

Abstract
Spectroscopic investigations of macromolecules generally attempt to interpret the measured spectra in terms of the summed contributions of the different molecular fragments. This is the basis of the local mode approximation in vibrational spectroscopy. In the case of resonance Raman spectroscopy independent contributions of molecular fragments require both a local mode-like behavior and the uncoupled electronic transitions. Here we show that the deep UV resonance Raman spectra of aqueous solution phase oligoglycines show independent peptide bond molecular fragment contributions indicating that peptide bonds electronic transitions and vibrational modes are uncoupled. We utilize this result to separately determine the conformational distributions of the internal and penultimate peptide bonds of oligoglycines. Our data indicate that in aqueous solution the oligoglycine terminal residues populate conformations similar to those found in crystals (31-helices and β-strands), but with a broader distribution, while the internal peptide bond conformations are centered around the 31-helix Ramachandran angles.
Keywords: UV Resonance Raman spectroscopy, peptides conformation analysis, oligoglycine, vibrational/electronic coupling
Spectroscopic methodologies are of a great importance in chemical analysis and for the determination of molecular structure. Interpretations of spectra generally use reductionist strategies whereby the spectrum of a macromolecule is first interpreted as the sum of the contributions of the individual molecular fragments. This first order analysis is then reanalyzed to take into account higher order phenomena such as the coupling of molecular dynamics between molecular fragments and the environment. For example, in UV resonance Raman (UVRR) spectra of polypeptides the vibrations could be localized within the individual peptide bond (PB), such that the PBs independently contribute. This would lead to spectra that are easily analyzed, where each PB vibration is not context dependent. Alternatively, spectra of polypeptides may result from coupled motion of the backbone atoms of adjacent PB.1 This would lead to vibrational modes which depend upon their adjacent PB secondary structures.
Our previous study of a mainly ala-peptide in H2O/D2O mixtures indicated that the spectra of the partially deuterated chains could be roughly modeled (except for the amide I band) as a statistically weighted sum of deuterated and protonated independent segments.2,3 This could only occur if the amide vibrations were essentially localized within the individual PB.
Here we show that the deep UV resonance Raman spectra of aqueous solution phase oligoglycines show independent peptide bond molecular fragment contributions; the peptide bonds independently resonance Raman scatter. The peptide bond electronic transitions and vibrational modes are uncoupled. We utilize this result to separately determine the conformational distributions of the internal and penultimate peptide bonds of oligoglycines.
Vibrational and Electronic Coupling
Fig. 1 compares the 204 nm UVRR difference spectrum between gly6 and gly5 to that of gly3. If there were negligible coupling between the PB vibrations the gly3 spectrum should closely approximate the two terminal PB spectra of oligopeptides, while the gly6-gly5 spectrum should approximate a single internal PB vibration.
Figure 1.
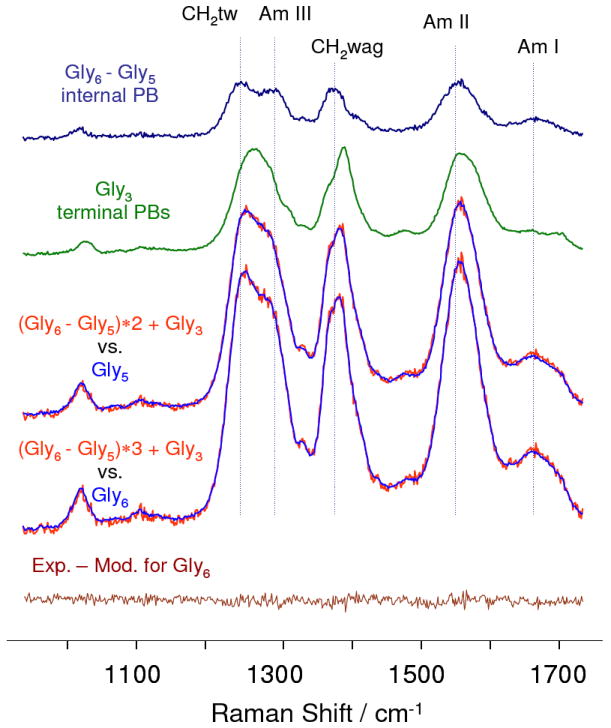
204 nm UVRR spectra of oligoglycines in aqueous solutions containing 0.5 M LiClO4, at neutral pH. The gly6-gly5 difference spectrum approximates the spectrum of an internal peptide bond, while that of gly3 approximates that of the two terminal peptide bonds. This is demonstrated by the fact that the summation of these spectra accurately model gly5 and gly6 spectra as demonstrated by the lack of features in the bottom gly6 difference spectrum between the experimental and modeled spectra. All the UVRRS were scaled relative to the internal standard, perchlorate 932 cm-1 band (not shown).
Summation of the gly6-gly5 difference spectra and the gly3 spectra essentially perfectly models the UVRRS of gly5 and gly6 (Fig. 1). The differences are below shot noise levels. This result indicates the lack of coupling of peptide bond vibrations between adjacent PB. The ability to accurately model the spectral intensities indicates that couplings between these vibrations and the resonant electronic transitions are localized within each PB.
Internal PB Conformation Preferences
The gly6-gly5 spectrum differs significantly from that of gly3 with a clear doublet spanning the CH2 twist/Am III spectral region, while gly3 shows a single complex bandshape. The CH2 wagging, as well as the Am II and Am I bands of gly6-gly5, also differ from those of gly3 which may imply different structural preferences of the internal and terminal residues.
In a separate study reported elsewhere4, we compared the internal residue oligoglycine UVRR spectra in solution to that of solid polyglycines of known conformations. These studies indicate that the internal PBs of oligoglycines occur as a broad ensemble of conformations centered around the 31-extended helix conformation.
Penultimate PB Conformation Preferences
The UVRR spectrum of gly3 closely approximates the spectrum of the terminal PB of longer oligoglycines. Gly3 shows a complex amide III band shape suggesting that multiple conformational states of the peptide are populated in solution. Subtraction of the spectrum of an internal PB (gly6-gly5) from the spectrum of gly3 produces a spectrum which is very similar to that of gly2 in water (Fig. 2) which should approximate the spectrum of the average of the two penultimate oligoglycine PBs.
Figure 2.
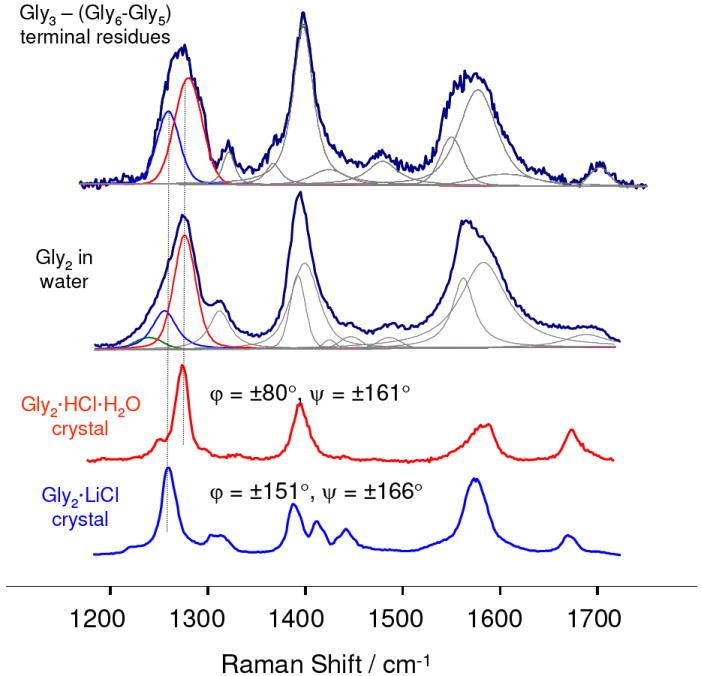
UVRR difference spectrum gly3-(gly6-gly5) approximates the spectrum of the two terminal residues of oligoglycines in solution. Also shown is the spectra of gly2 in solution and two gly2 crystal samples of known structure: Gly2·HCl·H2O (φ = ± 80°,ψ = ± 161°) and Gly2·LiCl (φ = ± 154°,ψ = ± 168°).5,6 The complex amide III bandshapes of the solution samples can be well modeled with the amide III bands of these crystal gly2 derivatives.
We characterized the conformations of the gly3-(gly6-gly5) PB and gly2 in solution by comparing their complex amide III bands to those of the amide III bands of crystalline gly2 derivatives of known structures.
The solution UVRR spectra of gly3-(gly6-gly5) and gly2 show amide III spectral features similar to those in the spectra of gly2 crystals with known φ; and ψ angles (Fig. 2). These frequencies and band widths are listed in Table 1.
Table 1.
Frequencies (ν) and bandwidths (w) of UVRR amide bands in the gly3-(gly6-gly5) difference spectrum, gly2 in solution and gly2 crystals
| Gly3-(Gly6-Gly5) difference spectrum | Gly2 in solution | Gly2·HCl·H2O crystals | Gly2·LiCl crystals | |
|---|---|---|---|---|
| ν (w) /cm-1 | ν (w) /cm-1 | ν (w) /cm-1 | ν (w) /cm-1 | |
| Am III | 1261(27), 1281(33) | 1245(34),1261(29), 1281(26), | 1286(15) | 1267(18) |
| CH2 wag | 1400(25) | 1395(18), 1402(34) | 1404(17) | 1393(16) |
| Am II | 1553(27), 1580(46) | 1562(27), 1584(57) | 1580(38), 1593(20) | 1575(29) |
| Am I | 1705(24) | 1686(51) | 1678(16) | 1670(17) |
Crystalline Gly2·HCl·H2O have φ = ± 80° and ψ = ± 161° dihedral angles that are close to those of a 31-extended helix.5 Crystals of Gly2·LiCl have φ = ± 154°,ψ = ± 168°, which are close to that of a β-strand.6 The amide III band of the β-strand gly2 is 19 cm-1 downshifted compare to that of the 31-extended helix indicating significant conformational sensitivity (Table 1). The amide III frequencies also depend upon the hydrogen bonding to the PB N-H since N-H bending significantly contributes to the amide III vibration. However both crystal structures have N-H hydrogen bonded to the Cl− which implies similar HB strengths.
The amide III region of the gly3-(gly6-gly5) difference spectrum, as well as, that of the gly2 spectrum have a complex shape indicating significant conformational inhomogeneity of the terminal residues.
We can estimate the conformational distribution of the solution oligoglycine terminal residues by modeling the amide III region of the gly3-(gly6-gly5) difference spectrum with the well defined amide III bands of crystal gly2 of known structures. Assuming that the integrated intensities of the amide III bands are proportional to the populations of corresponding conformations in solution, we estimate that in solution ~ 60% of terminal residues are in a 31-extended helix-like conformation while ~ 40% are in extended β-strand-like conformation. Modeling of the gly2 in solution gives similar results (~ 65% in a 31-extended helix-like conformation and ~ 35% in an extended β-strand-like conformation).
As discussed above the internal residues of oligoglycines in solution mainly populate a 31-extended helix-like conformation. The higher preferences of the terminal residues for a β-strand conformation may result from the favorable interaction between terminal -NH3 + and the adjacent PB carbonyl, as well as between the terminal -COO− and the adjacent PB N-H, which are maximized for the completely extended conformation.
The amide III bands of the crystalline samples show full widths at half height of ~15 - 18 cm-1 which is likely to be their homogeneous amide III bandwidth. The corresponding solution amide III bands show almost two-fold broader bandwidths of ~ 26 - 34 cm-1 which indicate that the terminal residues of oligoglycines in solution populate a broad ensemble of extended conformations with some preference for the 31-helix.
In conclusion, the UV resonance Raman spectrum of oligoglycines in aqueous solution can be accurately modeled as a linear sum of the spectra of their internal and terminal peptide bonds which indicates the absence of the vibrational and electronic transition coupling between adjacent PBs in polypeptides in solution. This result significantly simplifies polypeptide UVRR spectral analysis. The UVRR spectra of the oligoglycine internal PBs significantly differ from those of the terminal PBs which implies different structural preferences. Our data indicates that in aqueous solution oligoglycine terminal residues populate a broad range of extended conformations between 31-helices and β-strands while the internal PBs conformation are centered around the Ramachandran angles of the 31-helix.
Experimental Methods
Materials
Gly2, gly3, gly5 and gly6 were purchased from Bachem (King of Prussia, PA) and used as received. Gly2 hydrochloride (Sigma Inc.) was recrystallized from water to obtain Gly2·HCl·H2O crystals. Lithium chloride (T.J Baker Inc.) was used for cocrystallization with gly2 to grow Gly2·LiCl crystals. Lithium perchlorate (Fisher Scientific Inc.) was used to increase the solubility of gly5 and gly6 in water and as an internal Raman intensity standard.
Raman measurements
For solution samples we used 204 nm Raman excitation near the maximum absorbance of the peptide bond π→π* transition. The third harmonic of Nd:YAG laser operating at 100 Hz was anti-Stokes Raman shifted in hydrogen to 204 nm (fifth anti-Stokes) . The peptides were studied in a flow-stream to avoid any contribution from photochemical degradation processes. Scattered light was dispersed by a double spectrometer and was detected by a Princeton Instruments Spec- 10:400B CCD camera (Roper Scientific). A detailed description of the instrumentation is given elsewhere.7 For solid samples we use pre-resonance 229 nm excitation to minimize sample photodegradation. Raman measurements were performed using an intracavity doubled Ar-ion Laser (Coherent Inc.) A custom made, rotating metal cell was used for the solid powder samples to avoid light-induced sample degradation. The crystal powder was pressed into a circular groove in the rotating metal cylinder Typical accumulation times were less then 1 min.
Acknowledgments
We thank Dr. Steven Geib for x-ray crystal structure determinations and Bhavya Sharma for help in preparing this letter. This work was supported by NIH, Grant 5R01EB002053.
References
- 1.Wang Y, Spiro TG. Vibrational and Electronic Couplings in Ultraviolet Resonance Raman Spectra of Cyclic Peptides. Biophys Chem. 2003;105:461–470. doi: 10.1016/s0301-4622(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 2.Mikhonin AV, Asher SA. Uncoupled Peptide Bond Vibrations in alpha-Helical and Polyproline II Conformations of Polyalanine Peptides. J Phys Chem B. 2005;109:3047–3052. doi: 10.1021/jp0460442. [DOI] [PubMed] [Google Scholar]
- 3.Mix G, Schweitzer-Stenner R, Asher SA. Uncoupled Adjacent Amide Vibrations in Small Peptides. J Am Chem Soc. 2000;122:9028–9029. [Google Scholar]
- 4.Bykov SV, Asher SA. Conformation of Polyglycine in Solution. In preparation to be submitted. [Google Scholar]
- 5.Parthasarathy R. Crystal Structure of Glycylglycine Hydrochloride. Acta Crystallogr Sect B. 1969;25:509–518. [Google Scholar]
- 6.Mueller G, Maier G-M, Lutz M. Lithium Coordination to Amino Acids and Peptides. Synthesis, Spectroscopic Characterization and Structure Determination of Lithium Complexes of Neutral and Anionic Glycine and Diglycine. Inorg Chim Acta. 1994;218:121–131. [Google Scholar]
- 7.Bykov S, Lednev I, Ianoul A, Mikhonin A, Munro C, Asher SA. Steady-State and Transient Ultraviolet Resonance Raman Spectrometer for the 193-270 nm Spectral Region. Appl Spectrosc. 2005;59:1541–1552. doi: 10.1366/000370205775142511. [DOI] [PubMed] [Google Scholar]