

DNAs with catalytic function, or DNAzymes, have become important tools for a variety of biochemical applications.[1] Some of the most widely known DNAzymes, the 8–17 and the 10–23, were evolved by in vitro selection for RNA cleavage,[2] as a way of inhibiting gene expression.[3] Other DNAzymes have been used for catalyzing chemical reactions,[4] as biosensors[5,6] and in nanodevices.[7–9] Being able to photomodulate the function of DNAzymes with high spatial and temporal resolution could be useful for many applications in biotechnology and biology.
Photoactivatible (also known as ‘caged’) oligonucleotides have been used primarily to study gene expression,[10] and more recently, have also been used for controlling PCR product generation,[11] RNA folding,[12] oligonucleotide mismatch detection[13] and conformation switches used in nanomachines.[14] The focus of the current study is the 10–23 DNAzyme, a Mg2+-dependent enzyme that catalyzes RNA cleavage at a pyrimidine-purine junction, preferentially AU. The enzyme consists of a 15-base catalytic loop (GGCTAGCTACAACGA) and binding arms 6–12 bases long that hybridize to the target RNA. We report the design of caged 10–23 DNAzymes that either inactivate by breaking apart upon UV irradiation (Figures 1b, 1c) or are blocked from hybridizing before irradiation (Figures 1d, 1e), depending on the placement of a light-induced strand break (Figure 1f).
Figure 1.

a) General structure and function of the 10–23 DNAzyme; b,c) Photocleavable spacer (PS, red square) within DNAzyme core and binding arms causes enzyme to fragment upon UV photolysis. d,e) Circularized DNAzymes have impaired function until photolysis of PS restores linear form. f) structure of PS and product of photolysis
Caged versions of the 8–17 DNAzyme[15] and 10–23 DNAzyme[16] have been developed previously using phosphoramidite chemistry to incorporate photoactivatible bases at key positions in the enzyme. These modifications disrupted DNAzyme function until removal with UV light, which turned gene expression from “on” to “off”. Reversible 8–17 and 10–23 DNAzymes were created by replacing key bases with a nucleotide modified with the photoisomerizable azobenzene group, which adopts the cis conformation under UV irradiation and reverts to the trans isomer in visible light.[17] The 10–23 DNAzyme showed higher catalytic activity with cis-azobenzene, whereas the 8–17 DNAzyme was more active in trans form. This approach offers photo-reversibility but requires synthesis of azobenzene-modified nucleotides and remains to be optimized for bidirectional control.
Here, we used a 10–23 DNAzyme targeting the VEGFR2 receptor mRNA previously characterized by Zhang et al.[3c] The strand break was engineered site-specifically using a photocleavable spacer (PS, Figure 1f) which is commercially available as a phosphoramidite (Glen Research) and can be incorporated in a sequence anywhere during solid-phase synthesis.
The first site for PS incorporation was the 10–23 catalytic core (Figure 1b), which has been well studied through base substitutions and deletions. Changes to bases 7–12 of the core tend to cause the smallest loss of catalytic activity.[18] In particular, thymine in the 8th position (T8) is the most flexible to substitutions by all three other bases and inosine, as well as to deletion.[19] Cytosine in the 7th position (C7) can also be deleted with moderate loss of activity, and a double deletion mutant lacking both C7 and T8 retains some activity.[19] We therefore designed the first three caged DNAzymes (Table 1) to replace with PS either T8 (Dz1), C7 (Dz2), or both C7 and T8 (Dz3).
Table 1.
DNAzyme sequences, data fitting, and melting temperatures. First-order rate constants (k) were fit to to [S]=A*(1-e−kt), where [S] is the fraction of substrate uncleaved at timet, and A is the percentage of total RNA cleaved as time approaches infinity. Binding arms are in lowercase and catalytic loop in uppercase. PS indicates photocleavage site.
| Sequence |
k (−UV) (min−1) |
k (+UV) (min−1) |
Photo- modulation[a] |
A (−UV) (%) |
A (+UV) (%) |
Melting Temp. (°C) |
|
|---|---|---|---|---|---|---|---|
| Dz0 | tgctctccaGGCTAGCTACAACGAcctgcacct | 0.24(±0.03) | 0.21(±0.02) | 1:1 | 77(±2) | 80(±2) | 49 |
| Dz1 | tgctctccaGGCTAGC-PS-ACAACGAcctgcacct | 0.089(±0.003) | 0.039(±0.003) | 2:1 | 70(±1) | 76(±3) | 49 |
| Dz2 | tgctctccaGGCTAG-PS-TACAACGAcctgcacct | Inactive | Inactive | N/A | - | - | 49 |
| Dz3 | tgctctccaGGCTAG-PS-ACAACGAcctgcacct | Inactive | Inactive | N/A | - | - | 49 |
| Dz4 | tgctctccaGGCTAGCTACAACGAcct-PS-cacct | 0.055(±0.007) | 0.032(±0.01) | 2:1 | 53(±3) | 21(±4) | 39 |
| Dz5 | tgctc-PS-ccaGGCTAGCTACAACGAcctgcacct | 0.12(±0.02) | 0.077(±0.01) | 2:1 | 81(±3) | 31(±1) | 39 |
| Dz6 | tgctc-PS-ccaGGCTAGC-PS-ACAACGAcctgcacct | 0.069(±0.002) | Inactive | on→off | 74(±1) | - | 36 |
| Dz7 | 0.022(±0.007) | 0.21(±0.02) | 1:10 | 58(±10) | 79(±1) | 43 | |
| Dz8 |  |
Inactive | 0.024(±0.009) | off→on | - | 56(±10) | N/D[c] |
| Dz1a | tgctctccaGGCTAGC |
0.10(±0.02)[b] |
N/A |
N/A |
51(±2) |
N/A |
39 |
| Dz1b | 5′ – PO42− - ACAACGAcctgcacct | 39 |
Photomodulation is the ratio of rate constants (k) before and after UV photolysis.
Dz1a and Dz1b were inactive when tested alone. k value is for a 1:1 mixture of the two strands.
The internal melting temperature of Dz8 alone was 74 °C, so its melting temperature with the target RNA was not determined.
The DNAzymes were tested for their ability to catalyze cleavage of a 28-nucleotide radiolabeled RNA substrate in vitro, GCGCGAGGUGCAGGAUGGAGAGCAAGGC. Bases targeted by the binding arms are underlined and RNA cleavage site is highlighted in bold. Irradiation of the DNAzymes was carried out for 20 min with a UV transilluminator (9 mW at peak intensity, 365 nm). Gel electrophoresis of the DNAzymes containing PS anywhere in the sequence showed that after 20 min there was no intact DNAzyme remaining (Supporting Information).
Kinetic data from the RNA cleavage experiments were fit by a single exponential decay function, [S]=A*(1-e−kt), where [S] is the fraction of RNA uncleaved at time t. Apparent first-order rate constants (k) were determined, and A represents the percentage of RNA cleaved as time approaches infinity (Table 1). Near-UV irradiation did not appear to damage the unmodified DNAzyme, Dz0, as its rate coefficient was essentially unchanged by irradiation (k = 0.24 min−1 before and k = 0.21 min−1 after), with similar percentages of total RNA cleavage (A = 77% and 80%). Of the photoactive DNAzymes tested, Dz2 and Dz3 showed no cleavage after 1-h incubation, even without irradiation, indicating that replacement of C7 with PS abolishes activity.
Replacing T8 with PS within the catalytic core produced an active enzyme (Dz1) with lower efficiency (k = 0.089 min−1). Surprisingly, the enzyme activity decreased only 2-fold (k = 0.039 min−1) upon irradiation. Furthermore, the unirradiated and irradiated enzymes showed similar amounts of total RNA cleavage at later time points (A = 70% and 76%). To investigate these results for Dz1, the two halves of the DNAzyme were synthesized and tested. The 5′ half of the DNAzyme, Dz1a, consisted of the 5′ binding arm and bases 1–7 of the catalytic loop. Dz1b consisted of bases 9–15 of the catalytic loop and the 3′ binding arm. Dz1b was also phosphorylated at its 5′ end so that it would be the same as the product of Dz1 photolysis. When Dz1a, Dz1b, and RNA target were mixed simultaneously in the same reaction, significant RNA cleavage was observed (k = 0.10 min−1), which shows the two halves can reconstitute the active enzyme while lacking a phosphodiester linkage. After 1 h, the mixture of Dz1a and Dz1b cleaved 61% of the target RNA, almost as much as the 70% RNA cleaved by full-length Dz1 (Figure 2b).
Figure 2.
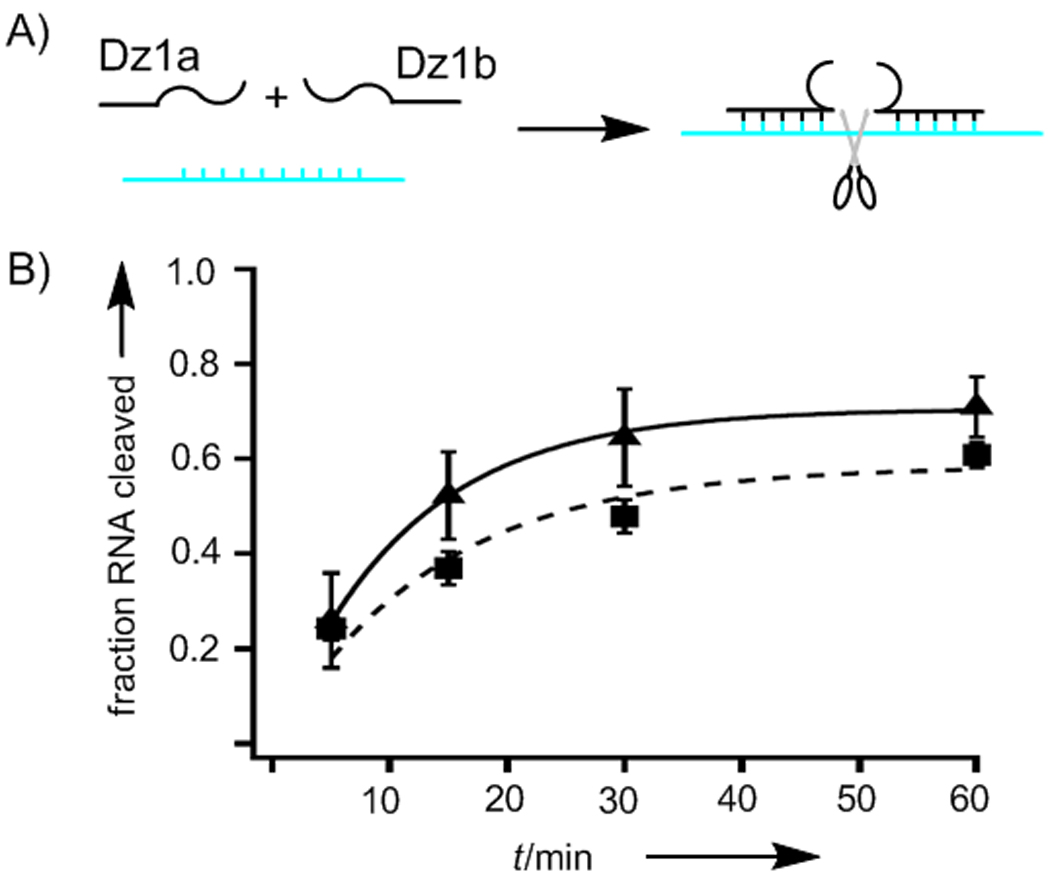
a) Scheme showing two DNA halves and RNA target mixed together to form the active enzyme; b) RNA cleavage vs. time for Dz1 (solid line) and the mixture of the two halves of the enzyme, Dz1a and Dz1b (broken line).
The Dz1a + Dz1b mixture performed as well as Dz1 in the early time points, suggesting that formation of the catalytic site occurs rapidly. Another DNAzyme consisting of two non-covalently linked oligonucleotides has been made previously by incorporating half a DNA aptamer in each sequence.[6] In the presence of hemin, the two halves of the aptamer assemble into a G quadruplex and the split DNAzyme becomes active. For the split 10–23 DNAzyme, further study needs to be conducted to determine whether the metal-binding properties and intramolecular contacts play roles in maintaining the structure and function of the enzyme after photolysis.
A second set of DNAzymes was then designed to incorporate strand breaks within the binding arms (Figure 1c). PS was placed three bases downstream from the catalytic site in the 3′ binding arm (Dz4) and three bases upstream from the catalytic site in the 5′ binding arm (Dz5). A guanosine in Dz4 was replaced while a thymine was replaced in Dz5. The sensitivity of DNAzyme arm length has been found to be highly sequence dependent.[20] Shortening each arm for different DNAzymes can decrease or increase the cleavage rate by varying degrees, making it difficult to predict whether photo-truncation should affect Dz4 and Dz5.
Substitution of PS in each binding arm produced enzymes with very different activities. Dz5 was slower than Dz0 (k = 0.12 min−1) but still cleaved the same amount of RNA as Dz0 during the experiment (A = 81%). Dz4 showed even less activity (k = 0.055 min−1 and A = 53%). Upon irradiation of Dz4 and Dz5, the activities of both decreased 2-fold. However, the binding arm truncations were clearly not sufficient to inactivate the enzyme.
To increase the degree of photomodulation, a new DNAzyme (Dz6) was designed to combine catalytic core and binding arm modifications, with PS at both T8 and in the 5′ binding arm. The robustness of the 10–23 DNAzyme to function as two halves or with shorter binding arms seemed to necessitate splitting the enzyme into three separate parts to achieve inactivation. Unphotolyzed Dz6 showed similar activity to Dz1 (k = 0.069 min−1 and A = 74%) and, unlike the other DNAzymes, RNA cleavage after UV irradiation was completely abolished (Figure 3a).
Figure 3.
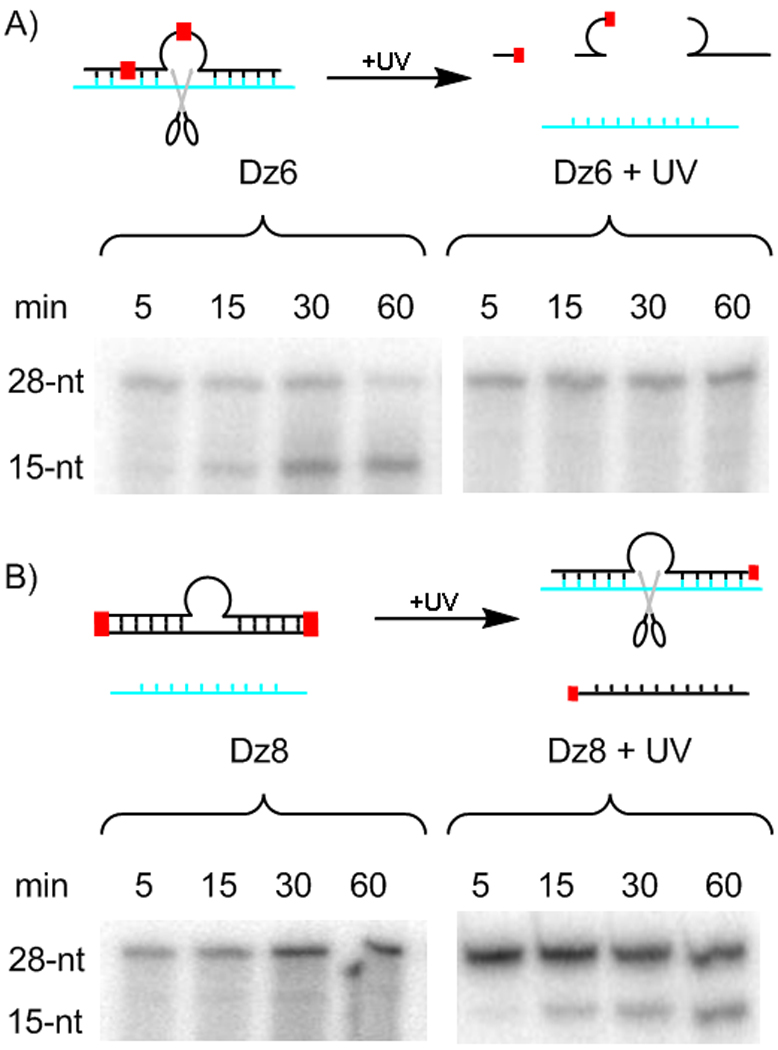
a) Dz6 hydrolyzes radiolabeled 28-nt RNA substrate to form a 15-nt product. In a parallel experiment, no Dz6 activity is seen after UV irradiation; b) In contrast, Dz8 shows no RNA hydrolysis until UV irradiation restores activity.
Having successfully made a DNAzyme that switched from “on” to “off” upon photolysis, we worked to develop a DNAzyme that was turned “on” by UV irradiation. To prevent the caged enzyme from functioning normally, we tried to block DNA hybridization to the RNA target. Modulating hybridization using photocleavable groups has been accomplished previously by several methods. One strategy has been to incorporate a number of photocleavable groups within the backbone to block binding sterically.[21] A similar approach has used caged nucleobases to prevent base pairing in the sequence.[22] Recently, a single photocleavable linker has been used to join the oligonucleotide to a complementary blocking strand that will dissociate upon photolysis.[23]
In order to achieve more complete inhibition of hybridization, the DNAzymes were circularized with PS included in the sequence. Previously, circular DNAzymes have been made by enzymatic ligation[24] or cloning into an expression vector[25] in order to make them more nuclease resistant and improve cellular delivery. These DNAzymes were 70-nt and 100-nt in length, thus their function was not impeded after circularization. In our design, we hypothesized that joining the 5′ and 3′ ends of this shorter 33-nt DNAzyme should create a compromised enzyme that gains activity upon UV irradiation and linearization.
For Dz7, PS was incorporated between the 5′ and 3′ ends of the DNAzyme to create a circular structure (Figure 1d). Because it is not known whether the structure of PS would be recognized by the ligase, the ligation site was chosen to be at a distance, between guanosine G6 and cytosine C7 of the catalytic loop. The linear sequence was synthesized using standard phosphoramidite chemistry with the phosphorylated G6 located at the 5′ end and C7 at the 3′ end, placing PS in the middle. The two ends were joined in 40% yield using Circligase, a single-stranded DNA ligase optimized for circularization of oligonucleotides with 5′ phosphate and 3′ hydroxyl groups. Any linear DNA remaining was digested using exonuclease I.
Dz7 was assayed for its ability to cleave RNA under the same conditions used for Dz1–6. Before photolysis, Dz7 cleaved RNA slowly, k = 0.022 min−1 (Table 1). After photolysis, activity was restored to the level observed for the unmodified enzyme Dz0, creating a 10-fold enhancement. The melting temperature of Dz7 hybridized to the RNA target is 43 °C, which, although 6 °C lower than the melting temperature of the linear DNAzyme (Table 1), indicates that the circularized DNA still hybridized to its target under the conditions of the enzymatic assay.
To ensure that hybridization could not occur with the RNA, we applied the circularization technique to create a DNAzyme (Dz8) that incorporated a self-complementary blocking strand. PS was incorporated at two positions: joining both the 5′ and 3′ ends of the DNAzyme to a DNA sequence identical to bases 7–23 of the RNA substrate, including the bulged A residue (Table 1). Like Dz7, the initial linear sequence of DNA was made by phosphoramidite chemistry including a 5′ phosphate with the ligation site between G6 and C7. PS was again incorporated in the middle of the linear sequence followed by the complementary region of DNA and a second photocleavable spacer. It was then enzymatically circularized under the same conditions as Dz7, with similar yields, to produce a circular structure with self-complementarity (Figure 1e).
In contrast to Dz7, Dz8 cleaved no RNA in a 1-h time course prior to UV irradiation. The complementary DNA successfully prevented Dz8 from having DNAzyme activity (Figure 3b). Post-photolysis, the enzyme gained modest activity (k = 0.024 min−1) and cleaved more than half of the RNA target during the course of the experiment (A = 56%). As in Dz6, reduced activity in the “on” state was traded for a complete shutoff of activity in the “off” state. Importantly, with Dz6 and Dz8, complete photomodulation was successfully achieved to create DNAzymes that switch “on” or “off”.
Caged DNAzymes of any sequence that will either activate or inactivate upon UV irradiation can be made using commercially available reagents without the need for synthesizing photoactive nucleobases. Photocleavable linkers similar to PS have been used for a variety of applications involving DNA strand breaks,[12a, 24a, 26] such as the study of strand repair, end-labeling of oligonucleotides, or control of hybridization. This work provides a novel use for PS that could be extended to other linkers.
In conclusion, these approaches should facilitate the development of caged DNAzymes for numerous biological applications. The circularization technique described provides a method for completely blocking oligonucleotide hybridization, which has many potential uses in biotechnology. In addition, this work demonstrates the robustness of the 10–23 DNAzyme, which can tolerate the chemically dissimilar substitution of PS for a nucleotide. It was also found to function as a binary DNAzyme divided within the catalytic core.
Experimental Section
RNA cleavage assay
Single turnover assays were performed at 37 °C with 0.83 µM DNAzyme and 0.083 µM P-32 labeled RNA substrate in 10 mM MgCl2, 10 mM Tris pH 7.5, and 83 mM NaCl. For experiments in which the DNAzyme was photolyzed, the reaction mixture without the RNA target was irradiated in an open 0.2 mL tube by a UV transilluminator (9 mW/cm2 at 365 nm) and the reaction was then initiated by addition of the RNA. Aliquots (5 µL) were removed at regular time intervals and the reaction quenched with RNA loading buffer II (Ambion). The aliquots were run on a 7 M urea, 20% polyacrylamide gel at 300 V for 40 min. Gels of RNA digestion were imaged using an Amersham Biosciences Storm 840 phosphorimager. Gels were analyzed using TotalLab Software (Nonlinear Dynamics) to detect the band intensities and correct for background using the rubber band subtraction function. Then the ratio of intensities of the uncleaved and cleaved RNA bands was calculated. First-order rate constants (k) were fit to a single exponential function using Igor (WaveMetrics), [S] = A*(1 - e−kt), where [S] is the fraction of substrate uncleaved at time t.
DNAzyme circularization
Dz7 and Dz8 were synthesized as linear sequences by standard phosphoramidite chemistry and chemically phosphorylated during synthesis. The linear sequences were respectively: GCTACAACGACCTGCACCT-PS-TGCTCTCCAGGCTA and GCTACAACGACCTGCACCT-PS-GGTGCAGGATGGAGAGC-PS-TGCTCTCCAGGCTA. Circligase (Epicentre) was used to create a phosphodiester linkage between these two ends using manufacturer’s protocols with 8 µM DNA and 5 U/µL ligase. After the reaction, exonuclease I was used to digest any remaining linear DNA. The product was purified by phenol/chloroform extraction and desalted on a NAP-10 column. Analysis by gel electrophoresis showed the circular product migrating more slowly than the linear sequence, and returning to its original position upon UV irradiation (Supporting Information).
Thermal denaturation methods
Solutions for the thermal denaturation studies contained 1 µM 19-base RNA with the AU cleavage site replaced with GU (AGGUGCAGGGUGGAGAGCA) and the complementary DNAzyme in 10 mM Tris-HCl, pH 8.0 and 100 mM NaCl. Melting studies were conducted on a Beckman Coulter DU800 UV-Vis spectrophotometer equipped with a programmable Peltier temperature controller. Samples were monitored at 260 nm while heating or cooling at a rate of 1.0 °C/min, with a 1-min hold per degree Celsius. Melting temperatures were determined from the peak of the first derivative plot of Abs260 vs. temperature.
Supplementary Material
{kind=link}
Acknowledgements
This work was supported by NIH R-01 GM083030 and a Camille and Henry Dreyfus Teacher-Scholar Award to IJD.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Baum DA, Silverman SK. Cell. Mol. Life Sci. 2008;65:2156. doi: 10.1007/s00018-008-8029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santoro SW, Joyce GF. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4262. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Mitchell A, Dass CR, Sun LQ, Khachigian LM. Nucleic Acids Res. 2004;32:3065. doi: 10.1093/nar/gkh626. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khachigian LM, Fahmy RG, Zhang GS, Bobryshev YV, Kaniaros A. J. Biol. Chem. 2002;277:22985. doi: 10.1074/jbc.M200977200. [DOI] [PubMed] [Google Scholar]; c) Zhang L, Gasper WJ, Stass SA, Ioffe OB, Davis MA, Mixson AJ. Cancer Res. 2002;62:5463. [PubMed] [Google Scholar]
- 4.Silverman SK. Chem. Commun. 2008:3467. doi: 10.1039/b807292m. [DOI] [PubMed] [Google Scholar]
- 5.Lee SE, Liu GL, Kim F, Lee LP. Nano Lett. 2009;9:562. doi: 10.1021/nl802689k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolpashchikov DM. J. Am. Chem. Soc. 2008;130:2934. doi: 10.1021/ja711192e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willner I, Shlyahovsky B, Zayats M, Willner B. Chem. Soc. Rev. 2008;37:1153. doi: 10.1039/b718428j. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Wang MS, Mao CD. Angew. Chem. 2004;27:3638. doi: 10.1002/anie.200453779. Angew. Chem. Int. Ed. 2004, 43, 3554. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Wang YF, Liu Q, Zhang ZZ, Fan CH, He L. Angew. Chem. 2006;118:1791. Angew. Chem. Int. Ed. 2006, 45, 1759. [Google Scholar]
- 10.a) Mayer G, Heckel A. Angew. Chem. 2006;118:5020. doi: 10.1002/anie.200600387. Angew. Chem. Int. Ed. 2006, 45, 4900. [DOI] [PubMed] [Google Scholar]; b) Dmochowski IJ, Tang XJ. BioTechniques. 2007;43:161. doi: 10.2144/000112519. [DOI] [PubMed] [Google Scholar]; c) Young DD, Deiters A. Angew. Chem. 2007;119:4368. Angew. Chem. Int. Ed. 2007, 46, 4290. [Google Scholar]
- 11.a) Tanaka K, Katada H, Shigi N, Kuzuya A, Komiyama M. ChemBioChem. 2008;9:2120. doi: 10.1002/cbic.200800285. [DOI] [PubMed] [Google Scholar]; b) Young DD, Deiters A. ChemBioChem. 2008;9:1225. doi: 10.1002/cbic.200800051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Hobartner C, Silverman SK. Angew. Chem. 2005;117:7471. doi: 10.1002/anie.200502928. Angew. Chem. Int. Ed. 2005, 44, 7305. [DOI] [PubMed] [Google Scholar]; b) Wenter P, Furtig B, Hainard A, Schwalbe H, Pitsch S. Angew. Chem. 2005;117:2656. doi: 10.1002/anie.200462724. Angew. Chem. Int. Ed. 2005, 44, 2600. [DOI] [PubMed] [Google Scholar]
- 13.Thoeni S, Kressierer CJ, Giese B. Angew. Chem. 2007;119:2160. doi: 10.1002/anie.200603092. Angew. Chem. Int. Ed. 2007, 46, 2112. [DOI] [PubMed] [Google Scholar]
- 14.Liu HJ, Xu Y, Li FY, Yang Y, Wang WX, Song YL, Liu DS. Angew. Chem. 2007;119:2467. Angew. Chem. Int. Ed. 2007, 46, 2515. [Google Scholar]
- 15.Ting R, Lermer L, Perrin DM. J. Am. Chem. Soc. 2004;126:12720. doi: 10.1021/ja046964y. [DOI] [PubMed] [Google Scholar]
- 16.Lusic H, Young DD, Lively MO, Deiters A. Org. Lett. 2007;9:1903. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Liu Y, Sen D. J. Mol. Biol. 2004;341:887. doi: 10.1016/j.jmb.2004.06.060. [DOI] [PubMed] [Google Scholar]; b) Keiper S, Vyle JS. Angew. Chem. 2006;118:3384. doi: 10.1002/anie.200600164. Angew. Chem. Int. Ed. 2006, 45, 3306. [DOI] [PubMed] [Google Scholar]
- 18.Zaborowska Z, Furste JP, Erdmann VA, Kurreck J. J. Biol. Chem. 2002;277:40617. doi: 10.1074/jbc.M207094200. [DOI] [PubMed] [Google Scholar]
- 19.Zaborowska Z, Schubert S, Kurreck J, Erdmann VA. FEBS Lett. 2005;579:554. doi: 10.1016/j.febslet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Cairns MJ, Hopkins TM, Witherington C, Sun LQ. Antisense Nucleic Acid Drug Dev. 2000;10:323. doi: 10.1089/oli.1.2000.10.323. [DOI] [PubMed] [Google Scholar]
- 21.Ghosn B, Haselton FR, Gee KR, Monroe WT. Photochem. Photobiol. 2005;81:953. doi: 10.1562/2004-11-15-RA-373. [DOI] [PubMed] [Google Scholar]
- 22.a) Krock L, Heckel A. Angew. Chem. 2005;117:475. doi: 10.1002/anie.200461779. Angew. Chem. Int. Ed. 2005, 44, 471. [DOI] [PubMed] [Google Scholar]; b) Matsunaga D, Asanuma H, Komiyama M. J. Am. Chem. Soc. 2004;126:11452. doi: 10.1021/ja0471976. [DOI] [PubMed] [Google Scholar]
- 23.a) Shah S, Friedman SH. Oligonucleotides. 2007;17:35. doi: 10.1089/oli.2006.0067. [DOI] [PubMed] [Google Scholar]; b) Tang XJ, Dmochowski IJ. Angew. Chem. 2006;118:3603. doi: 10.1002/anie.200600954. Angew. Chem. Int. Ed. 2006, 45, 3523. [DOI] [PubMed] [Google Scholar]; c) Shestopalov IA, Sinha S, Chen JK. Nat. Chem. Biol. 2007;3:650. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- 24.Seifert G, Taube T, Paal K, von Einsiedel HG, Wellmann S, Henze G, Seeger K, Schroff M, Wittig B. Nucleosides Nucleotides & Nucleic Acids. 2006;25:785. doi: 10.1080/15257770600726075. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, McMicken HW. Gene Ther. 2003;10:1776. doi: 10.1038/sj.gt.3302068. [DOI] [PubMed] [Google Scholar]
- 26.a) Zhang KJ, Taylor J-S.Biochemistry 20014015311141065 [Google Scholar]; b) Bai XP, Li ZM, Jockusch S, Turro NJ, Ju JY. Proc. Natl. Acad. Sci. U. S. A. 2003;100:409. doi: 10.1073/pnas.242729099. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tang XJ, Dmochowski IJ. Nature Protocols. 2006;1:3041. doi: 10.1038/nprot.2006.462. [DOI] [PubMed] [Google Scholar]; d) Ordoukhanian P, Taylor J-S. J. Am. Chem. Soc. 1995;117:9570. [Google Scholar]; e) Ordoukhanian P, Taylor J-S. Bioconjugate Chem. 2000;11:94. doi: 10.1021/bc9900993. [DOI] [PubMed] [Google Scholar]; f) Olejnik J, Krzymanska-Olejnik E, Rothschild KJ. Biophys. J. 1998;74:A296. [Google Scholar]; g) Pirrung MC, Zhao XD, Harris SV. J. Org. Chem. 2001;66:2067. doi: 10.1021/jo001594r. [DOI] [PubMed] [Google Scholar]; h) Crey-Desbiolles C, Lhomme J, Dumy P, Kotera M. J. Am. Chem. Soc. 2004;126:9532. doi: 10.1021/ja047976m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.