

Abstract
Healing of superficial skin wounds depends on the proliferation and migration of keratinocytes at the wound margin. When human epidermal keratinocytes were incubated on polymerized type I collagen, they rapidly attached and spread. The cells underwent a proliferative response and, over the subsequent 6-day period, covered the collagen surface with a monolayer of cells. When keratinocytes were plated on collagen that had been fragmented by exposure to matrix metalloproteinase-1 (MMP-1, collagenase-1), the cells attached as readily as to intact collagen but spread more slowly and less completely. Growth was reduced by approximately 50%. Instead of covering the collagen surface, the keratinocytes remained localized to the site of attachment. Keratinocytes on fragmented collagen expressed a more differentiated phenotype as indicated by a higher level of surface E-cadherin. Based on these findings, we suggest that damage to the underlying collagenous matrix may impede efficient keratinocyte function and retard wound closure.
Keywords: Keratinocyte, Collagen lattice, Collagenase, Matrix metalloproteinase-1, Wound healing, E-cadherin, Vinculin
Introduction
The dermal matrix is a three-dimensional structure in which type I collagen is the major component [22]. Rigidity of the matrix and the mechanical stress imparted on cells imbedded in the matrix is critical to fibroblast function [8]. In past studies, we demonstrated that when interstitial fibroblasts were imbedded in a three-dimensional lattice of polymerized type I collagen that had been subjected to proteolytic fragmentation with matrix metalloproteinase-1 (MMP-1; collagenase-1), mechanical tension on the imbedded cells was reduced [25]; under these conditions, collagen synthesis declined by greater than 80%. Fibroblasts are not unique in their response to fragmented collagen. In a recent study, it was shown that endothelial cells grew as a monolayer of cells on the surface of intact polymerized type I collagen but formed tubes when supported in the matrix following proteolytic cleavage [28]. Our conclusion from these studies is that alterations in the structure of the connective tissue resulting from proteolytic cleavage have the capacity to influence the behavior of multiple cell types in the dermis.
The present study continues our effort to understand how damage to the dermal matrix influences cell function. Here, we show that when epidermal keratinocytes are plated on the surface of polymerized type I collagen in a three-dimensional matrix, they proliferate, migrate over the collagen surface, and eventually cover the surface with a monolayer of cells. In contrast, when keratinocytes are plated on collagen that has been subjected to proteolytic fragmentation, there is a reduction in growth (approximately 50%) and a retardation in coverage of the collagen surface (approximately 80% reduction on Day 3 and 50% by Day 6).
Materials and methods
MMP-1 and other reagents
Human MMP-1 (collagenase-1) was obtained from Calbiochem (San Diego, CA). The enzyme was purified from human rheumatoid synovial fibroblasts as the naturally occurring proenzyme form. The MMP-1 preparation appeared as a doublet at 52 and 57 kDa in β-casein zymography, and was reactive with a rabbit polyclonal anti-MMP-1 antibody (AB806; Chemicon International, Temicula, CA) by western blotting. A mouse monoclonal antibody with neutralizing activity for MMP-1 (Ab-5; IM66) was obtained from Oncogene Research Products (San Diego, CA). Native pro-MMP-8 was obtained from stimulated human neutrophils, while MMP-2 (72-kDa gelatinase A) and MMP-9 (92-kDa gelatinase B) were obtained as recombinant proteins (active forms) produced in mammalian cells (Calbiochem). Clostridium histolyticum collagenase (Worthington Type I) and porcine pancreatic elastase (2× crystallized) were obtained from Worthington Biochemicals (Lakewood, NJ) and bovine pancreatic trypsin was obtained from Sigma Chemical Company (St. Louis, MO). Human recombinant tissue inhibitor of metalloproteinase-1 (TIMP-1) was obtained from Calbiochem, and soybean trypsin inhibitor (SBTI) was obtained from Sigma Chemical Co.
Mouse monoclonal antibodies to vinculin and E-cadherin were obtained from Chemicon and BD Biosciences (San Jose, CA), respectively. When used in immunofluorescence studies, the primary antibodies were visualized with a rabbit anti-mouse IgG antibody bound to Alexa Fluor 488 (Invitrogen, Carlsbad, CA) and further amplified with Alexa Fluor 488 goat anti-rabbit IgG. In some experiments, Alexa Fluor 546 phalloidin (Invitrogen) was used. [Note: Alexa Fluor 488 is spectrally similar to fluorescein, while Alexa Fluor 546 is spectrally similar to tetramethylrhodamine].
Human skin organ culture fluid
Human basal cell carcinoma tissue was used as a source of human skin organ culture fluid [24]. Pooled preparations of organ culture fluid were assessed for total and active MMP-1 by β-casein zymography and collagen fragmentation. Total and active MMP-2 and MMP-9 were determined by gelatin zymography and gelatin fragmentation. Consistent with past observations [24], the organ culture fluids contained a substantial amount of active MMP-1 but virtually no MMP-8 or MMP-13. Active forms of the two major gelatinolytic enzymes (e.g., MMP-2 and MMP-9) were also present in the conditioned media.
Preparation and degradation of polymerized collagen lattices
Three-dimensional lattices of reconstituted, polymerized type I collagen were prepared as described previously [25]. Rat tail collagen (3.7–4.7 mg/ml in 1 N HCl) (BD Biosciences) was diluted to 2 mg/ml in culture medium consisting of serum-free, Ca2+-supplemented keratinocyte basal medium (KBM). The collagen solution was made isotonic by addition of an appropriate amount of 10× concentrated Hanks’ balanced salt solution, and brought to pH 7.2. The collagen was added to wells of a 24-well plate (0.5 ml/well) and incubated for 2 h at 37°C, during which time a stiff lattice of polymerized collagen formed. In certain experiments, lattices with reduced amounts of collagen were prepared. When this was done, concentrations of other reagents were modified to maintain pH and isotonicity.
Degradation of the polymerized collagen was achieved by exposing the collagen lattice to human skin organ culture fluid or to human fibroblast-derived MMP-1. Activation of the proenzyme was accomplished by exposure of the latent enzyme to 1 μg of crystalline trypsin for 5 min at 37°C followed by 10 μg of SBTI. Intact collagen lattices exposed to buffer alone served as control. After enzyme treatment, the supernatant fluids from control or treated collagen lattices were removed. Collagen fragmentation was assessed by measuring the peptides (specifically, the ¾- and ¼-sized fragments) released into the supernatant fluid (resolved by SDS-PAGE and staining with Coomassie brilliant blue). In certain experiments, other enzymes were examined for ability to degrade polymerized type I collagen.
For quantitation purposes, lattices were treated with a sufficient amount of culture fluid (100 μl/lattice) or a sufficient amount of MMP-1 (75 ng/lattice) to obtain complete digestion of the lattice. A standard curve was then generated by SDS-PAGE resolution of increasing amounts of the digested material. By comparing the amount of ¾- and ¼- sized fragments recovered in the culture fluid during partial digestion with the standard curve, it was possible to precisely estimate the amount of degradation occurring under a given set of conditions. Using this approach, it was determined that between 30% and 45% of the initial collagen was converted into fragments under the conditions depicted in Figs. 1, 2, 3, and 4. Scanning electron microscopy (below) was used to assess the remaining collagen structure in control- and enzyme-treated lattices. After treatment, the lattices (still grossly intact) were rinsed carefully with Ca2+- supplemented KBM and were ready to use.
Fig. 1.
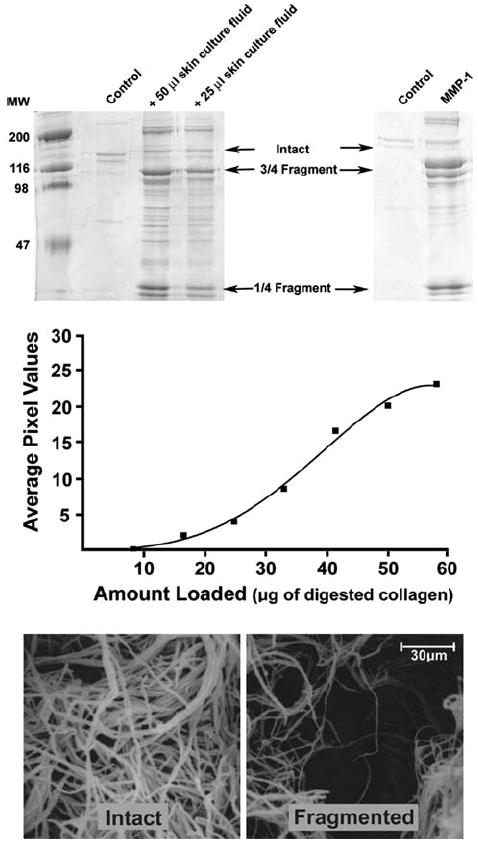
Collagen degradation upon exposure to skin organ culture fluid or fibroblast MMP-1. Upper panels: SDS-PAGE analysis of the collagen fragmentation pattern obtained following exposure of intact collagen lattices to control buffer alone, to two concentrations of skin organ culture fluid, or to activated MMP-1. The collagen lattices exposed to buffer alone (control lanes) show a faint doublet corresponding to the α1 and α2 chains of intact type I collagen. The bands are faint because most of the intact collagen remains polymerized in the lattice. The collagen lattices exposed to skin culture fluid show bands corresponding to the ¾- and ¼-sized fragments of type I collagen. Additional fragments are observed. The collagen lattice exposed to purified MMP-1 shows the ¾- and ¼-sized fragments. Middle panel: standard curve obtained by inducing complete hydrolysis of the collagen lattice with 75 ng of MMP-1 and resolving graded amounts of hydrolysate by SDS-PAGE. The ¾- and ¼-sized fragments were digitized and digital images quantified. Lower panels: scanning electron microscopic images of lattices exposed to buffer alone (intact) and to organ culture fluid (fragmented). Similar results were obtained with 10 different preparations of skin organ culture fluid
Fig. 2.
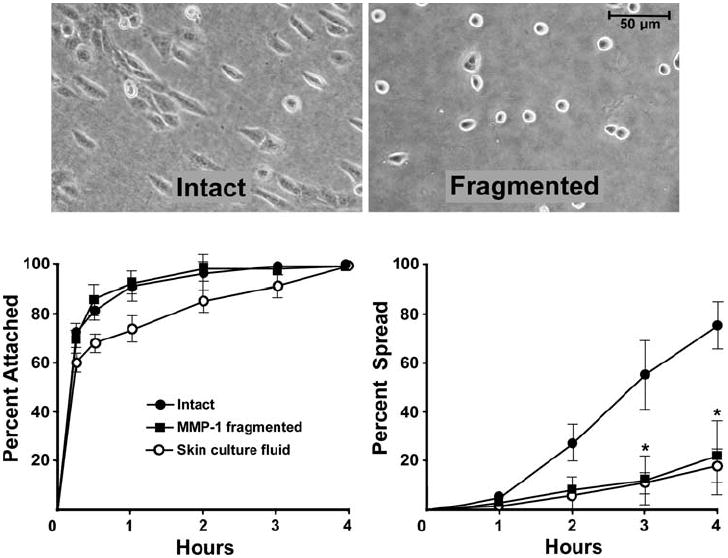
Attachment and spreading of human epidermal keratinocytes on intact and fragmented collagen lattices. Left panel: attachment. A similar rate of attachment is observed on intact and fragmented collagen. Right panel: spreading. This is significantly reduced on fragmented collagen. Values are means and standard deviations based on triplicate samples in a single experiment. Statistical significance was determined using the Student t-test, comparing each treatment group to its control. ‘*’ indicates statistical difference from control at P < 0.05 level. The experiment was repeated five times with similar results. Inset: appearance of keratinocytes after 2 h on intact and fragmented collagen lattices
Fig. 3.
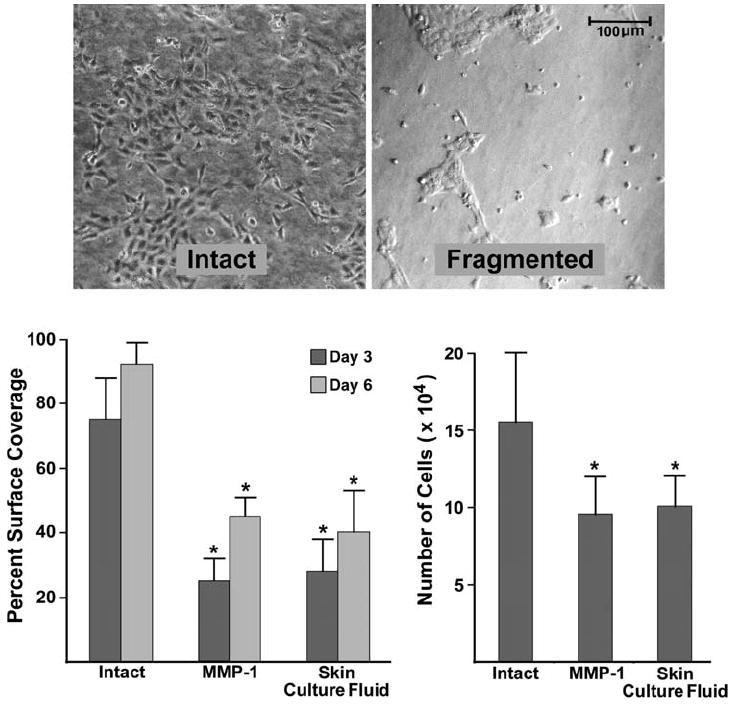
Keratinocyte coverage of the surface of intact and fragmented collagen lattices. Left panel: percentage of the collagen surface covered with cells on Days 3 and 6. Values shown are means and standard errors based on seven different experiments. Right panel: proliferation. Cells were harvested and counted on Day 3. Values shown are means and standard errors based on four separate experiments. Statistical significance was determined using the Student t-test. ‘*’ indicates statistical difference from control at P < 0.05 level. Inset: appearance of keratinocytes after 3 days on intact and fragmented collagen
Fig. 4.
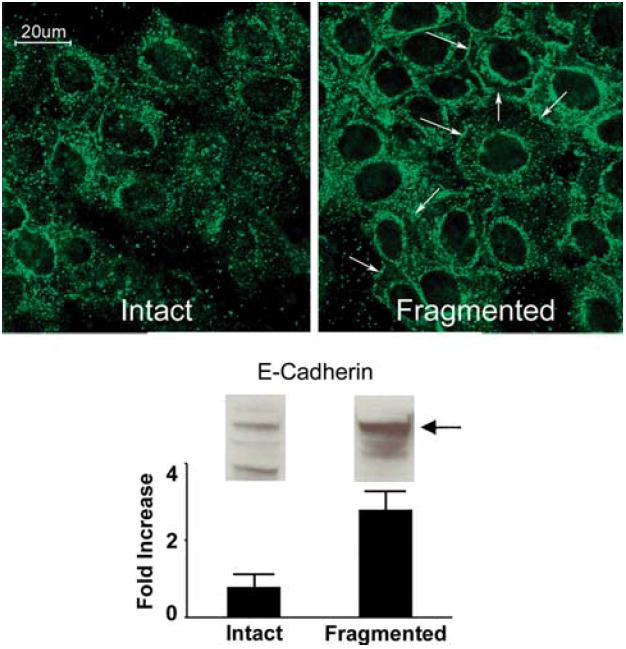
E-cadherin expression in keratinocytes on intact and fragmented collagen. Upper panels: confocal immunofluorescence microscopy of human epidermal keratinocytes after 48 h on a three-dimensional polymer of intact collagen or fragmented collagen. E-cadherin expression is more diffuse in the cells on the intact collagen substrate. E-cadherin is localized to the cell surface to a greater extent on damaged collagen. Expression is especially prominent at sites of cell-to-cell contact (arrows). Lower panel: western blot for E-cadherin. Increased E-cadherin was detected in cells on damaged collagen. Values shown in the bar graph represent digitized bands in western blots from two separate experiments and represent means and ranges. Confocal microscopic analysis was done with three separate preparations
Preparation and degradation of collagen films on polystyrene culture dishes
Monomeric collagen (1-50 μg/well) was dried onto the surface of 24-well polystyrene culture dishes from dilute acid. The film of dried collagen was then either exposed to MMP-1 (7.5 ng/well for 18 h) or left as control. Additional wells were coated with collagen that had been denatured by heating to 60°C for 5 min (i.e., gelatin) prior to use. The wells were rinsed carefully with Ca2+-supplemented KBM and used.
Scanning electron microscopy
Intact and partially degraded collagen lattices were fixed by mixing with an equal volume of 4% Sorenson’s buffered glutaraldehyde. Post fixation in 1% osmium tetroxide buffered in s-collidine was followed by en bloc staining with uranyl acetate. Dehydration was in a graded series of ethanol, followed by critical point drying from absolute ethanol through liquid carbon dioxide. Specimens were then mounted on stubs and conductive-coated with gold in a dc sputter coater. Following this, specimens were examined using an ISI Super IIIA scanning electron microscope.
Human epidermal keratinocytes
Human epidermal keratinocytes were isolated from neonatal foreskin and maintained in Keratinocyte growth medium (KGM) (Lonza) as reported previously [23]. KGM consists of the same basal medium as KBM, but is supplemented with a mixture of growth factors, including EGF, insulin, and bovine pituitary extract. For some experiments, the HaCat line of immortalized human epidermal keratinocytes was used [5]. The immortalized keratinocytes were handled exactly as low-passage keratinocytes. For experiments, the keratinocytes were harvested from culture by exposure to trypsin/EDTA, washed, and resuspended in either KGM or Dulbecco’s modified minimal essential medium with 10% fetal bovine serum (DMEM-FBS). Cells were added to lattices of intact or fragmented collagen (5 × 104 cells/well). Using these cultures, three sets of assays were carried out.
Attachment and spreading
Cells were added at time zero. At various times over the following 4-h period, nonattached cells were removed and counted. Counting was done with an automated particle counter after verifying that the cells were in single cell suspension. The dishes were then flooded with 10% buffered formalin and the percentage of remaining cells that were spread was determined microscopically. For this, photographs were taken under phase-contrast microscopy and magnified 254×. Cell length and width were determined. Cells were determined to be spread when the ratio of the two dimensions was greater than 1.5:1.
Proliferation
Cells were added at time zero. After incubation for 3 days, cells were harvested by exposure to trypsin/EDTA and counted. Counting was done with an automated particle counter after verifying that the cells were in single cell suspension.
Coverage of the collagen surface
Cells were added at time zero. Daily over the following 6 days, the cultures were examined by phase-contrast microscopy and the percentage of the surface covered with cells was estimated. Five high-power fields were examined in duplicate wells for each data point and the average was determined from this. To confirm the reliability of visual estimation, representative areas were photographed and the percentage of the surface area covered with cells determined from the photographs. The two methods for determining surface coverage gave similar results. To compare surface coverage on intact versus fragmented collagen, we used the same formula as used with proliferation. At time zero, surface coverage was 10-15%.
Data from experiments with multiple groups were analyzed using one-way analysis of variance (ANOVA) followed by the Bonferroni post-test for selected pairs (GraphPad Prism 4.0 for Windows, GraphPad Software, San Diego, CA). For experiments in which there were only two groups, the Student t-test was used to assess statistical significance of the differences. Data were considered significant at P < 0.05.
Cytotoxicity and apoptosis assays
Cytotoxicity and apoptosis analysis was done by staining the cells with Annexin V-FITC and propidium iodide and analyzing stained cells via flow cytometry [1]. Briefly, cells were added to the surface of intact and fragmented collagen as described above. After 48 h, cells were harvested, washed twice with ice cold PBS, and then resuspended in 1× binding buffer (BD Pharmingen, San Diego, CA) at a concentration of 1 × 106 cells/ml. Two hundred microliters of the above cell suspension was transferred to 96-well V bottom plates, and 10 μl of Annexin V-FITC (BD Pharmingen, San Diego, CA) and 5 μl of propidium iodide (Invitrogen Molecular Probes, Carlsbad, CA) were added to the wells. After incubation for 15 min in the dark, samples were then analyzed by flow cytometry (LSR II, BD Biosciences, San Diego, CA). Data acquisition and analysis were done using BD FACSDiva software.
Confocal immunofluorescence microscopy
Lattices of intact and fragmented collagen were prepared in the normal manner using Lab Tek II chamber slides in place of the 24-well culture dishes. Keratinocytes were added to the chamber slides and incubated overnight. The next day, cultures were washed and then fixed with 4% formaldehyde for 20 min. After fixation, cells were washed 2× with wash buffer [0.05% Tween-20 in Dulbecco’s phosphate buffered saline (DPBS)], followed by permeabilization with 0.1% Triton X-100 for 10 min. Cells were again washed and then exposed to a blocking solution consisting of 1% bovine serum albumin in DPBS for 30 min. Next, cells were treated with a monoclonal antibody to E-cadherin or vinculin in blocking solution for 1 h. After three subsequent washing steps with DPBS (5 min each), cells were treated with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA) in blocking solution and incubated for 45 min. Following three additional washing steps, the cells were rinsed once with wash buffer, and coverslips mounted with Prolong Gold Anti-fade Medium containing DAPI (Invitrogen). Cells were examined with a Zeiss LSM 510 confocal microscope using a 63× (C-Apochr) NA = 1.2 water immersion objective lens. Laser excitation wavelengths included 364, 488, and 543 nm scanned in sequence by the line method.
Preparation of cell lysates and immunoblot analysis
Keratinocytes were plated at 3 × 105 cells/well in 6-well tissue culture dishes containing intact or fragmented collagen and allowed to attach overnight. The next day, cultures were washed and then lysed in 1× cell lysis buffer consisting of 20 mM Tris–HCl (pH 7.4), 2 mM sodium vanadate, 1.0 mM sodium fluoride, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 25 μg/ml each of aprotinin, leupeptin and pepstatin, and 2 mM EDTA and EGTA. Lysis was performed by adding 200 μl of lysis buffer to each well and incubating the plate on ice for 5 min. After incubation, cells were scraped and samples were sonicated. Then, the extracts were cleared by microcentrifugation at 14,000g for 15 min. Supernatants were collected and protein concentrations estimated using the BioRad DC protein assay kit (BioRad, Hercules, CA).
Western blotting for E-cadherin was carried out as described previously [3]. Briefly, samples were separated in SDS-PAGE under denaturing and reducing conditions and transferred to nitrocellulose membranes. After blocking with a 5% nonfat milk solution in Tris-buffered saline with 0.1% Tween (TTBS) at 4°C overnight, membranes were incubated for 1 h at room temperature with the desired antibody, diluted 1:1,000 in 5% nonfat milk/0.1% TTBS. Thereafter, the membranes were washed with TTBS and bound antibody detected using the Phototope-HRP western blot detection kit (Cell Signaling Technology, Inc., Danvers, MA). A Kodak 1000 X-OMAT processor was used to capture the positive images of the western blots; these positive images were scanned and digitized. The digitized images were quantitated using NIH image analysis software.
Results
Keratinocyte dysfunction on three-dimensional lattices of type I collagen fragmented by human skin organ culture fluid or by MMP-1
In the first series of experiments, three-dimensional collagen lattices were exposed to human skin organ culture fluid obtained as described in the “Materials and methods” section. Past studies have shown that such a culture fluid contains a high level of active MMP-1 [24]. In parallel, collagen lattices were exposed to varying amounts of purified human fibroblast MMP-1. Both the purified MMP-1 and the skin organ culture fluid degraded the polymerized collagen, producing the expected ¾- and ¼-sized cleavage products (Fig. 1, upper panel). With concentrations of the purified enzyme above 50 ng/collagen lattice (1 mg of collagen) and with culture fluid concentrations above 100 μl/collagen lattice, the collagen lattices began to solubilize. However, with lower amounts, the lattices remained intact and appeared grossly indistinguishable from lattices not exposed to enzyme. By comparison with a standard curve obtained by converting 100% of the lattice into fragments and then resolving increasing amounts of the fragments by SDS-PAGE (middle panel), it was determined that 35–40% of the polymerized collagen could be converted into soluble fragments under conditions in which the lattice remained intact. In subsequent experiments, conditions were chosen that did not cause the polymerized matrix to solubilize but resulted in the conversion of 35–40% of the collagen into soluble fragments.
The lower panel of Fig. 1 presents scanning electron microscopic pictures of an intact collagen lattice and a similar lattice after exposure to skin organ culture fluid. Collagen fiber bundles are uniformly distributed throughout the intact lattice. After digestion, many thin and broken fiber bundles are detected. The collagen fiber bundles in the culture fluid-treated lattice are no longer uniformly distributed throughout the lattice. In some areas, the collagen is densely packed while in other areas there is a paucity of collagen. Similar changes were observed in collagen lattices exposed to purified MMP-1 alone.
Figure 2 demonstrates keratinocyte attachment and spreading on intact collagen lattices and lattices fragmented by exposure to purified MMP-1 or to human skin organ culture fluid. Cell attachment occurred equally well to both intact and fragmented collagen. Cell spreading, in contrast, was significantly slower and less complete on fragmented collagen.
Next, keratinocytes were plated on intact and fragmented collagen lattices and incubated for 6 days. The left-hand panel of Fig. 3 shows the percentage of the collagen surface covered by keratinocytes on Days 3 and 6. Coverage of the collagen surface was substantially inhibited on the substrate after fragmentation by either the purified enzyme or skin organ culture fluid. The right-hand panel shows the number of cells recovered from intact and fragmented collagen lattices on Day 3. The number of cells recovered from the fragmented collagen substrates was reduced by approximately 50% relative to control. The decrease in cell number primarily reflects reduced proliferation rather than increased cell death because, when cells were harvested and assessed with Annexin V-FITC and propidium iodide 1 day after plating, there were minimal differences in overall cytotoxicity (less than 5% overall) and apoptosis (less than 3% in all cases) between cells harvested from intact versus fragmented collagen substrates. Interestingly, when collagen lattices were exposed to higher enzyme concentrations such that solubilization occurred and the initially attached keratinocytes were essentially in suspension, apoptosis occurred rapidly (greater than 90% of the cells were nonviable after 4 h).
Correlation between collagen fragmentation and keratinocyte dysfunction
Table 1 summarizes experiments in which collagen fragmentation and keratinocyte function were assessed in parallel on collagen lattices treated with organ culture fluid and additional reagents. It can be seen that when the organ culture fluid was pre-incubated with SBTI (5 μg/50 μl of culture fluid), there was no reduction in collagen fragmentation. Concomitantly, keratinocyte coverage of the collagen surface was still reduced as compared to what was observed on intact collagen. In contrast, incubation of the organ culture fluid with TIMP-1 (150 ng/50 μl of culture fluid) suppressed collagen fragmentation and prevented the loss of keratinocyte function (i.e., keratinocytes behaved as on intact collagen). Pretreatment with a neutralizing antibody to MMP-1 (50 μg of antibody protein/50 μl of culture fluid) also suppressed collagen fragmentation and prevented the change in keratinocyte function, while a control IgG antibody had no effect. Taken together, these data strongly suggest that MMP-1 is the enzyme responsible for collagen damage in the organ culture fluid.
Table 1.
Collagen fragmentation and keratinocyte function
| Treatment | Collagen fragmentation (percent fragmented) | Percent of the collagen surface covered |
|---|---|---|
| EXP 1 (n = 5) | ||
| Control | <5 | 93 ± 7 |
| Skin culture fluid (alone) | 35 | 33 ± 10* |
| +SBTI | 40 | 30 ± 8* |
| +TIMP-1 | <5 | 88 ± 10 |
| +anti-MMP-1 | 10 | 85 ± 9 |
| +control IgG | 40 | 30 ± 14* |
| EXP 2 (n = 3-5) | ||
| Control | <5 | 90 ± 4 |
| MMP-8 | 45 | 42 ± 8* |
| Clostridium perfringens collagenase | ** | 27 ± 12* |
| MMP-9 | <5 | 90 ± 5 |
| MMP-2 | <5 | 94 ± 7 |
| Trypsin | <5 | 88 ± 7 |
| Elastase | <5 | 94 ± 10 |
In each experiment, collagen lattices were handled exactly as described in the “Materials and methods” section. At the end of the treatment phase, collagen fragmentation was assessed by SDS-PAGE and indicated by the presence of ¾- and ¼-sized fragments in the culture fluid. The percentage of the collagen digested in each condition was estimated based on quantification of SDS-PAGE-resolved ¾- and ¼-sized fragments and comparison with a standard curve. Keratinocytes were plated on the collagen lattices and the percentage of the surface covered on Day 6 assessed. Values are means and standard deviations except in the antibody experiment, in which the values represent means and ranges. Statistical significance was determined by ANOVA followed by the Bonferroni post-test for selected pairs
Indicates statistical difference from the control group at the P < 0.05 level
Since the ¾- and ¼-sized fragments do not build up in the presence of the bacterial collagenase, fragmentation was assumed based on the loss of intact collagen and altered cellular function on the enzyme-exposed collagen
Additional experiments were carried out in which a number of different enzymes were examined for ability to alter the collagen lattice structure and, concomitantly, for effects on keratinocyte function. These included two collagenases (human neutrophil MMP-8 and Clostridium perfringens collagenase) as well as MMP-2 and MMP-9 (an enzyme with gelatinolytic activity but lacking significant activity against type I collagen) and two serine proteinases, including bovine pancreatic trypsin and hog pancreatic elastase. With each enzyme preparation, polymerized collagen lattices were exposed to increasing amounts of enzyme until the collagen solubilized or until concentrations as high as 400 ng/lattice with MMP-2 and MMP-9 and 1 μg/lattice with the serine proteinases had been tested. Only the two enzymes with collagenolytic activity fragmented intact collagen lattices, and only those enzymes affected the ability of keratinocytes to cover the surface. With both enzymes, concentrations that produced collagen fragmentation were the same concentrations that inhibited keratinocyte coverage of the collagen surface (Table 1). Taken together, these data strongly suggest that while MMP-1 may be the major enzyme responsible for collagen damage in the organ culture fluid, other enzymes with collagenolytic activity have similar activity.
Altered expression of keratinocyte surface adhesion molecules on fragmented collagen
E-cadherin expression was assessed in keratinocytes on intact and fragmented collagen. The upper panel of Fig. 4 show results from confocal immunofluorescence microscopy. It can be seen from the left-hand photograph that in keratinocytes maintained on intact collagen, E-cadherin was diffusely expressed throughout the cell (similar to what is typically seen in rapidly proliferating basal keratinocytes on plastic). On damaged collagen (right-hand photograph), more of the E-cadherin was distributed around the periphery of the cell. Staining was especially prominent at sites of cell-to-cell contact (arrows). Consistent with these results, western blotting of whole cell extracts (lower panel of Fig. 4) demonstrated up-regulation of E-cadherin in cells on the fragmented matrix. In addition to assessing E-cadherin expression in whole cell extracts, we also used western blotting to assess E-cadherin released into the culture fluid. This was done to determine if shedding might account for the differences noted. As expected, there was no evidence of E-cadherin shedding into the culture fluid under either condition, i.e., there was no detectable E-cadherin in the culture fluid by western blotting (not shown). Finally, keratinocytes were exposed to MMP-1 for 18 h in monolayer culture. At the end of the incubation period, cells and media were assessed by western blotting for E-cadherin. No effects of MMP-1 were noted (not shown).
Keratinocytes on intact and fragmented collagen were also stained with an antibody to vinculin as a marker of focal adhesions (Fig. 5). Vinculin was broadly distributed throughout the cell layer in keratinocytes on intact collagen. Staining was observed at a distance from the nucleus, indicating that the cells were well spread. In contrast, on fragmented collagen, vinculin staining was predominantly observed in close association with the body of the cell. Western blotting showed no apparent change in the total amount of vinculin between the two conditions (lower panel). The cells were concomitantly stained with phalloidin as a marker for actin. Phalloidin staining was closely associated with cell bodies on both intact and damaged collagen. There were few stress fibers (typical in epithelial cells) and no differences between the two substrates.
Fig. 5.
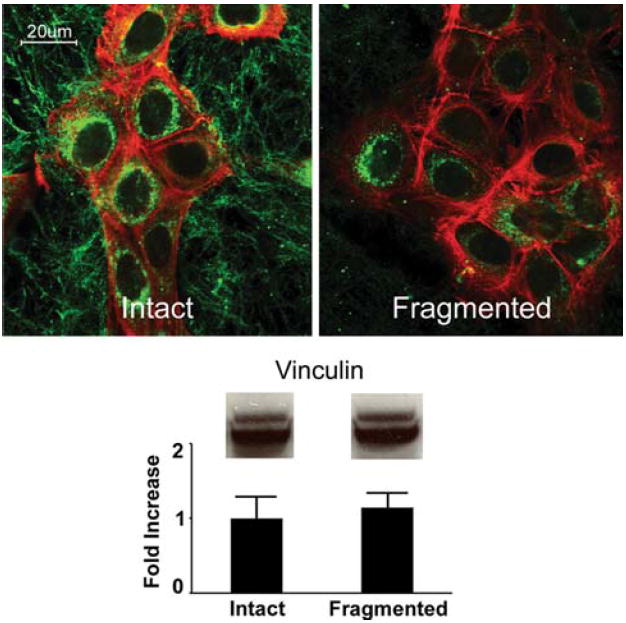
Vinculin and actin expression in keratinocytes on intact and fragmented collagen. Upper panels: confocal immunofluorescence microscopy of human epidermal keratinocytes after 48 h on a three-dimensional polymer of intact collagen or fragmented collagen. Vinculin expression (green fluorescence) is widespread and distant from cell bodies in keratinocytes on intact collagen (evidence that the cells are well spread and in contact with the substrate over a wide area). On fragmented collagen, distribution is predominantly observed in the immediate vicinity of the cell bodies. Actin expression (red fluorescence) is observed in the vicinity of the cell bodies on both substrates. Lower panel: western blot for vinculin. A similar level of vinculin is observed on both substrates. Values shown in the bar graph represent digitized vinculin bands in western blots from two separate experiments and represent means and ranges. Confocal microscopic analysis was done with three separate preparations
Keratinocyte function on plastic culture dishes coated with intact collagen or fragmented collagen
In a final set of studies, wells of a 24-well dish were coated with a film of type I collagen (20 μg/well). After deposition, the collagen in some wells was subjected to digestion with MMP-1 (7.5 ng/well for 18 h) while other wells were exposed to buffer alone. Following this, keratinocytes were added to the collagen films and the percentage of attached and spread cells determined at various times after plating. In contrast to what was seen in three-dimensional lattices, there was no significant difference in either attachment or spreading between the two substrates. Coverage of the collagen surface was also assessed. As with the other parameters, there was no difference between the two substrates (not shown).
Discussion
Superficial wounds in healthy skin are expected to heal rapidly and without incident. Keratinocytes at the wound margin respond to contact with the exposed dermal matrix by adhering to the collagen (primarily through the α2β1 integrin) and migrating across the exposed surface [9, 11, 13]. As the cells at the leading front migrate over the wound surface, they upregulate MMP-1 and cleave the collagen substrate beneath [16]. Keratinocytes immediately behind the leading front proliferate to fill in the space. Intrinsic and extrinsic factors that reduce keratinocyte proliferation and motility [4, 6, 10, 21, 30] are associated with impaired wound healing. The present study suggests that damage to the underlying connective tissue is another factor that contributes to epithelial dysfunction and failure to effectively cover the wound surface. Our findings may be relevant to understanding why superficial wounds often do not heal efficiently in aged/photoaged skin. While it is difficult to estimate precisely the loss of intact collagen in vivo, several past studies have utilized biochemical, immunohistochemical, and ultrastructural approaches to show convincingly that extensive collagen damage exists in aged/photoaged skin. While biochemical estimates range from 20% to 40% loss, immunohistochemical and ultrastructural approaches suggest that in some areas, virtually none of the remaining collagen is intact [12, 14, 15, 17, 19]. Our focus on the connective tissue is not meant to imply that other intrinsic and extrinsic factors do not also play a role in poor wound healing. Rather, the studies presented here simply point out the essential role of the connective tissue matrix as a regulator of keratinocyte function, regardless of the physiological state of the keratinocytes in the associated epithelium. Altered behavior of keratinocytes on fragmented collagen is not unique to these cells. Recent studies have shown that collagen damage also has a profound effect on fibroblast function [26, 27] and endothelial cell function [28].
How pre-existing fragmentation of the underlying collagen negatively impacts keratinocyte function is not fully understood, but we speculate that a loss of structural rigidity in the substrate and a corresponding decrease in mechanical stress imparted on the cells may be critical. This is based on the finding that while cell attachment occurred equally well to intact and fragmented collagen in the three-dimensional lattices, spreading was slower on the fragmented matrix. This was not observed when attachment and spreading were examined on intact versus MMP-1-exposed collagen when the substrate consisted of a collagen film coated onto the surface of a polystyrene culture dish. Our assumption is that if the enzyme-fragmented collagen fibrils are not “anchored,” the cells will not be able to generate the traction needed for movement over the substrate.
Of interest to this discussion are the observations by Pilcher et al. [16]. These investigators showed that elaboration of MMP-1 and fragmentation of the dermal matrix were critical for keratinocyte migration across the collagen surface during wound repair. Once the substrate was fragmented, adhesion to the fragmented collagen was reduced relative to intact collagen. Presumably, differences in cell–substrate adhesion favored continued contact with the intact collagen and facilitated migration over it. One might assume that if the collagen were substantially fragmented to begin with, the same factors would come into play, and reduction in migration would occur.
As an alternative hypothesis, the reduction in cell growth and inability to cover the collagen surface may relate directly to keratinocyte differentiation on the fragmented collagen. Previous studies have demonstrated that cell shape change (from well-spread to spherical) triggers terminal differentiation in human epidermal keratinocytes [29]. By preventing cell spreading, the damaged matrix may cause the cells to prematurely initiate events that lead to differentiation. Increased E-cadherin expression in keratinocytes on the damaged collagen is consistent with this. Of interest in this regard, past studies have shown that epithelial cell differentiation leads to β-catenin sequestration at the cell surface along with E-cadherin. As a result, there is less intranuclear β-catenin and a decrease in Wnt signaling [2, 7]. Since Wnt signaling is an important growth-regulating pathway in epithelial cells, it is likely that keratinocyte growth would be inhibited under these conditions.
A final issue concerns the enzymes that are responsible for the collagen damage leading to keratinocyte dysfunction. While our focus was primarily on MMP-1, this enzyme does not appear to be unique in its ability to damage collagen in such a manner that keratinocyte behavior is negatively impacted. One might suggest, in fact, that while MMP-1 appears to be the responsible enzyme in aged or photoaged skin, MMP-8 could be more important in situations where there is excessive inflammation. Excessive inflammation is well known to retard wound healing. Likewise, bacterial collagenolytic enzymes could play a role in situations where there is infection [18, 20].
In summary, the studies described here demonstrate that when keratinocytes are maintained on a three-dimensional collagen lattice, cell function is significantly affected by the condition of the collagen in the lattice. When the collagen is fragmented by exposure to collagenolytic enzymes, the keratinocytes fail to cover the surface of the collagen and instead grow as dense colonies of isolated (differentiated) cells. We suggest that impaired re-epithelialization, seen in a variety of conditions in which connective tissue damage also occurs, may reflect a similar mechanism.
Acknowledgments
This study was supported in part by grants GM077724 and GM080779 from the United Stated Public Health Service.
Contributor Information
James Varani, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA, varani@umich.edu.
Patricia Perone, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA.
Monica O’Brien Deming, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA.
Roscoe L. Warner, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA
Muhammad N. Aslam, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA
Narasimharao Bhagavathula, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA.
Michael K. Dame, Department of Pathology, University of Michigan Medical School, 1301 Catherine Street, SPC 5602, Ann Arbor, MI 48109, USA
John J. Voorhees, Department of Dermatology, University of Michigan Medical School, Ann Arbor, MI 48109, USA
References
- 1.Aubry JP, Blaecke A, Lecoanet-Henchoz S, Jeannin P, Herbault N, Caron G, Moine V, Bonnefoy JY. Annexin V used for measuring apoptosis in the early events of cellular cytotoxicity. Cytometry. 1999;37(3):197–204. doi: 10.1002/(sici)1097-0320(19991101)37:3<197::aid-cyto6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 2.Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 3.Bhagavathula N, Hanosh AW, Nerusu KC, Appelman H, Chakrabarty S, Varani J. Regulation of E-cadherin and beta-catenin by Ca2+ in colon carcinoma is dependent on calcium-sensing receptor expression and function. Int J Cancer. 2007;121(7):1455–1462. doi: 10.1002/ijc.22858. [DOI] [PubMed] [Google Scholar]
- 4.Bosset S, Bonnet-Duquennoy M, Barré P, Chalon A, Lazou K, Kurfurst R, Bonté F, Schnébert S, Disant F, Le Varlet B, Nicolas JF. Decreased expression of keratinocyte beta1 integrins in chronically sun-exposed skin in vivo. Br J Dermatol. 2003;148(4):770–778. doi: 10.1046/j.1365-2133.2003.05159.x. [DOI] [PubMed] [Google Scholar]
- 5.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106(3):761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilchrest BA, Garmyn M, Yaar M. Aging and photoaging affect gene expression in cultured human keratinocytes. Arch Dermatol. 1994;130(1):82–86. [PubMed] [Google Scholar]
- 7.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153(5):1049–1060. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13(5):264–269. doi: 10.1016/s0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 9.Guo M, Toda K, Grinnell F. Activation of human keratinocyte migration on type I collagen and fibronectin. J Cell Sci. 1990;96(Pt 2):197–205. doi: 10.1242/jcs.96.2.197. [DOI] [PubMed] [Google Scholar]
- 10.Haratake A, Uchida Y, Mimura K, Elias PM, Holleran WM. Intrinsically aged epidermis displays diminished UVB-induced alterations in barrier function associated with decreased proliferation. J Invest Dermatol. 1997;108(3):319–323. doi: 10.1111/1523-1747.ep12286474. [DOI] [PubMed] [Google Scholar]
- 11.Harrison CA, Heaton MJ, Layton CM, MacNeil S. Use of an in vitro model of tissue-engineered skin to study keratinocyte attachment and migration in the process of reepithelialization. Wound Repair Regen. 2006;14(2):203–209. doi: 10.1111/j.1743-6109.2006.00111.x. [DOI] [PubMed] [Google Scholar]
- 12.Kligman LH, Schwartz E, Sapadin AN, Kligman AM. Collagen loss in photoaged human skin is overestimated by histochemistry. Photodermatol Photoimmunol Photomed. 2000;16(5):224–228. doi: 10.1034/j.1600-0781.2000.160506.x. [DOI] [PubMed] [Google Scholar]
- 13.Langholz O, Röckel D, Mauch C, Kozlowska E, Bank I, Krieg T, Eckes B. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by alpha 1 beta 1 and alpha 2 beta 1 integrins. J Cell Biol. 1995;131(6 Pt 2):1903–1915. doi: 10.1083/jcb.131.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavker RM. Structural alterations in exposed and unexposed aged skin. J Invest Dermatol. 1979;73(1):59–66. doi: 10.1111/1523-1747.ep12532763. [DOI] [PubMed] [Google Scholar]
- 15.Lavker RM. Cutaneous aging: chronologic versus photoaging. In: Gilchrest BA, editor. Photoaging. Blackwell Science; Cambridge, MA: 1995. pp. 123–135. [Google Scholar]
- 16.Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, Parks WC. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol. 1997;137(6):1445–1457. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz E, Cruickshank FA, Christensen CC, Perlish JS, Lebwohl M. Collagen alterations in chronically sun-damaged human skin. Photochem Photobiol. 1993;58(6):841–844. doi: 10.1111/j.1751-1097.1993.tb04981.x. [DOI] [PubMed] [Google Scholar]
- 18.Singer AJ, Clark RA. Cutaneous wound healing. New Engl J Med. 1999;341(10):738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 19.Smith JG, Jr, Davidson EA, Sams WM, Jr, Clark RD. Alterations in human dermal connective tissue with age and chronic sun damage. J Invest Dermatol. 1962;39:347–350. doi: 10.1038/jid.1962.122. [DOI] [PubMed] [Google Scholar]
- 20.Stadelmann WK, Digenis AG, Tobin GR. Impediments to wound healing. Am J Surg. 1998;176(2A Suppl):39S–47S. doi: 10.1016/s0002-9610(98)00184-6. [DOI] [PubMed] [Google Scholar]
- 21.Stanulis-Praeger BM, Gilchrest BA. Growth factor responsiveness declines during adulthood for human skin-derived cells. Mech Ageing Dev. 1986;35(2):185–198. doi: 10.1016/0047-6374(86)90009-6. [DOI] [PubMed] [Google Scholar]
- 22.Uitto J. Collagen. In: Fitzpatrick TB, Eisen AZ, Wolff K, Freedberg IM, Austen KF, editors. Dermatology in general medicine. 4. McGraw-Hill; New York: 1993. pp. 299–315. [Google Scholar]
- 23.Varani J, Perone P, Griffiths CE, Inman DR, Fligiel SE, Voorhees JJ. All-trans retinoic acid (RA) stimulates events in organ-cultured human skin that underlie repair. Adult skin from sun-protected and sun-exposed sites responds in an identical manner to RA while neonatal foreskin responds differently. J Clin Invest. 1994;94(5):1747–1756. doi: 10.1172/JCI117522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varani J, Hattori Y, Chi Y, Schmidt T, Perone P, Zeigler ME, Fader DJ, Johnson TM. Collagenolytic and gelatinolytic matrix metalloproteinases and their inhibitors in basal cell carcinoma of skin: comparison with normal skin. Br J Cancer. 2000;82(3):657–665. doi: 10.1054/bjoc.1999.0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varani J, Spearman D, Perone P, Fligiel SE, Datta SC, Wang ZQ, Shao Y, Kang S, Fisher GJ, Voorhees JJ. Inhibition of type I procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degraded collagen in vitro. Am J Pathol. 2001;158(3):931–942. doi: 10.1016/S0002-9440(10)64040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varani J, Schuger L, Dame MK, Leonard C, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Reduced fibroblast interaction with intact collagen as a mechanism for depressed collagen synthesis in photodamaged skin. J Invest Dermatol. 2004;122(6):1471–1479. doi: 10.1111/j.0022-202X.2004.22614.x. [DOI] [PubMed] [Google Scholar]
- 27.Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168(6):1861–1868. doi: 10.2353/ajpath.2006.051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varani J, Perone P, Warner RL, Dame MK, Kang S, Fisher GJ, Voorhees JJ. Vascular tube formation on matrix metalloproteinase- 1-damaged collagen. Br J Cancer. 2008;98(10):1646–1652. doi: 10.1038/sj.bjc.6604357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watt FM, Jordan PW, O’Neill CH. Cell shape controls terminal differentiation of human epidermal keratinocytes. Proc Natl Acad Sci USA. 1988;85(15):5576–5580. doi: 10.1073/pnas.85.15.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia YP, Zhao Y, Tyrone JW, Chen A, Mustoe TA. Differential activation of migration by hypoxia in keratinocytes isolated from donors of increasing age: implication for chronic wounds in the elderly. J Invest Dermatol. 2001;116(1):50–56. doi: 10.1046/j.1523-1747.2001.00209.x. [DOI] [PubMed] [Google Scholar]