

Abstract
Experimental studies have shown that the C-H oxidation of ibuprofen and methylcyclohexane acetic acid can be carried out with high selectivies using [(terpy’)Mn(OH2)(μ-O)2Mn(OH2)(terpy’)]3+ as catalyst, where terpy’ is a terpyridine ligand functionalized with a phenylene linker and a Kemp’s triacid serving to recognize the reactant via H-bonding. Experiments, described here, suggest that the sulfate counter anion, present in stochiometric amounts, coordinates to manganese in place of water. DFT calculations have been carried out using [(terpy’)Mn(O)(μ-O)2Mn(SO4)(terpy’)]+ as model catalyst, to analyze the origin of selectivity and its relation to molecular recognition, as well as the mechanism of catalyst inhibition by tert-butyl benzoic acid. The calculations show that a number of spin states, all having radical oxygen character, are energetically accessible. All these spin states promote C-H oxidation via a rebound mechanism. The catalyst recognizes the substrate by a double H bond. This interaction orients the substrate inducing highly selective C-H oxidation. The double hydrogen bond stabilizes the reactant, the transition state and the product to the same extent. Consequently, the reaction occurs at lower energy than without molecular recognition. The association of the catalyst with tert-butyl benzoic acid is shown to shield the access of unbound substrate to the reactive oxo site, hence preventing non-selective hydroxylation. It is shown that the two recognition sites of the catalyst can be used in a cooperative manner to control the access to the reactive centre.
Introduction
Activation of inert C-H bonds is at the heart of several chemical processes of high current interest, including the functionalization of hydrocarbons.1 This transformation is specially challenging when it involves substrates containing multiple reactive functional groups, which can all take part in the reaction decreasing the selectivity.
This problem can be solved with a “bio-inspired” approach,2 by using a catalyst in which a recognition site is bound to the reactive centre through a linker (Scheme 1). The recognition site binds to a functional group of the substrate by means of weak non-covalent interactions, like π-stacking and H-bonds. This prevents the reaction on the functional group and orients a specific part of the substrate to make it approach the reactive centre, promoting the reaction in a selective fashion. This molecular recognition approach is widely used in nature by enzymes,3 like monooxygenases and fatty acid desaturases. Nevertheless, its implementation in much smaller and well characterized synthetic catalysts is very challenging, and few successful examples have been reported to date.4
Scheme 1.

The molecular recognition strategy in selective catalysts.
The C-H oxidation of ibuprofen can involve either the benzyl carbon bound to the iso-propyl group, leading to ketone A, or the benzyl carbon bound to the carboxylic group, leading to ketone B (Scheme 2). Using our manganese oxidation catalyst, [(terpy’)Mn(OH2)(μ-O)2Mn(OH2)(terpy’)]3+, product A is obtained with a high selectivity of 98.5%.5 In the terpy’ ligand, terpyridine is functionalized with a phenylene linker attached to a Kemp’s triacid fragment. Our hypothesis for the origin of selectivity is that the Kemp’s triacid recognizes the carboxylic acid group of ibuprofen through a double H-bond interaction, which prevents the formation of product B (Scheme 3). The benzylic methylene group is oriented so that it approaches the reactive centre leading to the selective formation of product A. Without molecular recognition, selectivity is dramatically reduced by a 10-fold factor. The same strategy also proved applicable to aliphatic substrates such as methylcyclohexane acetic acid.
Scheme 2.

Catalytic selective oxidation of ibuprofen.
Scheme 3.

Molecular recognition model for the selective oxidation of ibuprofen.
For efficient recognition, the substrate must fit the molecular pocket spanning the reactive centre and the recognition site. The binding of the substrate should be strong enough to promote the desired reaction, but not so strong as to cause catalyst poisoning by the final product.6 In addition, steric exclusion of unbound substrate by the recognized substrate may be needed to reach high selectivies. In line with this idea, we have previously shown that tert-butyl benzoic acid acts as a potent inhibitor of the catalyst.7 The COOH group binds to the Kemp’s triacid fragment and the bulky t-Bu group shields the reactive centre, preventing the attack of unbound substrate.
In this article, we present a computational study on the mechanism of C-H oxidation by our dinuclear manganese catalyst (Scheme 2). The reaction mechanism and the electronic nature of the active species are explored in a model system. The origin of the selectivity, associated with the molecular recognition process, is studied by fully modeling the real system.
The rebound mechanism proposed by Groves,8 is generally accepted for C-H oxidation by metal-oxo complexes. The key steps of this mechanism are: 1) H abstraction by the oxo active species, yielding a radical and a metal-hydroxo intermediate and 2) OH rebound, in which the radical attacks the hydroxo ligand of the intermediate, yielding the oxidation product. Several studies support this mechanism9 and suggest that oxygen radical character is needed in the catalyst in order to favour step 1.10 To the best of our knowledge, the rebound mechanism in dinuclear manganese systems has not yet been studied.
Several issues concerning the molecular recognition process remain unclear: 1) is the molecular recognition model of Scheme 3 correct? 2) how far is the pathway involving molecular recognition favoured energetically over the pathway lacking recognition? 3) is there a perfect fit of the substrate, or is some flexibility needed in the catalyst? 4) on which side does the substrate bind, on the Mn1 side, as shown in Scheme 3, or on the Mn2 side? 5) is it possible that two molecules of the substrate bind simultaneously to the catalyst, involving both the Mn1 and Mn2 sides? 6) does the model of Scheme 3 account for the steric exclusion of unbound substrate and the inhibition experiments? These questions will be answered and rationalized with the molecular-level description given by our DFT calculations.
Experimental Details
Chemicals
All reagents were from Aldrich Chemicals (Milwaukee, WI), and used as received. Ph-terpy (4′-phenyl-2,2′:6′,2″-terpyridine) and the dimanganese(III,IV) di-μ-oxo dimer of Ph-terpy ([MnIII/IV2(μ-O)2(C21H15N3)2(H2O)2](NO3)3 were synthesized according to literature procedures.11
Tetraphenylphosphonium sulfate
This acetonitrile-soluble compound was prepared according to the following procedure. Tetraphenylphosphonium chloride (1082 mg, 2.88 mmol) and sodium sulfate (409 mg, 2.88 mol) were added to 8 mL of doubly distilled water. After stirring vigorously for 20 min, the solution was extracted with 9 mL of dichloromethane. Upon drying over magnesium sulfate, the solvent was removed with rotary evaporation under reduced pressure, giving the desired compound as a white solid. Chloride was absent (Ag+ test) and the IR spectra showed characteristic bands for both ions. (Yield: 63%, 790 mg, 1.81 mmol)
Electrochemistry
Electrochemical measurements were collected with an EG&G Princeton Applied Research Model 273 potentiostat/galvanostat using a standard three-electrode configuration. Experiments were carried out in acetonitrile solution containing 0.1 M tetrabutylammonium perchlorate (Fluka, electrochemical grade) as the supporting electrolyte. A platinum wire was used as the counter electrode, and a nonaqueous reference electrode of Ag/AgNO3 in acetonitrile with saturated potassium chloride (Princeton Applied Research) was used as the reference.
A basal plane carbon electrode was used as the working electrode to reduce background oxidation. The electrode was composed of a brass cylinder, sheathed in a teflon tube; at the tip of the brass, a two-part silver conducting epoxy (Alfa Aesar) was used to firmly attach the basal plane carbon electrode surface to the brass. Finally, the tip was sealed with the organic solvent-resistant, electrically-insulating, two-part epoxy Tra-bond 2151 (Emerson and Cuming, Canton, Mass., USA). Immediately prior to experiments, the working electrode was polished with 1 micron alumina paste, washed with copious amounts of water, and allowed to dry completely. The surface of the working electrode was then resurfaced with tape to restore the gray, basal surface.
Computational Details
Methodology
Unrestricted DFT calculations are performed using the B3LYP functional12 as implemented in Gaussian03.13 Geometry optimizations are carried out for each electronic state without any symmetry or geometry constraints. All transition states are relaxed towards reactants and products using the vibrational data to confirm their nature. The geometries are optimized with basis set I (Stuttgart-Bonn scalar relativistic RECP with the associated basis set14 for Mn and S, and the 6-31G basis set15 for O, N, C and H). The energies given in the text result from single-point calculations with basis set II (basis set I plus polarization functions16 on all atoms). The spin densities are obtained from NPA17 (Natural Population Analysis). The zero-point energy and entropy corrections are not considered.
The system under study is large but quite rigid. Nevertheless, the rotation around some σ bonds, like the C-iPr bond, the terpy-Ph bond or the C-COOH bonds, may generate several isomers, which are most likely to be close in energy. A full and extensive conformational search has not been performed. Molecular recognition and C-H oxidation are either promoted or avoided by selecting the appropriate structure, which is subsequently fully optimized. These geometries are built using chemical sense, trying to keep the energy of the system low. For instance, preliminary calculations showed that the rotation around the C-iPr bond of the substrate yields three different conformations, all within an energy range of only 2 kcal mol−1. The most stable conformation is used in all calculations. These calculations show that the rotation around these σ bonds will probably not have a large effect on the energy of the system.
Models
The study of mixed valence dinuclear Mn species requires the challenging calculation of several electronic states, with different spin distributions. The antiferromagnetic states are optimized by means of broken-symmetry DFT calculations,18 using the ferromagnetic wave functions as the initial guess. These calculations are computationally demanding due to the large size of the system. The reaction mechanism is therefore studied in a model system, in which the molecular recognition part of the catalyst, the phenylene linker and the Kemp’s triacid, is removed (Figure 1). This simplification of the catalyst saves computational time but keeps the essential electronic features of the system, which govern the reaction mechanism. The apical sites of Mn1 and Mn2 are occupied by an oxo and a sulfate group respectively, as in the real active species (vide infra). The substrate, ibuprofen, is modeled by toluene. The nature of all stationary points found in the model study, is confirmed by analytical calculation of their frequencies.
Figure 1.

Model system with the labels used in the text.
The molecular recognition process is studied in the full real system, considering the association of ibuprofen with the terpy’ ligand (Figure 2). The approach of the substrate to the catalyst was explored from both the Mn1 and Mn2 sides. We assume that the ligand on the nonreactive side does not bind to the substrate and therefore, we replace it by terpyridine, as in the model system, in order to save computational time. The inhibition experiments were modeled with the real inhibitor, tert-butyl benzoic acid. In this case, the simultaneous recognition of two molecules of substrate is explored, by considering the two real terpy’ ligands. As in the study of the model system, all geometries are fully optimized at the DFT(B3LYP) level. Due to the large size of the system (up to 1339 basis functions), the calculation of the antiferromagnetic states becomes extremely difficult and the analytic calculation of the frequencies has a high computational cost. The calculations on the smallest system show that the ferromagnetic octet has the same qualitative reactivity as the antiferromagnetic states. The more easily computed ferromagnetic octet state is, therefore, the only one considered (vide infra) and frequencies are only calculated for the transition states.
Figure 2.

Models and labels used in the study on the origin of the selectivity: molecular recognition from the a) Mn1 and b) Mn2 sides and c) steric exclusion of unbound substrate.
Labeling of the stationary points
In the model system, all species are labeled taking into account their electronic state: AFD for the antiferromagnetic doublet, FS for the ferromagnetic sextet, AFD2 for the alternative anti-ferromagnetic doublet and FO for the ferromagnetic octet (Figure 1).
In the real system, the geometries involving molecular recognition (R) are labeled depending on which side the substrate approaches the catalyst: RMn1 for the Mn1 side and RMn2 for the Mn2 side (Figure 2). In the absence of recognition (NR) the following labels are used: NRA for the pathway leading to product A and NRB for the pathway leading to product B (Scheme 2). In the inhibition (I) study, the structures are labeled considering the nature and the number of bound substrates: IBA for one molecule of inhibitor (benzoic acid), IBA2 for two molecules of inhibitor and IIP2 for two molecules of substrate (ibuprofen).
For both the model and real systems, the stationary points found along the oxidation pathway are identified with an additional label: 1 for the H-bound complex, TS1 for the H abstraction transition state, 2 for the hydroxo intermediate, TS2 for the OH rebound transition state and 3 for the alcohol coordinated intermediate.
Results. Experimental Section
Nature of the Axial Ligand
In the precatalyst, the axial positions of the Mn atoms are occupied by water molecules (Scheme 3). In the reaction mixture, these water ligands are probably replaced by counteranions which bind more strongly to the metal and reduce the unreasonably high tripositive charge of the system. Sulfate is a satisfactory axial ligand because it is a signification anion component of the solution. This counteranion is present in stoichiometric amounts and its concentration grows as the oxidant, oxone (KHSO5), is reduced. The presence of sulphate as axial ligand is supported by previous X-ray studies on this catalyst.19 This hypothesis is also supported by CV experiments (Figure 3).
Figure 3.

Cyclic voltammograms for the MnIII/MnIV dimer in MeCN solution (blue trace) and in MeCN solution with sulfate (red trace).
The aqueous electrochemistry of the dimanganese(III,IV) di-μ-oxo complex ([MnIII/IV2(μ-O)2(C15H11N3)2(H2O)2](NO3)3) has been well-studied.20 In aqueous solution, this complex shows no detectable interaction with sulfate ions.21 In acetonitrile, it was necessary to move from the unsubstituted terpyridine complex to the more soluble 4′-phenyl terpyridine complex to obtain good data. Cyclic voltammetry was performed to determine the effect of sulfate binding on the oxidation potential of the corresponding MnIII/MnIV mixed-valence dimer. As for the unsubstituted case, the phenyl terpyridine complex gave no indication of sulfate binding in water. In acetonitrile, however, the presence of sulfate in the form of the PPh4+ salt greatly modifies the CV, consistent with sulfate binding. The results can be seen in Figure 3.
The electrochemistry of the 4′-phenyl terpyridine complex is quite similar to that of the unsubstituted case. In MeCN solution without sulfate present, the results are shown in the blue trace above in Figure 3. The dimer is oxidized from the MnIII/MnIV state to the MnIV/MnIV state at ca. 860 mV. The corresponding quasi-reversible reduction from MnIV/MnIV to MnIII/MnIV is located at ca. 740 mV. These processes are labeled as Pa1 and Pc1, respectively. As has been reported previously, the cathodic wave at ca. 520 mV (in systems both with and without sulfate present, labeled Pc2) corresponds to reduction of a tetrameric Mn4IV species, formed from the MnIV/MnIV dimer in solution, back to the MnIII/MnIV dimer.
Upon addition of sulfate (5 equiv), there is a pronounced decrease in the electrochemical potential needed to oxidize the MnIII/MnIV dimer. The anodic peak corresponding to MnIII/MnIV → MnIV/MnIV now occurs at ca. 715 mV; this is a shift of ca. 145 mV. Similarly, the cathodic reduction of the MnIV/MnIV species occurs at a lower potential (ca. 680 mV). The anodic shoulder (denoted S in Figure 3) which occurs at ca. 615 mV corresponds to an oxidation process of tetraphenyl-phosphonium sulfate. It is interesting to note that sulfate does not interact with the tetrameric Mn4IV species, as the cathodic wave at ca. 520 mV (labeled Pc2) does not shift upon addition of sulfate.
Results. Theoretical Section
Model System. Electronic Nature of the Active Species
Several studies9 suggest that the active species in the rebound mechanism for C-H oxidation, is an oxo complex generated in-situ by the oxidation of the catalyst (Eq. 1). In our system, considering sulphate as axial ligand, this complex can be formulated as [(terpy’)Mn(O)(μ-O)2Mn(SO4)(terpy’)]+.
| (1) |
The electronic nature of this complex is explored considering the model system (Figure 1). The formal oxidation states of Mn1 and Mn2 are V and IV respectively, pointing to the mixed valence nature of this species. A similar system, with water as axial ligand bound to Mn2 instead of sulfate, has been previously studied by Siegbahn.22 This study showed that the MnV=O oxo group has an essentially MnIV–O• oxyl configuration (Figure 4). Thus, the dinuclear fragment is better described as MnIV(O•)MnIV, rather than MnV(O)MnIV. The six metal spins of this system can be arranged either in a parallel, ferromagnetic, or anti-parallel, antiferromagnetic, fashion generating several electronic configurations. The calculations showed that the most stable states are two antiferromagnetic doublets (AFD and AFD2), a ferromagnetic sextet (FS) and an octet (FO).
Figure 4.

Oxo/oxyl configurations and spin distribution in the different electronic states.
These four electronic states are fully optimized in our chosen sulfate model (Table 1). The most stable configuration is a doublet state, AFD, in which both Mn1,O1 and Mn1,Mn2 pairs are antiferromagnetically coupled, as shown by the spin densities (Table 1). The simultaneous flipping of the metal spins generates an alternative doublet state, AFD2, in which Mn1 and O1 are ferromagnetically coupled. This change in the spin distribution, destabilizes the system by 5.7 kcal mol−1 and causes a significant elongation of the Mn=O bond (1.80 Å in AFD, 1.88 Å in AFD2). A ferromagnetic sextet state, FS, is also accessible. This state is only 1.9 kcal mol−1 above the the ground state, AFD, and has an antiparallel alignement of the Mn1,O1 spins. The least stable state, FO, corresponds to a ferromagnetic octet configuration with all spins aligned in parallel. This state is 10.8 kcal mol−1 above AFD.
Table 1.
Relative energies (kcal mol−1), spin densities (ρ) and bond distances (Å) for the different electronic states of [(terpy)Mn(O)(μ-O)2Mn(SO4)(terpy)]+. See Figure 1 for labels.
| AFD | FS | AFD2 | FO | |
|---|---|---|---|---|
| Energy | 0.0 | 1.9 | 5.7 | 10.8 |
| O1 spin ρ | 0.84 | −0.82 | 1.11 | 1.11 |
| Mn1 spin ρ | −2.42 | 2.47 | 2.38 | 2.45 |
| Mn2 spin ρ | 2.51 | 2.55 | −2.51 | 2.51 |
| d(Mn1-O1) | 1.80 | 1.80 | 1.88 | 1.91 |
| d(Mn1-O2) | 1.76 | 1.76 | 1.75 | 1.78 |
| d(Mn1-O3) | 1.90 | 1.91 | 1.89 | 1.87 |
| d(Mn1-N1) | 2.05 | 2.05 | 2.05 | 2.03 |
| d(Mn1-N2) | 2.02 | 2.02 | 2.02 | 2.01 |
| d(Mn1-N3) | 2.05 | 2.05 | 2.04 | 2.02 |
| d(Mn2-O2) | 1.97 | 1.98 | 2.01 | 1.96 |
| d(Mn2-O3) | 1.80 | 1.81 | 1.80 | 1.83 |
| d(Mn2-O4) | 1.80 | 1.80 | 1.80 | 1.80 |
| d(Mn2-N4) | 2.03 | 2.03 | 2.03 | 2.03 |
| d(Mn2-N5) | 2.00 | 2.00 | 2.00 | 1.99 |
| d(Mn2-N6) | 2.03 | 2.03 | 2.03 | 2.03 |
In the four electronic states, the spin density on O1 is high, varying from 0.82 to 1.11, and in all cases close to 1.00. In addition, the Mn1-O1 bond distance, varying from 1.80 Å to 1.91 Å, is clearly longer than expected for a normal oxo group, which is ca. 1.60 Å in similar compounds.23 The high oxygen spin density and long Mn-O distances support the Mn-O• oxyl configuration, in contrast to the Mn=O oxo.
Interestingly, the electronic nature of the Mn(O)(μ-O)2Mn core has a low impact on the geometry of the complex, as shown by the other bond distances involving Mn1 and Mn2, with the sole exception noted above of Mn1-O1. Otherwise, the maximum distorsion caused by the excitation of AFD to the other electronic states is 0.04 Å in the bridging Mn-O distances and 0.03 Å in the Mn-N distances (Table 1).
Model System. Rebound Mechanism
The rebound mechanism has been postulated for C-H oxidation by metal oxo compounds.9 Under catalytic conditions, the first step of the mechanism is the generation of the oxo active species (Eq. 1). This species undergoes radical H abstraction from the substrate, yielding a metal hydroxo intermediate and a carbon radical (Eq. 2). In the final step, the OH rebound, the radical attacks the hydroxo ligand, leading to the formation of the product (Eq 3).
| (2) |
| (3) |
Several theoretical studies have suggested that radical character on oxygen, i.e. oxyl character, is needed for H abstraction.10 In our system, this electronic requirement is fulfilled by all four electronic states, including the ground state, AFD, because they have high oxygen spin densities on the oxo group. In all cases, the O spin densities are close to unity (e.g., 0.84 in AFD).
The H abstraction and OH rebound steps were explored in the ground state of the model catalyst (Figure 5). The approach of toluene to AFD yields a pre-reaction complex, AFD-1, in which a benzylic C-H is H-bonded to the oxyl group (d(CH···O1) = 2.55 Å). This species undergoes H abstraction through the transition state AFD-TS1 yielding intermediate AFD-2 (Figure 6). In AFD-TS1, a benzylic H of toluene is transferred to the oxyl group; the cleavage of the C-H bond (1.24 Å) is accompained by the formation of an O1-H bond (1.34 Å). The resulting product, AFD-2, is a manganese hydroxo complex bound to a PhCH2 radical, with a spin density on the benzyl carbon of 0.71. The transformation of the oxyl group into a hydroxo group causes the elongation of the Mn1-O1 bond (1.81 Å in AFD-1; 1.85 Å in AFD-2). The H abstraction step, AFD-1 → AFD-TS1 → AFD-2, has an energy barrier of 6.5 kcal mol−1 and is exothermic by 13.9 kcal mol−1.
Figure 5.

Energy profile, in kcal mol−1, for toluene oxidation by AFD.
Figure 6.

Optimized geometries of selected stationary points in the oxidation of toluene by afd.
The AFD-2 intermediate undergoes OH rebound with the benzyl radical. In the transition state, AFD-TS2, the formation of the C-O1 bond (2.37 Å) is concerted with the cleavage of the Mn1-O1 bond (1.89 Å). The relaxation of AFD-TS2 towards products yields intermediate AFD-2, in which benzyl alcohol (d(C-O1 = 1.50 Å) remains coordinated to Mn1 (d(Mn1···O1 = 2.09 Å). The OH rebound step, AFD-2 → AFD-TS2 → AFD-3, has an energy barrier, ΔE‡ = 6.0 kcal mol−1, similar to the that of the H abstraction, but it is much more exothermic, ΔE = −21.8 kcal mol−1. Once the substrate has been oxidized to the alcohol, further oxidation to the experimentally observed carbonyl compound is a fast step and, therefore, has not been studied.
The highest energy point along the C-H oxidation pathway is AFD-TS1, which is 3.0 kcal mol−1 above AFD + toluene. This suggests that H abstraction is the most difficult step. In addition, this step has a strong influence on the selectivity, because it defines which carbon of the substrate becomes the radical centre subsequently oxidized. This critical step was, therefore, further explored considering the other possible electronic states of the catalyst: FS, AFD2 and FO.
The H abstraction pathway for FS runs parallel to that of AFD, being ca. 2 kcal mol−1 above in energy (Figure 7). In the AFD2 pathway, the transition state, AFD2-TS1, is 3.0 kcal mol−1 above that associated with the AFD state. The energy profiles suggest that if the system starts reacting on the AFD2 surface, it may undergo spin-crossover to the FS surface and after reaching the transition state, FS-TS1, it may cross back to the AFD2 surface ending up in AFD2-2. Nevertheless, this more complex spin-crossover pathway probably has little advantage, given the small energy difference of ca. 2 kcal mol−1 or less between the FS and AFD2 states. The highest energy pathway corresponds to the FO state; the maximum energy difference, ca. 7 kcal mol−1, is located between the AFD-1 and FO-1 intermediates.
Figure 7.

Energy profiles, in kcal mol−1, for H abstraction in the different electronic states.
The geometries of the stationary points involved in H abstraction by AFD, FS, AFD2 and FO are compared (Table 2). In general, the key bond distances reflecting the H abstraction process, d(Mn1-O1), d(O1-H) and d(C-H), are not affected to any great extent by changing the electronic state. In the H-bonded intermediate, 1, these distances do not change by more than 0.01 Å when the system is excited from AFD to the other states. The only exception is FO-1, in which the O1-H distance is 0.18 Å shorter than in AFD-1. In the transition state, TS1, the maximum variation, 0.05 Å, occurs between the AFD and AFD2 states. This variation goes down to only 0.01 Å in the hydroxo intermediate, 2, which is essentially unaffected by the change of the electronic state.
Table 2.
Selected bond distances (Å) for the stationary points involved in H abstraction in the different electronic states. See Figure 1 for labels.
| Mn1-O1 | O1-H | C-Ha | |
|---|---|---|---|
| AFD-1 | 1.81 | 2.55 | 1.10 |
| FS-1 | 1.81 | 2.57 | 1.10 |
| AFD2-1 | 1.81 | 2.56 | 1.10 |
| FO-1 | 1.88 | 2.37 | 1.10 |
| AFD-TS1 | 1.81 | 1.34 | 1.24 |
| FS-TS1 | 1.81 | 1.34 | 1.24 |
| AFD2-TS1 | 1.85 | 1.39 | 1.21 |
| FO-TS1 | 1.85 | 1.39 | 1.21 |
| AFD-2 | 1.85 | 0.98 | 2.38 |
| FS-2 | 1.85 | 0.98 | 2.38 |
| AFD2-2 | 1.85 | 0.98 | 2.38 |
| FO-2 | 1.85 | 0.98 | 2.37 |
CH distance for the reactive C-H bond.
These calculations show that the four electronic states of the catalyst studied by us have similar reactivities in C-H bond oxidation. This is probably because all states have a similar radical character on the apical oxygen, which determines the reactivity. The difference in energy between the potential energy surfaces is in great part imposed by the difference in energy between the electronic states of the catalyst.
Real System. Molecular Recognition
The selective oxidation of ibuprofen is explored considering the real catalyst, [(terpy’)Mn(O)(μ-O)2Mn(SO4)(terpy’)]+ (Figure 2). The calculations on the FS state and specially on the anti-ferromagnetically coupled states, AFD and AFD2, are very difficult due to the large size of the system. In several cases, despite numerous attempts, the convergence of these states could not be achieved. We have shown above that the geometry and energy of the model system does not depend significantly on the electronic nature of the Mn(O)(μ-O)2Mn(SO4) core. This suggests that this factor probably does not affect the molecular recognition process to any great extent. The origin of the selectivity is, therefore, only studied in the FO state, which is the easiest to calculate. Furthermore, only the H abstraction step, 1 → TS1 → 2, which governs selectivity, is studied.
The catalyst binds to ibuprofen from the Mn1 side (Figure 2) leading to the formation of RMn1-1 (Figure 8). In the recognition site, the carboxylic groups of the catalyst and the substrate are bound with a double H-bond, with (C=)O···H(-O) distances of 1.60 Å and 1.62 Å (Table 3). This species undergoes radical H abstraction through the transition state RMn1-TS1, yielding the intermediate RMn1-2. In RMn1-TS1, a benzylic H is transferred from the CH2(i-Pr) group of ibuprofen to the oxyl group (d(O1···H = 1.37 Å, d(C···H = 1.23 Å). The transition state is reached without even weakening the double H-bond between the substrate and the catalyst, as shown by the (C=)O···H(-O) distances of 1.62 Å and 1.63 Å. The recognition is also maintained in the product, RMn1-2, in which the ibuprofen radical remains anchored to the recognition site by two H-bonds (1.62 Å and 1.62 Å). The reaction step RMn1-1 → RMn1-TS1 → RMn1-2 is exothermic by 18.9 kcal mol−1 and has a low barrier of 5.1 kcal mol−1 (Figure 9). The subsequent OH rebound to the ibuprofen radical and further oxidation, yields the final reaction product A.
Figure 8.

Optimized geometries for H abstraction from recognized ibuprofen.
Table 3.
Selected bond distances (Å) for the stationary points involved in H abstraction in the real system. See Figure 2 for labels.
| O1-H | O/C-Ha | H-Bond1b | H-Bond2c | |
|---|---|---|---|---|
| RMn1-1 | 2.63 | 1.10 | 1.60 | 1.62 |
| RMn1-TS1 | 1.37 | 1.23 | 1.63 | 1.62 |
| RMn1-2 | 0.98 | 2.77 | 1.62 | 1.62 |
| RMn2-1 | 3.35 | 1.10 | 1.59 | 1.62 |
| RMn2-TS1 | 1.40 | 1.21 | 1.62 | 1.62 |
| RMn2-2 | 0.98 | 3.56 | 1.61 | 1.64 |
| NRA-1 | 2.37 | 1.10 | — | — |
| NRA-TS1 | 1.38 | 1.22 | — | — |
| NRA-2 | 0.98 | 2.54 | — | — |
| NRB-1 | 1.65 | 1.02 | — | — |
| NRB-TS1 | 1.17 | 1.27 | — | — |
| NRB-2 | 1.00 | 1.73 | — | — |
C-H for product A formation and O-H for product B formation;
O···H distance between the H of the catalyst and the O of the substrate in the double H bond bridge;
O···H distance between the H of the substrate and the O of the catalyst in the double H bond bridge
Figure 9.

Energy profiles, in kcal mol−1, for H abstraction in the real system.
These calculations confirm the molecular recognition model represented in Scheme 3. Interestingly, before H abstraction, in RMn1-1, the benzylic C-H bond that needs to be oxidized is relatively far from the reactive centre (d(O1···H = 2.63 Å). Nevertheless, the transition state, RMn1-TS1, with a much shorter O1···H distance (1.37 Å) is reached without loosing recognition. In addition, the energy barrier, 5.1 kcal mol−1, is only slightly higher than that found for the FO state in the model system, 4.4 kcal mol−1, in the absence of molecular recognition. These results suggest that the catalyst has a certain degree of flexibility, which not only allows binding of the substrate but also attainement of the transition state, without weakening the double H bond serving to anchor the substrate to the catalyst.
The approach of ibuprofen from the Mn2 side was also explored. The associated reaction pathway is analogous to that described for the Mn1 side. In the starting intermediate, RMn2-1, ibuprofen binds to the recognition site by a double H-bond. A H radical is transferred to the oxyl group through the transition state RMn2-TS1 (Figure 10), leading to the formation of a Mn hydroxo intermediate, RMn2-2.
Figure 10.
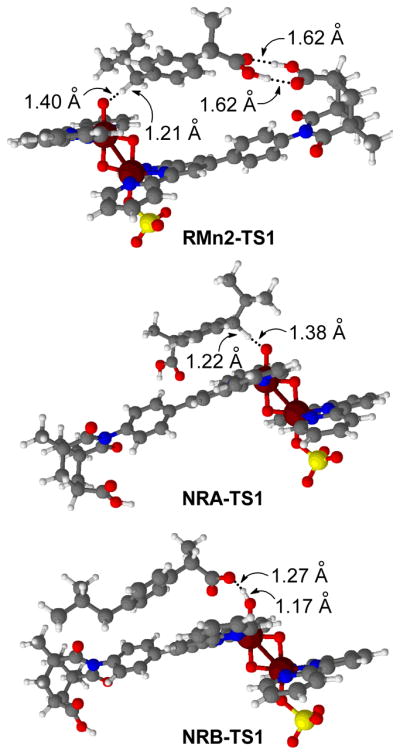
Optimized geometries of H abstraction transition states in the real system.
The recognition interaction is conserved all along the reaction pathway, RMn2-1 → RMn2-TS1 → RMn2-2. Nevertheless, this pathway is higher in energy than that seen for attack on the Mn1 side (Figure 9). The maximum energy difference, 4.3 kcal mol−1, is found between the transitions states. The higher relative energy of RMn2-TS1, is probably associated with the higher degree of flexibility needed to reach the transition state. In the Mn2 side approach the difference in the O1-H distances between 1 and TS1, 1.95 Å, is clearly larger than in the Mn1 side approach, 1.26 Å (Table 3).
The substrate can also approach the catalyst without being recognized. Both benzylic positions can then react yielding either product A (NRA pathway) or B (NRB pathway). In our calculations, molecular recognition was switched off by a 180° rotation of the N-C bond connecting the phenylene linker and the Kemp’s triacid fragment.
In the starting intermediate, NRA-1, ibuprofen binds to the reactive centre only by a H-bond between O1 and the oxidizible C-H bond. Due to the absence of the recognition constraint, the distance between H and O1 (2.37 Å) is much shorter than in RMn1-1 (2.63 Å) and RMn2-1 (3.35 Å); Table 3. In the transition state, NRA-TS1 (Figure 10), a benzylic H of the CH2(i-Pr) group is transferred to the oxyl group. The relaxation of NRA-TS1 towards products yields the hydroxo intermediate, NRA-2. The COOH group of ibuprofen is not recognized at any point.
The energy profile for the NRA pathway, NRA-1 → NRA-TS1 → NRA-2, with ΔE‡ = 4.1 kcal mol−1 and ΔE = −16.4 kcal mol−1, is very similar to that found for the molecular recognition pathway, RMn1 (Figure 9). These results indicate that molecular recognition does not affect the intrinsic reactivity of the substrate. Nevertheless, the whole NRA pathway is shifted above the RMn1 pathway in energy. The pathway with recognition is stabilized by the double H-bond, which does not change much along the reaction pathway. The energy difference between both pathways is 14.2 kcal mol−1 for 1, 13.2 kcal mol−1 for TS1 and 16.7 kcal mol−1 for 2. The mean value, 14.7 kcal mol−1, is a good estimate of the double H-bond strength24 and, indeed, is close to the value found for the acetic acid dimer.25 The strength of this interaction will be reduced by the polar solvents used experimentally, CH3CN or CH3CN/H2O mixtures. These calculations show that ibuprofen can react without being recognized and yield product A. Nevertheless, the associated H abstraction pathway is ca. 15 kcal mol−1 higher in energy than that with molecular recognition, due to the absence of the stabilization by the double H-bond.
The approach of ibuprofen yielding the alternative reaction product, B, is also explored. In this case, the mechanism of the reaction is different because it involves decarboxylation. In the postulated mechanism,26 the oxidation is initiated by radical H abstraction from the COOH group of the substrate (Eq. 4). In a subsequent step, a C radical is formed by CO2 release (Eq. 5). As in the classical OH rebound mechanism, this radical reacts with the hydroxo intermediate yielding the oxidized product (Eq. 6).
| (4) |
| (5) |
| (6) |
The first step has a great impact on selectivity because it determines which carbon of the substrate is oxidized. The energy pathway associated with this reaction, NRB, is thus studied in our catalytic system. In the pre-reaction complex, NRB-1, the COOH group of ibuprofen is bound to the oxyl group by a H-bond. This OH···O1 bond is much shorter, 1.65 Å, than the CH···O bonds found in the other pathways (Table 3). The H involved in this bond is abstracted by the oxyl group in the transition state, NRB-TS1 (Figure 10). In NRB-TS1, the transferred H lies in between the carboxylic and oxyl oxygens (d(O···H = 1.27 Å, d(H···O1 = 1.17 Å)). The resulting intermediate, NRB-2, contains the O radical product H-bonded (1.73 Å) to the Mn hydroxo centre. This H abstraction process, NRB-1 → NRB-TS1 → NRB-2, has a low energy barrier, ΔE‡ = 4.3 kcal mol−1, similar to those found for the other pathways. Nevertheless, the reaction is less favoured thermodynamically, with ΔE = 1.3 kcal mol−1, due to the poor stability of the O radical product.
In the absence of recognition, the transition state yielding product B, NRB-TS1, is 9.1 kcal mol−1 below that yielding A, NRA-TS1. This suggests that without molecular recognition the selectivity for product A will decrease dramatically, as observed experimentally.27 Nevertheless, NRB-TS1 is still 4.1 kcal mol−1 above the transition state involving recognition, RMn1-TS1. This energy difference is small, but enough to account for the high selectivities, >99%, obtained experimentally at room temperature.5
Real System. Inhibition
The inhibition experiments are modeled by analyzing the binding of the catalyst with the inhibitor, tert-butyl benzoic acid (Figure 2). The recognition of one molecule of inhibitor, leads to the formation of IBA-1 (Figure 11). In this complex, the COOH groups of the inhibitor and the catalyst are bound through a double H-bond (1.56 Å and 1.62 Å). The t-Bu group of the inhibitor stands above the reactive centre, but relatively far from it; the shortest H···O1 distance is 5.36 Å.
Figure 11.

Side and top views of the catalyst bound to a single molecule of inhibitor.
The spacefilling models of Figure 11, show that the bulky t-Bu group of the inhibitor is able to block the attack of unbound substrate from the Mn1 side. Nevertheless, such attack can still happen from the Mn2 side, where there is a sterically free open pocket.
The calculations on the reaction pathways for selective C-H oxidation described above, have shown that the approach and recognition of the substrate can happen on both the Mn1 and Mn2 sides. This suggests that, given the high concentration of inhibitor with respect to the catalyst, two molecules of tert-butyl benzoic acid may be recognized simultaneously, yielding IBA2-1 (Figure 12). In this complex, there is a double H-bond in each recognition centre, with (C=)O···H(-O) distances of 1.57 Å and 1.62 Å in both sides.
Figure 12.

Side and top views of the catalyst bound to two molecules of inhibitor.
The spacefilling models show that the steric exclusion of unbound substrate is much more efficient with two molecules of inhibitor, IBA2-1, than with only one, IBA-1. In IBA2-1, the approach of unrecognized molecules to the oxyl centre is sterically hindered on both the Mn1 and Mn2 sides.
Discussion
The calculations on the electronic nature of the model active species, [(terpy)Mn(O)(μ-O)2Mn(SO4)(terpy)]+, show that the reactive centre, formally a MnV=O oxo moiety, is indeed a MnIV-O• radical oxyl moiety. This is clearly indicated by the spin density on the apical oxygen, close to 1.00 (Table 1). This feature is observed in all four electronic states characterized by us, namely AFD, FS, AFD2 and FO (Figure 4). Thus, the presence of radical oxyl character does not depend on the spin distribution over the two metal centres.
The oxyl character of the active species is associated with the presence of a high lying singly occupied π*(Mn=O) orbital (Figure 13). The O contribution to this antibonding orbital accounts for the radical character of oxygen. The partial occupation of π*(Mn=O) also accounts for the long Mn=O distances. The radical character on oxygen promotes the cleavage of the benzyl C-H bond in a homolytic fashion. In addition, the electron vacancy in the π*(Mn=O) orbital, increases the electrophilic character of the oxyl group. In fact, several experimental studies have suggested that this reaction has some of the character of a nucleophilic attack of the C-H bond on the electrophilic Mn=O group.23
Figure 13.
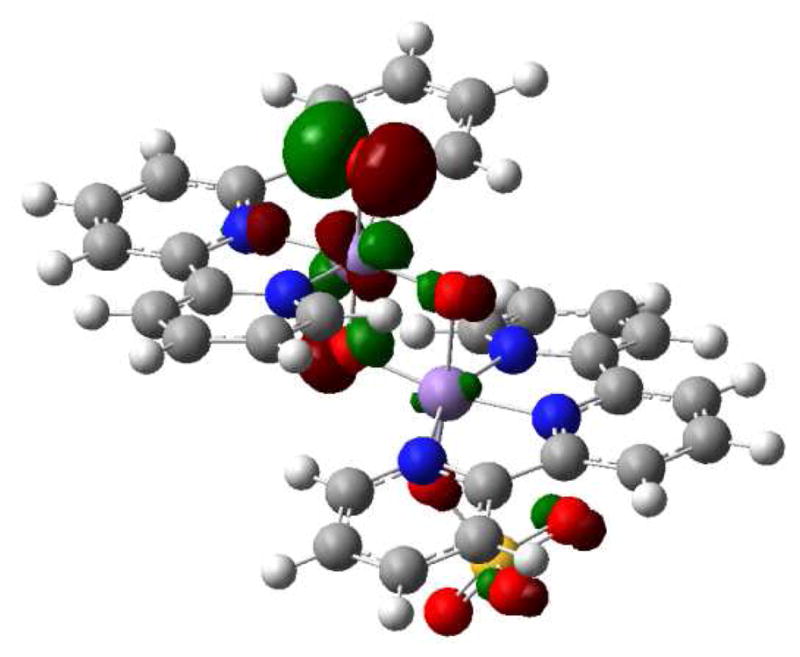
Singly occupied π*(Mn=O) orbital in [(terpy)Mn(O)(μ-O)2Mn(SO4)(terpy)]+.
The two metal centres, having a formal oxidation state of IV, are d3. These three spins can yield two alternative configurations: a high spin configuration, Mn(↑↑↑), with a formal spin density of 3.00, and a low spin configuration, Mn(↑↓↑), with a formal spin density of 1.00. The spin densities calculated for Mn1 and Mn2 are close to 2.50 in all cases (Table 1), showing that the high spin configuration is preferred because of the very small energy splitting of the orbitals involved.
The calculations show that the four electronic states follow the energy ordering: AFD < FS < AFD2 < FO. Nevertheless, the energy differences are small and the three most stable states lie in a range of only ca. 5 kcal mol−1. Of course, the calculations are carried out at the unrestricted DFT(B3LYP) level, which is not the most accurate method for these systems, specially considering their potential multireference character. The identification of AFD as the ground state, should thus be taken with precaution. The calculations do show that the four states, being close in energy, are all accessible.
All relative energies, spin densities and bond distances found for [(terpy)Mn(O)(μ-O)2Mn(SO4)(terpy)]+ are very similar to those reported by Siegbahn for [(terpy)Mn(O)(μ-O)2Mn(OH2)(terpy)]3+,22b in the four most stable electronic states. This suggests that the overall charge and the nature of the axial ligand bound to Mn2, only have a small influence on the electronic nature of the bimetalic core.
The calculations on the hydroxylation of toluene by [(terpy)Mn(O)(μ-O)2Mn(SO4)(terpy)]+, show that the rebound mechanism is feasible (Figure 5). Both the H abstraction and OH rebound steps are exothermic and involve low energy barriers. For the lowest energy state, AFD, the highest energy barrier, which corresponds to the H abstraction step, is only 6.5 kcal mol−1, and the overall oxidation process is exothermic by 39.2 kcal mol−1.
Study of the H abstraction step for the other electronic states, FS, AFD2 and FO, shows that all these states are also capable of promoting homolytic C-H cleavage. This is probably associated with the radical oxyl character, which is present in the four states. Previous theoretical studies have shown that oxyl character is required for efficient C-H oxidation10 and oxygen evolution from water.22
The H abstraction pathways associated to AFD, FS, AFD2 and FS are very close, both in terms of geometry (Table 2) and energy (Figure 7). The highest energy difference is 7.3 kcal mol−1. Toluene hydroxylation can be thus promoted by any of these electronic states.
The calculations on the selective oxidation of ibuprofen by the real catalyst (Figure 8), confirm the molecular recognition model postulated in Scheme 3. The Kemp’s triacid fragment of the catalyst binds to the COOH group of the substrate with a double H bond, thus protecting it from decarboxylative oxidation (product B). At the same time, the benzyl carbon of the CH2(i-Pr) group is exposed to the reactive centre, where it undergoes selective oxidation to product A. We also find that ibuprofen can be recognized and react from either the Mn1 or the Mn2 side.
Interestingly, in the initial intermediate, before H abstraction occurs, the oxidizable C-H bond is located far from the oxyl site. Nevertheless, the catalyst is flexible enough to allow the approach of the substrate to the reactive centre, keeping the double H bond of the recognition site. Indeed, this interaction is maintained through the whole reaction pathway. Without molecular recognition (Figures 9 and 10), the same reaction may also occur but it then involves an energy pathway ca. 15 kcal mol−1 higher in energy, due to the absence of the double H bond. An alternative view to see this selectivity is that the recognized substrate benefits from the entropic advantage of intramolecularity, paid for by forming a double H bond.
In principle, the oxidation of ibuprofen could also involve the primary and tertiary C-H bonds of the iso-butyl fragment.28 This is not favored due to the intrisic reactivity of these bonds, which is lower than that of the benzylic bond. Further, in RMn1-1, the benzylic C-H is the closest to the reactive Mn=O moiety. The oxidation of the primary and tertiary positions may be achieved by re-designing the molecular recognition part of the catalyst and by re-optimizing the reaction conditions, such as temperature and concentration of the catalyst and the substrate.
In the absence of molecular recognition, ibuprofen can undergo decarboxylative oxidation to product B, decreasing the selectivity of the reaction (Figures 9 and 10). The H abstraction transition state associated with this process, which involves homolytic O-H cleavage, is only 4.1 kcal mol−1 higher than that with molecular recognition. This small difference in energy is probably due to the following two factors: 1) the presence of a strong H bond between the COOH group of ibuprofen and the oxyl centre and 2) the lower energy barrier associated with the cleavage of an O-H bond (product B), with respect to that associated with the cleavage of a C-H bond (product A).
The spacefilling models of the real system, show that catalyst inhibition experimentally observed with tert-butyl benzoic acid7 is likely to involve two molecules of inhibitor (Figures 11 and 12). With this molecular configuration, the approach of unbound substrate to the reactive centre is effectively shielded by the bulky t-Bu groups on both the Mn1 and Mn2 sides.
Both substrates could be located on one side of the complex because the Kemp triacid unit enjoys free rotation. If these substrates were both located on the anion binding site, unrecognized substrate would have free access to the oxyl and undergo unselective reaction, contrary to experiment.5–7 If the anion side of the complex is hydrated by trace water or even specially strongly solvated by MeCN, it is possible that the preferred conformation is represented by IBA2-1 (Figure 12), where both hydrophobic substrate molecules are located on the hydrophobic side of the catalyst. This would ensure strong steric protection of the oxyl by recognized substrate.
All these results indicate that the catalyst is probably able to recognize two molecules of ibuprofen simultaneously. This could lead to the formation of IIP2-1 (Figure 14). In this complex, each molecule of substrate is recognized by the formation of a double H-bond on the oxyl face of the catalyst (1.59 Å and 1.62 Å in both cases).
Figure 14.

Top views of the catalyst bound to one and two molecules of ibuprofen.
The spacefilling model of RMn1-1 (Figure 14) shows that in the presence of only a single molecule of ibuprofen, the Mn2 side of the catalyst would be open enough to allow the attack of unbound molecules of substrate, which will diminish the selectivity of the reaction contrary to experiment.5 As found for catalyst inhibition, the binding of an additional molecule of substrate to the Mn2 recognition site in IIP2-1, leads to a much more effective protection of the reactive oxyl site; the approach of unbound substrate is sterically hindered on both the Mn1 and Mn2 sides.
Our calculations thus suggest that there is a cooperative effect between the two recognition sites of the catalyst. The Mn1 site recognizes the substrate and promotes selective reaction with the oxyl group. In conjunction with this, the Mn2 site recognizes an additional molecule of substrate, which blocks the attack of unbound substrate keeping the selectivity high. In addition, the ibuprofen molecule bound to the Mn2 site may also react, but without eroding selectivity since it is also recognized.
Conclusions
A combined experimental/theoretical study of the C-H oxidation of ibuprofen catalyzed by [(terpy’)Mn(OH2)(μ-O)2Mn(OH2)(terpy’)]3+ has allowed us to identify the origin of the high selectivity found experimentally. Cyclic voltammetry experiments, show that the sulfate anion present in stochiometric amounts can replace water in the coordation sphere of the MnIII/MnIV core, most likely at the more electrophilic MnIV site. Oxidation at the other site would then lead to formation of the formally MnV=O group. Consequently, [(terpy’)Mn(O)(μ-O)2Mn(SO4)(terpy’)]+ is considered as active species in the computational study. The electronic structure of the catalyst is studied at the DFT(B3LYP) level in a simplified model, in which the molecular recognition part of the terpy’ ligand is removed. Two antiferromagnetic doublets and a ferromagnetic sextet and octet are calculated to be relatively close in energy. All these electronic states have significant radical character on oxygen and are all reactive in C-H oxidation. The rebound mechanism (H-abstraction followed by OH rebound) has low energy barriers for all the states in the oxidation of toluene. The similarity of the reactivity of these electronic states suggests that the detailed spin distribution in the bimetallic core and the spin coupling between the two metals, have little influence on the C-H oxidation energy profile. The relative energy ranking of the possible reaction pathways is essentially determined by the energy ordering of the electronic states.
The molecular recognition has been studied in the full reaction system. It is shown that the double H bond between the catalyst and the substrate stabilizes the entire potential energy surface, because this interaction is preserved all along the reaction pathway. Consequently, selective C-H oxidation occurs at lower energy than without molecular recognition. The flexibility of the catalyst allows the bound substrate to approach the reactive centre, keeping intact the double H bond of the recognition site.
The association of the catalyst with tert-butyl benzoic acid is shown to shield the access of unbound substrate to the reactive centre, hence preventing non selective hydroxylation. It is also shown that the two recognition sites can be used in a cooperative manner to control the access to the reactive centre.
Supplementary Material
Acknowledgments
D.B., C.R. and O.E. thank the Institut Charles Gerhardt, the Ministère de l’Enseignement Supérieur et de la Recherche and the CNRS for funding. D.B. thanks Sanofi-Aventis for a postdoctoral fellowship. R.H.C. thanks the NSF (Grant 0614403) for funding.
Footnotes
Electronic Supplementary Information (ESI) available: Optimized structures of all stationary points with their absolute energies with basis sets I and II.
Contributor Information
Pamela Moles, Email: pmoles@qfa.uji.es.
Robert H. Crabtree, Email: robert.crabtree@yale.edu.
Odile Eisenstein, Email: odile.eisenstein@univmontp2.fr.
Notes and references
- 1.Shilov AE, Shul’pin GB. Chem Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886.Dyker G. Angew Chem, Int Ed. 1999;38:1699–1712. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6.Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a.Pamplin CB, Legzdins P. Acc Chem Res. 2003;36:223–233. doi: 10.1021/ar0202215.Goldberg KI, Goldman AS, editors. Activation and Functionalization of C-H Bonds. Oxford University Press; Washington, DC: 2004. Lersch M, Tilset M. Chem Rev. 2005;105:2471–2526. doi: 10.1021/cr030710y.Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731.Bergman RG. Nature. 2007;446:391–393. doi: 10.1038/446391a.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Muller J, Lippard SJ. Angew Chem, Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P.Que L, Dong YH. Acc Chem Res. 1996;29:190–196.
- 2.Meunier B. Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Imperial College Press; London: 2000. [Google Scholar]
- 3.(a) Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]; (b) Dawson JH, Sono M. Chem Rev. 1987;87:1255–1276. [Google Scholar]; (c) Behrouzian B, Savile CK, Dawson B, Buist PH, Shanklin J. J Am Chem Soc. 2002;124:3277–3283. doi: 10.1021/ja012252l. [DOI] [PubMed] [Google Scholar]; (d) Moche M, Shanklin J, Ghoshal A, Lindqvist Y. J Biol Chem. 2003;278:25072–25080. doi: 10.1074/jbc.M301662200. [DOI] [PubMed] [Google Scholar]; (e) Broadwater JA, Whittle E, Shanklin J. J Biol Chem. 2002;277:15613–15620. doi: 10.1074/jbc.M200231200. [DOI] [PubMed] [Google Scholar]; (f) Fox BG, Lyle KS, Rogge CE. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 4.(a) Breslow R, Zhang XJ, Huang Y. J Am Chem Soc. 1997;119:4535–4536. [Google Scholar]; (b) Breslow R, Huang Y, Zhang XJ, Yang J. Proc Natl Acad Sci USA. 1997;94:11156–11158. doi: 10.1073/pnas.94.21.11156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang J, Breslow R. Angew Chem, Int Ed. 2000;39:2692–2694. doi: 10.1002/1521-3773(20000804)39:15<2692::aid-anie2692>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]; (d) Yang J, Gabriele B, Belvedere S, Huang Y, Breslow R. J Org Chem. 2002;67:5057–5067. doi: 10.1021/jo020174u. [DOI] [PubMed] [Google Scholar]
- 5.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941–1943. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- 6.Das S, Brudvig GW, Crabtree RH. Chem Commun. 2008:413–424. doi: 10.1039/b710355g. [DOI] [PubMed] [Google Scholar]
- 7.Das S, Brudvig GW, Crabtree RH. J Am Chem Soc. 2008;130:1628–1637. doi: 10.1021/ja076039m. [DOI] [PubMed] [Google Scholar]
- 8.(a) Groves JT, Mcclusky GA. J Am Chem Soc. 1976;98:859–861. [Google Scholar]; (b) Groves JT. J Chem Educ. 1985;62:928–931. [Google Scholar]
- 9.(a) Groves JT. J Inorg Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]; (b) Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]; (c) Shaik S, de Visser SP, Ogliaro F, Schwarz H, Schroder D. Curr Opin Chem Biol. 2002;6:556–567. doi: 10.1016/s1367-5931(02)00363-0. [DOI] [PubMed] [Google Scholar]; (d) Shaik S, Kumar D, de Visser SP. J Am Chem Soc. 2008;130:10128–10140. doi: 10.1021/ja8019615. [DOI] [PubMed] [Google Scholar]; (e) Yoshizawa K. Coord Chem Rev. 2002;226:251–259. [Google Scholar]; (f) Kamachi T, Yoshizawa K. J Am Chem Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]; (g) Bathelt CM, Ridder L, Mulholland AJ, Harvey JN. J Am Chem Soc. 2003;125:15004–1500. doi: 10.1021/ja035590q. [DOI] [PubMed] [Google Scholar]; (h) Slaughter LM, Collman JP, Eberspacher TA, Brauman JI. Inorg Chem. 2004;43:5198–5204. doi: 10.1021/ic049922j. [DOI] [PubMed] [Google Scholar]; (i) Sharma PK, de Visser SP, Ogliaro F, Shaik S. J Am Chem Soc. 2003;125:2291–2300. doi: 10.1021/ja0282487. [DOI] [PubMed] [Google Scholar]; (j) Dhuri SN, Seo MS, Lee YM, Hirao H, Wang Y, Nam W, Shaik S. Angew Chem, Int Ed. 2008;47:3356–3359. doi: 10.1002/anie.200705880. [DOI] [PubMed] [Google Scholar]
- 10.(a) Balcells D, Raynaud C, Crabtree RH, Eisenstein O. Chem Commun. 2008:744–746. doi: 10.1039/b715939k. [DOI] [PubMed] [Google Scholar]; (b) Balcells D, Raynaud C, Crabtree RH, Eisenstein O. Inorg Chem. 2008;47:10090–10099. doi: 10.1021/ic8013706. [DOI] [PubMed] [Google Scholar]; (c) Balcells D, Raynaud C, Crabtree RH, Eisenstein O. Chem Commun. 2009 doi: 10.1039/b821029b. [DOI] [PubMed] [Google Scholar]
- 11.(a) Constable EC, Lewis J, Liptrot MC, Raithby PR. Inorg Chim Acta. 1990;178:47–54. [Google Scholar]; (b) Chen H, Tagore R, Das S, Incarvito C, Faller JW, Crabtree RH, Brudvig GW. Inorg Chem. 2005;44:7661–7670. doi: 10.1021/ic0509940. [DOI] [PubMed] [Google Scholar]
- 12.(a) Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Miehlich B, Savin A, Stoll H, Preuss H. Chem Phys Lett. 1989;157:200–206. [Google Scholar]; (d) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 13.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski J, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03 (Revision D.01) Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 14.For Mn: Andrae D, Haüssermann U, Dolg M, Stoll H, Preuss H. Theor Chim Acta. 1990;77:123–141.For S: Bergner A, Dolg M, Küchle W, Stoll H, Preuss H. Mol Phys. 1993;30:1431–1441.
- 15.Hehre WJ, Ditchfield R, Pople JA. J Phys Chem. 1972;56:2257–2261. [Google Scholar]
- 16.(a) Ehlers AW, Böhme M, Dapprich S, Gobbi A, Hollwarth A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G. Chem Phys Lett. 1993;208:111–114. [Google Scholar]; (b) Höllwarth A, Böhme H, Dapprich S, Ehlers AW, Gobbi A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G. Chem Phys Lett. 1993;203:237–240. [Google Scholar]; (c) Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213–222. [Google Scholar]
- 17.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899–926. [Google Scholar]
- 18.(a) Noodleman L, Baerends EJ. J Am Chem Soc. 1984;106:2316–2327. [Google Scholar]; (b) Han WG, Liu T, Lovell T, Noodleman L. J Am Chem Soc. 2005;127:15778–15790. doi: 10.1021/ja050904q. [DOI] [PubMed] [Google Scholar]
- 19.Limburg J, Vrettos JS, Chen H, de Paula JC, Crabtree RH, Brudvig GW. J Am Chem Soc. 2001;123:423–430. doi: 10.1021/ja001090a. [DOI] [PubMed] [Google Scholar]
- 20.Baffert C, Romain S, Richardot A, Leprêtre J, Lefebvre B, Deronzier A, Collomb MN. J Am Chem Soc. 2005;127:13694–13704. doi: 10.1021/ja052595+. [DOI] [PubMed] [Google Scholar]
- 21.Cady CW, Gao Y, Shinopoulos K, Crabtree RH, Brudvig GW. manuscript in preparation [Google Scholar]
- 22.(a) Siegbahn PEM, Crabtree RH. J Am Chem Soc. 1999;121:117–127. [Google Scholar]; (b) Lundberg M, Blomberg MRA, Siegbahn PEM. Inorg Chem. 2004;43:264–274. doi: 10.1021/ic0348188. [DOI] [PubMed] [Google Scholar]
- 23.(a) Song WJ, Seo MS, George SD, Ohta T, Song R, Kang MJ, Tosha T, Kitagawa T, Solomon EI, Nam W. J Am Chem Soc. 2007;129:1268–1277. doi: 10.1021/ja066460v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin N, Ibrahim M, Spiro TG, Groves JT. J Am Chem Soc. 2007;129:12416–12417. doi: 10.1021/ja0761737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The strength of the double H bond can be also evaluated as the energy associated with the following reaction: ibuprofen + catalyst → RMn1-1. Nevertheless, the accurate calculation of this energy is challenging due to the presence of significant charge redistribution and entropy change. The modelization of these effects requires the inclusion of the solvent and the calculation of frequencies for all stationary points, which were not considered in our study.
- 25.(a) Colominas C, Teixidó J, Cemelí J, Luque FJ, Orozco M. J Phys Chem B. 1998;102:2269–2276. [Google Scholar]; (b) Frurip DJ, Curtiss LA, Blander M. J Am Chem Soc. 1980;102:2610–2616. [Google Scholar]; (c) Winkler A, Hess P. J Am Chem Soc. 1994;116:9233–9240. [Google Scholar]
- 26.(a) Bockman TM, Hubig SM, Kochi JK. J Am Chem Soc. 1996;118:4502–4503. [Google Scholar]; (b) Balahura RJ, Sorokin A, Bernadou J, Meunier B. Inorg Chem. 1997;36:3488–3492. doi: 10.1021/ic9700765. [DOI] [PubMed] [Google Scholar]
- 27.Our results suggest that selectivity would be even reversed, which is not observed experimentally. In the reaction pathway leading to B, the decarboxylation step may have a high energy barrier. In such case, decarboxylation will have a strong influence in the kinetic competition between the A and B reaction pathways, thus affecting the final selectivity. This is not considered in our simplified picture of the reaction.
- 28.Hamman MA, Thompson GA, Hall SD. Biochem Pharmacol. 1997;54:33–41. doi: 10.1016/s0006-2952(97)00143-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.