprotective “effectors” induced by ischemic and other delayed preconditioning approaches (Carmel et al., 2004; Gidday, 2006). In that regard, significant induction of hsp70 (Belmadani et al., 2004) and hsp27 occurred in HEC slices after ~6 d of moderate alcohol exposure—but not at 4 d—which correlates with the onset of significant neuroprotection. Furthermore, phosphorylated hsp27 (p-hsp27), known to colocalize with cytoskeletal elements and particularly actin, was increased at 6 d of alcohol preconditioning both in HEC slices and cerebellar cultures (Sivaswamy et al., 2008). The results from co-treatment with the pharmacologic heat shock inhibitor, quercetin (5 uM, a nontoxic dose that blocks ~80% of hsp induction), indicated that induced hsps are critical components of neuroprotection against gp120, since quercetin abolished the preconditioning neuroprotection while concomitantly restoring the superoxide potentiation.
Studies with more common preconditioning modalities—in particular, ischemic tolerance—have elucidated the involvement of early cellular “sensors” that trigger subsequent “transducer” mechanisms such as kinases, cytokines and transcription factors upstream of hsp effectors (Dirnagl et al., 2003). Sensors include glutamate receptors (notably the NMDA receptor channel), adenosine receptors, and perhaps others coupled to Gi proteins; transducers include PKC isoforms and possibly other kinases. Alcohol preconditioning experiments with mixed cerebellar cultures using receptor antagonists for NMDA receptors, memantine or AP-5, indicated that neuroprotective activation of this “sensor” receptor is important during the first day or so of alcohol exposure (Mitchell et al., 2007). Consistent with these findings is that NMDA receptor stimulation directly with (sub-neurotoxic) NMDA preconditions the cultures, neuroprotecting against β-amyloid. In the case of alcohol, western blotting data indicate significant changes in NMDAR subunit (NR1, NR2A, NR2C) ratios during the early part of preconditioning that could be important in this receptor’s involvement. Parenthetically, there is evidence that subchronic alcohol, albeit at high concentrations, perturbs NMDAR subunit composition in similar brain cultures (Snell et al., 2001).
Examining signal transduction events potentially downstream of NMDAR and upstream of hsp induction, the activity of PKC and changes in PKC isoform expression were determined in HEC slices and/or cerebellar cultures during moderate alcohol exposure. Consistent with ischemic preconditioning results (Uchiyama et al., 2003), PKC activity, moderately elevated ~15% after 2 d of alcohol, was increased ~40% after 6 d of exposure. Correlated with the activity changes were relatively large increases (~200%) in PKCε levels along with modest increases in PKCα and PKCδ (but not PKCβ) expression (Sivaswamy et al., 2008). Furthermore, in alcohol-exposed cerebellar cells, focal adhesion kinase (FAK) and activated p-FAK were modestly increased after 4 d and significantly higher after 6 d (Sivaswamy et al., 2008), and transfection experiments using dominant negative FAK (“FRNK”) have confirmed that FAK activation is upstream of hsp27 phosphorylation (S. Sivaswamy, unpubl. results). FAK, a non-receptor tyrosine kinase activated by phosphorylation, has a key role in transducing cytoskeletal-derived survival signals. Of importance, activation of focal adhesion kinase (FAK) has been linked to cytoprotective elevations in hsp70 and hsp27 (two hsps elevated by alcohol preconditioning) in severely stressed myocytes (Wei et al., 2006). Overall, an emerging hypothesis is that alcohol (preconditioning)-induced neuronal survival mechanisms involving hsp-dependent protection is intimately linked to selective PKC and FAK activation, the focal adhesion complex, and stabilization of the cytoskeleton.
CONCLUDING REMARKS
There is convincing evidence that light-moderate, non-binge alcohol intake reduces the risk of coronary heart disease and, in agreement with an NIAAA position paper on moderate alcohol consumption (Gunzerath et al., 2004), that it does no apparent harm to cognition during aging, even possibly reducing the risk of cognitive decline/dementia. The studies reviewed here indicate that potential common anti-inflammatory mechanisms are initiated by low-moderate alcohol levels and that these mechanisms have the capability of promoting a cytoprotective state or condition. Notably, alcohol-related anti-inflammatory heat shock protein and protein kinase changes in the brain bear similarities to those elucidated in heart, particularly with respect to PKCε and quite likely other signal transduction kinases.
While alcohol levels achieved in periodic social consumption among older adults are no doubt lower (albeit more prolonged) than those inducing neuroprotection in the brain cultures summarized above, it is nonetheless conceivable that light-to-moderate, stable alcohol ingestion over years exerts “preconditioning-like” effects on glia and neurons akin to that which have been observed, leading to consequent dampening of the endogenous neuroinflammatory processes associated with sometimes accelerated or “pathological” brain aging (Chung et al., 2006). Further experimental studies are necessary to examine the potential underlying mechanisms. In view of facts that cardiovascular disease and stroke are two of the top three leading causes of mortality in developed countries and, according to a Lancet review (Ferri et al., 2005), new cases of dementia are being diagnosed about every seven seconds worldwide, it seems imperative that as much as possible is learned about the apparent cytoprotective mechanisms engendered by low-moderate alcohol intake and levels.
Acknowledgments
Research supported by NIH AA013568, NIH AA13361 and NIH AA011723 (MAC), NIH AA11135 and the ABMRF (MOG), and NIH AA014945 and NIH DK043785 (RJK). The roundtable was supported in part by the Loyola University Neuroscience Institute and the Alcohol Beverage Medical Research Foundation (ABMRF).
Abbreviations
- AD
Alzheimer’s disease
- CGRP
calcitonin gene related peptide
- CHS
Cardiovascular Health Study
- COX
cyclooxygenase
- EC
endothelial cell
- FAK
focal adhesion kinase
- HDL-C
high-density lipoprotein cholesterol
- HEC
hippocampal-entorhinal cortical
- HIV
human immunodeficiency virus
- HO-1
heme oxygenase-1
- HPFS
Health Professionals Follow-up Study
- hsp
heat shock protein
- I/R
ischemia/reperfusion
- MI
myocardial infarction
- NMDAR
N-methyl-D-aspartate receptor
- NO
nitric oxide
- NOS
nitric oxide synthase
- PC
preconditioning
- PKC
protein kinase C
- SOD
superoxide dismutase
References
- Abou-Agag LH, Khoo NK, Binsack R, White CR, Darley-Usmar V, Grenett HE, Booyse FM, Digerness SB, Zhou F, Parks DA. Evidence of cardiovascular protection by moderate alcohol: role of nitric oxide. Free Radic Biol Med. 2005;39(4):540–8. doi: 10.1016/j.freeradbiomed.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Agarwal DP. Cardioprotective effects of light-moderate consumption of alcohol: a review of putative mechanisms. Alcohol & Alcoholism. 2002;37(5):409–15. doi: 10.1093/alcalc/37.5.409. [DOI] [PubMed] [Google Scholar]
- Aggoun-Zouaoui D, Charriaut-Marlangue C, Rivera S, Jorquera I, Ben-Ari Y, Represa A, Aggoun-Zouaoui D, Charriaut-Marlangue C, Rivera S, Jorquera I, Ben-Ari Y, Represa A. The HIV-1 envelope protein gp120 induces neuronal apoptosis in hippocampal slices. Neuroreport. 1996;7(2):433–6. doi: 10.1097/00001756-199601310-00014. [DOI] [PubMed] [Google Scholar]
- Aikens ML, Grenett HE, Benza RL, Tabengwa EM, Davis GC, Booyse FM. Alcohol-induced upregulation of plasminogen activators and fibrinolytic activity in cultured human endothelial cells. Alcohol Clin Exp Res. 1998;22(2):375–81. [PubMed] [Google Scholar]
- Al-Abed Y, Mitsuhashi T, Li H, Lawson JA, FitzGerald GA, Founds H, Donnelly T, Cerami A, Ulrich P, Bucala R. Inhibition of advanced glycation endproduct formation by acetaldehyde: role in the cardioprotective effect of ethanol. Proc Natl Acad Sci U S A. 1999;96(5):2385–90. doi: 10.1073/pnas.96.5.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MA, Glynn RJ, Ridker PM. Alcohol consumption and plasma concentration of C-reactive protein. Circulation. 2003;107(3):443–7. doi: 10.1161/01.cir.0000045669.16499.ec. [DOI] [PubMed] [Google Scholar]
- Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase C epsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92(8):873–80. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmadani A, Kumar S, Schipma M, Collins MA, Neafsey EJ. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport. 2004;15(13):2093–6. doi: 10.1097/00001756-200409150-00019. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Neafsey EJ, Collins MA. Human immunodeficiency virus type 1 gp120 and ethanol coexposure in rat organotypic brain slice cultures: Curtailment of gp120-induced neurotoxicity and neurotoxic mediators by moderate but not high ethanol concentrations. J Neurovirol. 2003;9(1):45–54. doi: 10.1080/13550280390173409. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Zou JY, Schipma MJ, Neafsey EJ, Collins MA. Ethanol pre-exposure suppresses HIV-1 glycoprotein 120-induced neuronal degeneration by abrogating endogenous glutamate/Ca2+-mediated neurotoxicity. Neuroscience. 2001;104(3):769–81. doi: 10.1016/s0306-4522(01)00139-7. [DOI] [PubMed] [Google Scholar]
- Best N, Sundstrom LE, Mitchell J, Wheal HV. Pre-exposure to subtoxic levels prevents kainic acid lesions in organotypic hippocampal slice cultures: effects of kainic acid on parvalbumin-immunoreactive neurons and expression of heat shock protein 72 following the induction of tolerance. Eur J Neurosci. 1996;8(6):1209–19. doi: 10.1111/j.1460-9568.1996.tb01289.x. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Pert CB, Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Pert CB. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. 1988;335(6191):639–42. doi: 10.1038/335639a0. [DOI] [PubMed] [Google Scholar]
- Brenner H, Rothenbacher D, Bode G, Marz W, Hoffmeister A, Koenig W. Coronary heart disease risk reduction in a predominantly beer-drinking population. Epidemiology. 2001;12(4):390–5. doi: 10.1097/00001648-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Callemien D, Jerkovic V, Rozenberg R, Collin S. Hop as an interesting source of resveratrol for brewers: optimization of the extraction and quantitative study by liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry. J Agric Food Chem. 2005;53(2):424–9. doi: 10.1021/jf040179n. [DOI] [PubMed] [Google Scholar]
- Carmel JB, Kakinohana O, Mestril R, Young W, Marsala M, Hart RP. Mediators of ischemic preconditioning identified by microarray analysis of rat spinal cord. Exp Neurol. 2004;185(1):81–96. doi: 10.1016/j.expneurol.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Cebere A, Liljequist S. Ethanol differentially inhibits homoquinolinic acid- and NMDA-induced neurotoxicity in primary cultures of cerebellar granule cells. Neurochem Res. 2003;28(8):1193–9. doi: 10.1023/a:1024228412198. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sumners C, Crews FT. Ethanol inhibits NMDA receptor-mediated excitotoxicity in rat primary neuronal cultures. Alcohol Clin Exp Res. 1993;17(1):54–60. doi: 10.1111/j.1530-0277.1993.tb00726.x. [DOI] [PubMed] [Google Scholar]
- Chen CH, Gray MO, Mochly-Rosen D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: role of epsilon protein kinase C. Proc Natl Acad Sci U S A. 1999;96(22):12784–9. doi: 10.1073/pnas.96.22.12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Davis-Gorman G, Watson RR, McDonagh PF. Chronic ethanol consumption modulates myocardial ischaemia-reperfusion injury in murine AIDS. Alcohol & Alcoholism. 2003;38(1):18–24. doi: 10.1093/alcalc/agg014. [DOI] [PubMed] [Google Scholar]
- Chopp M, Chen H, Ho KL, Dereski MO, Brown E, Hetzel FW, Welch KM, Chopp M, Chen H, Ho KL, Dereski MO, Brown E, Hetzel FW, Welch KM. Transient hyperthermia protects against subsequent forebrain ischemic cell damage in the rat. Neurology. 1989;39(10):1396–8. doi: 10.1212/wnl.39.10.1396. [DOI] [PubMed] [Google Scholar]
- Chung HY, Sung B, Jung KJ, Zou Y, Yu BP. The molecular inflammatory process in aging. Antioxidants & Redox Signaling. 2006;8(3–4):572–81. doi: 10.1089/ars.2006.8.572. [DOI] [PubMed] [Google Scholar]
- Cleophas TJ. Wine, beer and spirits and the risk of myocardial infarction: a systematic review. Biomed Pharmacother. 1999;53(9):417–23. doi: 10.1016/S0753-3322(99)80121-8. [DOI] [PubMed] [Google Scholar]
- Cohen G, Heikkila RE, Allis B, Cabbat F, Dembiec D, MacNamee D, Mytilineou C, Winston B. Destruction of sympathetic nerve terminals by 6-hydroxydopamine: protection by 1-phenyl-3-(2-thiazolyl)-2-thiourea, diethyldithiocarbamate, methimazole, cysteamine, ethanol and n-butanol. J Pharmacol Exp Ther. 1976;199(2):336–52. [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ, Zou JY. HIV-1 gp120 neurotoxicity in brain cultures is prevented by moderate ethanol pretreatment. Neuroreport. 2000;11(6):1219–22. doi: 10.1097/00001756-200004270-00015. [DOI] [PubMed] [Google Scholar]
- Corrao G, Rubbiati L, Bagnardi V, Zambon A, Poikolainen K. Alcohol and coronary heart disease: a meta-analysis. Addiction. 2000;95(10):1505–23. doi: 10.1046/j.1360-0443.2000.951015056.x. [DOI] [PubMed] [Google Scholar]
- Das DK, Maulik N. Resveratrol in cardioprotection: a therapeutic promise of alternative medicine. Molecular Interventions. 2006;6(1):36–47. doi: 10.1124/mi.6.1.7. [DOI] [PubMed] [Google Scholar]
- Das DK, Ursini F. Alcohol and wine in health and disease. Ann N Y Acad Sci. 2002;957:1–350. [PubMed] [Google Scholar]
- Das S, Khan N, Mukherjee S, Bagchi D, Gurusamy N, Swartz H, Das DK. Redox regulation of resveratrol-mediated switching of death signal into survival signal. Free Radic Biol Med. 2008;44(1):82–90. doi: 10.1016/j.freeradbiomed.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Das SK, Vasudevan DM. Alcohol-induced oxidative stress. Life Sci. 2007;81(3):177–87. doi: 10.1016/j.lfs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Davies MJ, Baer DJ, Judd JT, Brown ED, Campbell WS, Taylor PR. Effects of moderate alcohol intake on fasting insulin and glucose concentrations and insulin sensitivity in postmenopausal women: a randomized controlled trial. JAMA. 2002;287(19):2559–62. doi: 10.1001/jama.287.19.2559. [DOI] [PubMed] [Google Scholar]
- Dayton C, Yamaguchi T, Kamada K, Carter P, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic leukocyte adhesion and P-selectin expression by a protein kinase C-dependent mechanism. Dig Dis Sci. 2005;50(4):684–90. doi: 10.1007/s10620-005-2557-1. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26(5):248–54. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Djousse L, Ellison RC, Beiser A, Scaramucci A, D’Agostino RB, Wolf PA. Alcohol consumption and risk of ischemic stroke: The Framingham Study. Stroke. 2002;33(4):907–12. doi: 10.1161/hs0402.105245. [DOI] [PubMed] [Google Scholar]
- Doll R. One for the heart. BMJ. 1997;315(7123):1664–8. doi: 10.1136/bmj.315.7123.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufouil C, Tzourio C, Brayne C, Berr C, Amouyel P, Alperovitch A. Influence of apolipoprotein E genotype on the risk of cognitive deterioration in moderate drinkers and smokers. Epidemiology. 2000;11(3):280–4. doi: 10.1097/00001648-200005000-00009. [DOI] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366(9503):2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberg MS, Samet JH. Alcohol and coronary heart disease: the answer awaits a randomized controlled trial. Circulation. 2005;112(10):1379–81. doi: 10.1161/CIRCULATIONAHA.105.568030. [DOI] [PubMed] [Google Scholar]
- Gaskin FS, Kamada K, Yusof M, Korthuis RJ. 5′-AMP-activated protein kinase activation prevents postischemic leukocyte-endothelial cell adhesive interactions. American Journal of Physiology - Heart & Circulatory Physiology. 2007;292(1):H326–32. doi: 10.1152/ajpheart.00744.2006. [DOI] [PubMed] [Google Scholar]
- Gaziano JM, Hennekens CH, Godfried SL, Sesso HD, Glynn RJ, Breslow JL, Buring JE. Type of alcoholic beverage and risk of myocardial infarction. Am J Cardiol. 1999;83(1):52–7. doi: 10.1016/s0002-9149(98)00782-6. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nature Reviews Neuroscience. 2006;7(6):437–48. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem. 2004;279(5):3596–604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- Gunzerath L, Faden V, Zakhari S, Warren K. National Institute on Alcohol Abuse and Alcoholism report on moderate drinking. Alcohol Clin Exp Res. 2004;28(6):829–47. doi: 10.1097/01.alc.0000128382.79375.b6. [DOI] [PubMed] [Google Scholar]
- Hillbom M, Haapaniemi H, Juvela S, Palomaki H, Numminen H, Kaste M. Recent alcohol consumption, cigarette smoking, and cerebral infarction in young adults. Stroke. 1995;26(1):40–5. doi: 10.1161/01.str.26.1.40. [DOI] [PubMed] [Google Scholar]
- Juric D, Wojciechowski P, Das DK, Netticadan T. Prevention of concentric hypertrophy and diastolic impairment in aortic-banded rats treated with resveratrol. American Journal of Physiology - Heart & Circulatory Physiology. 2007;292(5):H2138–43. doi: 10.1152/ajpheart.00852.2006. [DOI] [PubMed] [Google Scholar]
- Kamada K, Dayton CB, Yamaguchi T, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic microvascular dysfunction. Pathophysiology. 2004;10(2):131–7. doi: 10.1016/j.pathophys.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kanai AJ, Mesaros S, Finkel MS, Oddis CV, Birder LA, Malinski T. Beta-adrenergic regulation of constitutive nitric oxide synthase in cardiac myocytes. Am J Physiol. 1997;273(4 Pt 1):C1371–7. doi: 10.1152/ajpcell.1997.273.4.C1371. [DOI] [PubMed] [Google Scholar]
- Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL, 2nd, Billiar TR. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation. 2000;101(23):2742–8. doi: 10.1161/01.cir.101.23.2742. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Current HIV Research. 2006;4(3):307–18. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Khadour FH, O’Brien DW, Fu Y, Armstrong PW, Schulz R. Endothelial nitric oxide synthase increases in left atria of dogs with pacing-induced heart failure. Am J Physiol. 1998;275(6 Pt 2):H1971–8. doi: 10.1152/ajpheart.1998.275.6.H1971. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528(1):21–4. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Kitakaze M, Node K, Minamino T, Kosaka H, Shinozaki Y, Mori H, Inoue M, Hori M, Kamada T. Role of nitric oxide in regulation of coronary blood flow during myocardial ischemia in dogs. J Am Coll Cardiol. 1996;27(7):1804–12. doi: 10.1016/0735-1097(96)00064-2. [DOI] [PubMed] [Google Scholar]
- Klatsky AL. Alcohol, coronary disease, and hypertension. Annu Rev Med. 1996;47:149–60. doi: 10.1146/annurev.med.47.1.149. [DOI] [PubMed] [Google Scholar]
- Klatsky AL, Friedman GD, Siegelaub AB. Alcohol consumption before myocardial infarction. Results from the Kaiser-Permanente epidemiologic study of myocardial infarction. Ann Intern Med. 1974;81:294–301. doi: 10.7326/0003-4819-81-3-294. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovasc Res. 1999;41(3):514–23. doi: 10.1016/s0008-6363(98)00314-9. [DOI] [PubMed] [Google Scholar]
- Korthuis RJ. Alcohol and cardioprotection. Pathophysiology. 2004;10(2):1–145. doi: 10.1016/j.pathophys.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Korthuis RJ. Antecedent ethanol ingestion prevents postischemic microvascular dysfunction. In: Shepro D, editor. Microvascular Research: Biology and Pathology. 2006. pp. 777–782. [Google Scholar]
- Krenz M, Cohen MV, Downey JM. Protective and anti-protective effects of acute ethanol exposure in myocardial ischemia/reperfusion. Pathophysiology. 2004;10(2):113–9. doi: 10.1016/j.pathophys.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Kuller LH, Lopez OL, Newman A, Beauchamp NJ, Burke G, Dulberg C, Fitzpatrick A, Fried L, Haan MN. Risk factors for dementia in the cardiovascular health cognition study. Neuroepidemiology. 2003;22(1):13–22. doi: 10.1159/000067109. [DOI] [PubMed] [Google Scholar]
- Lamas S, Perez-Sala D, Moncada S. Nitric oxide: from discovery to the clinic. Trends Pharmacol Sci. 1998;19(11):436–8. doi: 10.1016/s0165-6147(98)01265-6. [DOI] [PubMed] [Google Scholar]
- Larrieu S, Letenneur L, Helmer C, Dartigues JF, Barberger-Gateau P. Nutritional factors and risk of incident dementia in the PAQUID longitudinal cohort. J Nutr Health Aging. 2004;8(3):150–4. [PubMed] [Google Scholar]
- Leak RK, Liou AK, Zigmond MJ. Effect of sublethal 6-hydroxydopamine on the response to subsequent oxidative stress in dopaminergic cells: evidence for preconditioning. J Neurochem. 2006;99(4):1151–63. doi: 10.1111/j.1471-4159.2006.04149.x. [DOI] [PubMed] [Google Scholar]
- Lekli I, Szabo G, Juhasz B, Das S, Das M, Varga E, Szendrei L, Gesztelyi R, Varadi J, Bak I, Das DK, Tosaki A. Protective mechanisms of resveratrol against ischemia-reperfusion-induced damage in hearts obtained from Zucker obese rats: the role of GLUT-4 and endothelin. American Journal of Physiology - Heart & Circulatory Physiology. 2008;294(2):H859–66. doi: 10.1152/ajpheart.01048.2007. [DOI] [PubMed] [Google Scholar]
- Liao SL, Chen WY, Raung SL, Chen CJ. Ethanol attenuates ischemic and hypoxic injury in rat brain and cultured neurons. Neuroreport. 2003;14(16):2089–94. doi: 10.1097/00001756-200311140-00016. [DOI] [PubMed] [Google Scholar]
- Loke KE, McConnell PI, Tuzman JM, Shesely EG, Smith CJ, Stackpole CJ, Thompson CI, Kaley G, Wolin MS, Hintze TH. Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ Res. 1999;84(7):840–5. doi: 10.1161/01.res.84.7.840. [DOI] [PubMed] [Google Scholar]
- Lucas DL, Brown RA, Wassef M, Giles TD. Alcohol and the cardiovascular system research challenges and opportunities. J Am Coll Cardiol. 2005;45(12):1916–24. doi: 10.1016/j.jacc.2005.02.075. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Tang MX, Siddiqui M, Shea S, Mayeux R. Alcohol intake and risk of dementia. J Am Geriatr Soc. 2004;52(4):540–6. doi: 10.1111/j.1532-5415.2004.52159.x. [DOI] [PubMed] [Google Scholar]
- Maclure M. Demonstration of deductive meta-analysis: ethanol intake and risk of myocardial infarction. Epidemiol Rev. 1993;15(2):328–51. doi: 10.1093/oxfordjournals.epirev.a036124. [DOI] [PubMed] [Google Scholar]
- Marfella R, Cacciapuoti F, Siniscalchi M, Sasso FC, Marchese F, Cinone F, Musacchio E, Marfella MA, Ruggiero L, Chiorazzo G, Liberti D, Chiorazzo G, Nicoletti GF, Saron C, D’Andrea F, Ammendola C, Verza M, Coppola L. Effect of moderate red wine intake on cardiac prognosis after recent acute myocardial infarction of subjects with Type 2 diabetes mellitus. Diabet Med. 2006;23(9):974–81. doi: 10.1111/j.1464-5491.2006.01886.x. [DOI] [PubMed] [Google Scholar]
- McConnell MV, Vavouranakis I, Wu LL, Vaughan DE, Ridker PM. Effects of a single, daily alcoholic beverage on lipid and hemostatic markers of cardiovascular risk. Am J Cardiol. 1997;80(9):1226–8. doi: 10.1016/s0002-9149(97)00647-4. [DOI] [PubMed] [Google Scholar]
- Mehlig K, Skoog I, Guo X, Schutze M, Gustafson D, Waern M, Ostling S, Bjorkelund C, Lissner L. Alcoholic beverages and incidence of dementia: 34-year follow-up of the prospective population study of women in Goteborg. Am J Epidemiol. 2008;167(6):684–91. doi: 10.1093/aje/kwm366. [DOI] [PubMed] [Google Scholar]
- Mitchell RM, Neafsey EJ, Collins MA. Neuroprotection by moderate ethanol preconditioning: Involvement of NMDA receptor signaling. Alcohol Clin Exp Res. 2007;31(s2):196A. [Google Scholar]
- Miyamae M, Diamond I, Weiner MW, Camacho SA, Figueredo VM. Regular alcohol consumption mimics cardiac preconditioning by protecting against ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 1997;94(7):3235–9. doi: 10.1073/pnas.94.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokni M, Limam F, Elkahoui S, Amri M, Aouani E. Strong cardioprotective effect of resveratrol, a red wine polyphenol, on isolated rat hearts after ischemia/reperfusion injury. Arch Biochem Biophys. 2007;457(1):1–6. doi: 10.1016/j.abb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Ascherio A, Mittleman MA, Conigrave KM, Camargo CA, Jr, Kawachi I, Stampfer MJ, Willett WC, Rimm EB. Alcohol and risk for ischemic stroke in men: the role of drinking patterns and usual beverage. Ann Intern Med. 2005a;142(1):11–9. doi: 10.7326/0003-4819-142-1-200501040-00007. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Chung H, Jenny NS, Kuller LH, Longstreth WT, Jr, Mittleman MA, Burke GL, Cushman M, Beauchamp NJ, Jr, Siscovick DS. Alcohol use and risk of ischemic stroke among older adults: the cardiovascular health study. Stroke. 2005b;36(9):1830–4. doi: 10.1161/01.STR.0000177587.76846.89. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Chung H, Jenny NS, Kuller LH, Longstreth WT, Jr, Mittleman MA, Burke GL, Cushman M, Psaty BM, Siscovick DS. Alcohol consumption and risk of coronary heart disease in older adults: the Cardiovascular Health Study. J Am Geriatr Soc. 2006;54(1):30–7. doi: 10.1111/j.1532-5415.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Conigrave KM, Mittleman MA, Camargo CA, Jr, Stampfer MJ, Willett WC, Rimm EB. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med. 2003a;348(2):109–18. doi: 10.1056/NEJMoa022095. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Jensen MK, Gronbaek M, Stampfer MJ, Manson JE, Pischon T, Rimm EB. Drinking frequency, mediating biomarkers, and risk of myocardial infarction in women and men. Circulation. 2005c;112(10):1406–13. doi: 10.1161/CIRCULATIONAHA.105.537704. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Jensen MK, Gronbaek M, Stampfer MJ, Manson JE, Pischon T, Rimm EB. Drinking frequency, mediating biomarkers, and risk of myocardial infarction in women and men. Circulation. 2005d;112(10):1406–13. doi: 10.1161/CIRCULATIONAHA.105.537704. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Kuller LH, Fitzpatrick AL, Longstreth WT, Jr, Mittleman MA, Siscovick DS. Prospective study of alcohol consumption and risk of dementia in older adults. JAMA. 2003b;289(11):1405–13. doi: 10.1001/jama.289.11.1405. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Longstreth WT, Jr, Mittleman MA, Crum RM, Siscovick DS. Alcohol consumption and subclinical findings on magnetic resonance imaging of the brain in older adults: the cardiovascular health study. Stroke. 2001;32(9):1939–46. doi: 10.1161/hs0901.095723. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Muller JE, Maclure M, Sherwood JB, Mittleman MA. Lack of effect of recent alcohol consumption on the course of acute myocardial infarction. Am Heart J. 1999;138(5 Pt 1):926–33. doi: 10.1016/s0002-8703(99)70019-0. [DOI] [PubMed] [Google Scholar]
- Nvue R, Gorianov V, Best N, Sundstrom LE, Pringle AK. Time window and pharmacological characterisation of kainate-mediated preconditioning in organotypic rat hippocampal slice cultures. Neurosci Lett. 2004;367(3):365–8. doi: 10.1016/j.neulet.2004.06.033. [DOI] [PubMed] [Google Scholar]
- Okubo T, Suto N, Kudo S, Hanada H, Mikuniya A, Okumura K. A study on the effects of endogenous nitric oxide on coronary blood flow, myocardial oxygen extraction and cardiac contractility. Fundam Clin Pharmacol. 1999;13(1):34–42. doi: 10.1111/j.1472-8206.1999.tb00318.x. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, Renaud S, Breteler MB. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997;153(3):185–92. [PubMed] [Google Scholar]
- Pagel PS, Krolikowski JG, Kehl F, Mraovic B, Kersten JR, Warltier DC. The role of mitochondrial and sarcolemmal K(ATP) channels in canine ethanol-induced preconditioning in vivo. Anesth Analg. 2002;94(4):841–8. doi: 10.1097/00000539-200204000-00012. [DOI] [PubMed] [Google Scholar]
- Patel RP, McAndrew J, Sellak H, White CR, Jo H, Freeman BA, Darley-Usmar VM. Biological aspects of reactive nitrogen species. Biochim Biophys Acta. 1999;1411(2–3):385–400. doi: 10.1016/s0005-2728(99)00028-6. [DOI] [PubMed] [Google Scholar]
- Pinder RM, Sandler M. Alcohol, wine and mental health: focus on dementia and stroke. J Psychopharmacol (Oxf) 2004;18(4):449–56. doi: 10.1177/0269881104047272. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88(1):59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Moncada S. Regulation of vascular homeostasis by nitric oxide. Thromb Haemost. 1993;70(1):36–41. [PubMed] [Google Scholar]
- Renaud SC, Ruf JC. Effects of alcohol on platelet functions. Clin Chim Acta. 1996;246(1–2):77–89. doi: 10.1016/0009-8981(96)06228-6. [DOI] [PubMed] [Google Scholar]
- Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. JAMA. 2003;289(5):579–88. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Vaughan DE, Stampfer MJ, Glynn RJ, Hennekens CH. Association of moderate alcohol consumption and plasma concentration of endogenous tissue-type plasminogen activator. JAMA. 1994;272(12):929–33. doi: 10.1001/jama.1994.03520120039028. [DOI] [PubMed] [Google Scholar]
- Rimm EB, Williams P, Fosher K, Criqui M, Stampfer MJ. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319(7224):1523–8. doi: 10.1136/bmj.319.7224.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Ruitenberg A, van Swieten JC, Witteman JC, Mehta KM, van Duijn CM, Hofman A, Breteler MM. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet. 2002;359(9303):281–6. doi: 10.1016/S0140-6736(02)07493-7. [DOI] [PubMed] [Google Scholar]
- Sato M, Fraga C, Das DK. Induction of the expression of cardioprotective proteins after mild-to-moderate consumption of alcohol. Pathophysiology. 2004;10(2):139–45. doi: 10.1016/j.pathophys.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Ishida S, Hara M, Takahashi N, Yoshimatsu H, Sakata T, Korthuis RJ. Resveratrol, a red wine constituent polyphenol, prevents superoxide-dependent inflammatory responses induced by ischemia/reperfusion, platelet-activating factor, or oxidants. Free Radic Biol Med. 2003;34(7):810–7. doi: 10.1016/s0891-5849(02)01430-2. [DOI] [PubMed] [Google Scholar]
- Sierksma A, van der Gaag MS, Kluft C, Hendriks HF. Moderate alcohol consumption reduces plasma C-reactive protein and fibrinogen levels; a randomized, diet-controlled intervention study. Eur J Clin Nutr. 2002a;56(11):1130–6. doi: 10.1038/sj.ejcn.1601459. [DOI] [PubMed] [Google Scholar]
- Sierksma A, van der Gaag MS, Kluft C, Hendriks HF. Moderate alcohol consumption reduces plasma C-reactive protein and fibrinogen levels; a randomized, diet-controlled intervention study. Eur J Clin Nutr. 2002b;56(11):1130–6. doi: 10.1038/sj.ejcn.1601459. [DOI] [PubMed] [Google Scholar]
- Singh AK, Jiang Y, Gupta S, Benlhabib E. Effects of chronic ethanol drinking on the blood brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol & Alcoholism. 2007;42(5):385–99. doi: 10.1093/alcalc/agl120. [DOI] [PubMed] [Google Scholar]
- Sivaswamy S, Neafsey EJ, Collins MA. PKC and Focal Adhesion Kinase (FAK): Possible Transducer Roles in Ethanol Preconditioning-Induced Neuroprotection from HIV-1 gp120. J Neurochem. 2008;104(Suppl 1):32. [Google Scholar]
- Snell LD, Bhave SV, Tabakoff B, Hoffman PL, Snell LD, Bhave SV, Tabakoff B, Hoffman PL. Chronic ethanol exposure delays the ‘developmental switch’ of the NMDA receptor 2A and 2B subunits in cultured cerebellar granule neurons. J Neurochem. 2001;78(2):396–405. doi: 10.1046/j.1471-4159.2001.00424.x. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Kang JH, Chen J, Cherry R, Grodstein F. Effects of moderate alcohol consumption on cognitive function in women. N Engl J Med. 2005;352(3):245–53. doi: 10.1056/NEJMoa041152. [DOI] [PubMed] [Google Scholar]
- Sumeray MS, Rees DD, Yellon DM. Infarct size and nitric oxide synthase in murine myocardium. J Mol Cell Cardiol. 2000;32(1):35–42. doi: 10.1006/jmcc.1999.1050. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW, Jr, Doll R. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. 1997;337(24):1705–14. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- Truelsen T, Thudium D, Gronbaek M. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology. 2002;59(9):1313–9. doi: 10.1212/01.wnl.0000031421.50369.e7. [DOI] [PubMed] [Google Scholar]
- Tsai SK, Hung LM, Fu YT, Cheng H, Nien MW, Liu HY, Zhang FB, Huang SS. Resveratrol neuroprotective effects during focal cerebral ischemia injury via nitric oxide mechanism in rats. J Vasc Surg. 2007;46(2):346–53. doi: 10.1016/j.jvs.2007.04.044. [DOI] [PubMed] [Google Scholar]
- Uchiyama Y, Otani H, Wakeno M, Okada T, Uchiyama T, Sumida T, Kido M, Imamura H, Nakao S, Shingu K. Role of mitochondrial KATP channels and protein kinase C in ischaemic preconditioning. Clin Exp Pharmacol Physiol. 2003;30(5–6):426–36. doi: 10.1046/j.1440-1681.2003.03853.x. [DOI] [PubMed] [Google Scholar]
- Wallerath T, Poleo D, Li H, Forstermann U. Red wine increases the expression of human endothelial nitric oxide synthase: a mechanism that may contribute to its beneficial cardiovascular effects. J Am Coll Cardiol. 2003;41(3):471–8. doi: 10.1016/s0735-1097(02)02826-7. [DOI] [PubMed] [Google Scholar]
- Wang Q, Sun AY, Simonyi A, Kalogeris TJ, Miller DK, Sun GY, Korthuis RJ. Ethanol preconditioning protects against ischemia/reperfusion-induced brain damage: role of NADPH oxidase-derived ROS. Free Radic Biol Med. 2007;43(7):1048–60. doi: 10.1016/j.freeradbiomed.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegelius K, Korpi ER. Ethanol inhibits NMDA-induced toxicity and trophism in cultured cerebellar granule cells. Acta Physiol Scand. 1995;154(1):25–34. doi: 10.1111/j.1748-1716.1995.tb09882.x. [DOI] [PubMed] [Google Scholar]
- Wei H, Campbell W, Vander Heide RS. Heat shock-induced cardioprotection activates cytoskeletal-based cell survival pathways. American Journal of Physiology - Heart & Circulatory Physiology. 2006;291(2):H638–47. doi: 10.1152/ajpheart.00144.2006. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Dayton C, Shigematsu T, Carter P, Yoshikawa T, Gute DC, Korthuis RJ. Preconditioning with ethanol prevents postischemic leukocyte-endothelial cell adhesive interactions. American Journal of Physiology - Heart & Circulatory Physiology. 2002;283(3):H1019–30. doi: 10.1152/ajpheart.00173.2002. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Dayton CB, Ross CR, Yoshikawa T, Gute DC, Korthuis RJ. Late preconditioning by ethanol is initiated via an oxidant-dependent signaling pathway. Free Radic Biol Med. 2003;34(3):365–76. doi: 10.1016/s0891-5849(02)01292-3. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kamada K, Dayton C, Gaskin FS, Yusof M, Yoshikawa T, Carter P, Korthuis RJ. Role of eNOS-derived NO in the postischemic anti-inflammatory effects of antecedent ethanol ingestion in murine small intestine. American Journal of Physiology - Heart & Circulatory Physiology. 2007;292(3):H1435–42. doi: 10.1152/ajpheart.00282.2006. [DOI] [PubMed] [Google Scholar]
- Yuan JM, Ross RK, Gao YT, Henderson BE, Yu MC. Follow up study of moderate alcohol intake and mortality among middle aged men in Shanghai, China. BMJ. 1997;314:18–23. doi: 10.1136/bmj.314.7073.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhari S. Molecular mechanisms underlying alcohol-induced cardioprotection: contribution of hemostatic components. Introduction to the symposium. Alcohol Clin Exp Res. 1999;23(6):1108–10. doi: 10.1111/j.1530-0277.1999.tb04232.x. [DOI] [PubMed] [Google Scholar]
- Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. 1993;92(2):652–62. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HZ, Karliner JS, Gray MO. Moderate alcohol consumption induces sustained cardiac protection by activating PKC-epsilon and Akt. American Journal of Physiology - Heart & Circulatory Physiology. 2002;283(1):H165–74. doi: 10.1152/ajpheart.00408.2001. [DOI] [PubMed] [Google Scholar]
- Zhou HZ, Ma X, Cecchini G, Simonis U, Gray MO. Moderate ethanol consumption improves mitochondrial function and cardiac energetics during oxidative stress: roles of PKCepsilon and Akt. Circulation. 2004;110(17):III–203. [Google Scholar]
- Zhu P, Zhou HZ, Gray MO. Chronic ethanol-induced myocardial protection requires activation of mitochondrial K(ATP) channels. J Mol Cell Cardiol. 2000;32(11):2091–5. doi: 10.1006/jmcc.2000.1233. [DOI] [PubMed] [Google Scholar]