

Abstract
A simple and modular synthetic approach, based on miniemulsion polymerization, has been developed for the fabrication of composite polymer-metal nanoparticle materials. The procedure produces well-defined composite structures consisting of gold, silver or MnFe2O4 nanoparticles (∼10 nm in diameter) encapsulated within larger spherical nanoparticles of poly(divinylbenzene) (∼100 nm in diameter). This methodology readily permits the incorporation of multiple metal domains into a single polymeric particle, while still preserving the useful optical and magnetic properties of the metal nanoparticles. The morphology of the composite particles is retained upon increasing the inorganic content, and also upon redispersion in organic solvents. Finally, the ability to tailor the surface chemistry of the composite nanoparticles and incorporate steric stabilizing groups using simple thiol-ene chemistry is demonstrated.
Keywords: gold nanoparticles, silver nanoparticles, gold nanorods, composite nanoparticles, hybrid nanoparticles, miniemulsion polymerization, thiol-ene
Introduction
Metallic nanostructures are presently finding application across a broad range of scientific disciplines, due to their remarkable optical, electronic and catalytic properties.1 Among these structures, gold, silver and magnetic nanoparticles are particularly well studied, and have already shown promise for use in a range of device applications2 and in medical diagnosis and treatment.3 However, the practical use of metal nanoparticles is strongly governed by their sensitivity to environmental factors such as solvent composition, salt concentration and temperature. As such, there is a continuing need for reliable methods to modify and deliver metal particles with improved solvent affinity, stability against aggregation, and tailorable functionality. This is commonly achieved by preparing composite nanoparticles, which combine metallic and non-metallic (e.g., polymeric4) components.
A survey of the literature reveals a variety of methods for generating composite nanoparticles, each with particular advantages and limitations. A straightforward and widely-used approach has been to attach ligands (either small molecules or polymers) to the nanoparticle surface. For gold and silver particles in particular, a vast number of nanoparticle-ligand systems has been reported, based on various groups that are capable of binding to metal surfaces: amines,5, 6 thiols,7 disulfides,8, 9 thioethers,10, 11 thioesters, 12 thiocarbonates,12 and thiocarbamates.13 Commonly, these ligands are incorporated during the metal nanoparticle synthesis or at the surface of pre-formed nanoparticles by ligand exchange.14 While the simplicity of this approach is highly appealing, the ligated metal nanoparticles may remain susceptible to ligand displacement and other reactions with solvated species (e.g., etchants). Also, by definition, this approach is limited to producing functionalized nanostructures containing only a single metal nanoparticle domain—a feature which may not be optimal for certain applications, such as sensing based on surface enhanced Raman spectroscopy (SERS) or for multi-modal, dual functional materials.15
A more robust protecting layer may be created by silica coating of inorganic nanoparticles, using either seeded-growth16 or microemulsion methods.17 A particular advantage of this approach is that, in addition to its stabilizing function, the silica shell of these composite nanoparticles is also highly amenable to attachment of surface functional groups through siloxy coupling reactions.18 However, these procedures are typically time-consuming and require careful control over pH, solvent composition and concentration of silica precursors at each stage of the process, in order to obtain the desired composite nanoparticle product. Depending on the thickness of the cross-linked silica shell, these core-shell nanostructures can be porous, allowing infiltration by small molecules, or for thicker systems can provide structural isolation.19
In contrast to the ligation and silica coating approaches, in which the metal nanoparticle core acts as a template for the composite nanoparticle structure, a non-metallic particle can also act as a template for incorporation of metallic domains. Straightforward examples include the attachment of metal nanoparticles to the surface or pores of a solid polymer or silica seed particle—either by adsorption of pre-formed metal particles,20 or by in situ reduction of a metal salt.21 In a related strategy, Crooks has demonstrated that well-defined nanoparticles can be incorporated into the internal free volume of dendrimers by in-situ reduction of ions chelated within the dendrimer.22
A promising alternative to solid template approaches is the formation of composite nanoparticles in solution through self-assembly or dispersion processes. Several recent reports have described composite nanoparticles driven by co-assembly of amphiphilic block copolymers into micelles or vesicles.23 Taton and co-workers have further demonstrated that block copolymer self-assembly, followed by chemical cross-linking, yields composite nanoparticles that are dispersible in a variety of solvent media and impermeable to metal etching agents.24-26
Another dispersion-based approach for composite nanoparticle formation is miniemulsion polymerization.27, 28 An emulsion, consisting of water and immiscible monomer droplets containing the inorganic material, is polymerized to yield polymer-inorganic composite nanoparticles. This simple method, which uses predominantly commercially available reagents, is appealing when compared with block copolymer self-assembly methods. Various literature reports describe the use of miniemulsion polymerization to prepare composite particles based on titania,29-31 calcium carbonate,32 magnetite,33-37 carbon black,38 alumina,39 silica,40, 41 zinc oxide42 and yttrium oxysulfide.43 However, in these cases the composite particles formed were subject to a variety of challenges: low inorganic content, inhomogeneous distribution of inorganic material, incomplete encapsulation of inorganic material, and large populations of pure (non-composite) polymer particles. Moreover, to our knowledge, there have been no literature reports of successful miniemulsion polymerization incorporating gold or silver nanoparticles—both important materials for use in emerging nanoparticle applications.
This work aims to develop a versatile synthetic platform which combines many of the desired features for composite nanoparticles and overcomes the above challenges to give a methodology that is robust, reliable and scalable; requires only a limited number of steps with little or no purification; and allows facile tailoring of composite functionality and properties. In a previous Communication,44 we reported the initial development of a new synthetic procedure for composite nanoparticles, based on miniemulsion polymerization, which is depicted schematically in Figure 1. First, metal nanoparticles are prepared and grafted with short, hydrophobic polymer chains, which render the nanoparticles dispersible in organic solvents. The grafted metal particles are then dispersed in a suitable monomer for miniemulsion polymerization, and the nanoparticle/monomer mixture is emulsified with an aqueous solution of surfactant and free radical initiator. Polymerization within the miniemulsion droplets yields composite nanostructures comprising metal domains encapsulated within larger polymer nanoparticles.
Figure 1.

Schematic description of composite nanoparticle synthesis: (A) polystyrene-grafted gold nanoparticles are dispersed in monomer (divinylbenzene) and emulsified with aqueous surfactant solution (B) to form small, uniform monomer droplets (C), which are then polymerized to yield composite polymer-gold nanoparticles (D); hydrophilic polymer chains (poly(ethylene glycol)) are grafted to the surface of composite nanoparticles by thiol-ene chemistry (E).
A key criterion for tailoring composite nanoparticles to a wide range of applications is the ability to conveniently modify all aspects of their structure from the size, shape and nature of the inorganic building block to the surface chemistry of the polymeric nanoparticle. To this end, the present synthetic strategy employs thiol-ene chemistry which has emerged as a powerful tool for the modification of polymeric and dendritic materials45 due to its high efficiency, tolerance of functional groups, and mild conditions.46 In this work, the efficacy of thiol-ene chemistry for the functionalization of composite nanoparticles is exploited for surface-grafting of hydrophilic polymer chains—as illustrated in Figure 1.
This modular synthetic approach—combining miniemulsion polymerization with thiol-ene chemistry—allows a wide variety of composite nanoparticles consisting of multiple inorganic nanoparticle cores, within a cross-linked polymer matrix, surrounded by a diffuse polymeric corona to be prepared. The particles are highly impermeable to external chemical species, are stable against aggregation, and can be dispersed in a wide range of solvents. This method is simple, scalable, and employs reagents that are generally commercially available. Moreover, it involves only a limited number of synthetic steps, relatively short reaction times and facile purification steps.
Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich and used as received, unless otherwise noted. HAuCl4·3H2O was purchased from Strem Chemicals. 2,2′-azobis-isobutyronitrile (AIBN, Sigma-Aldrich) was recrystallized from methanol before use. Divinylbenzene (Sigma-Aldrich, 80%, mixture of isomers) was percolated through basic alumina prior to use. Thiol-terminated poly(ethylene glycol) (Mn = 2,000) was purchased from Polymer Source. Silver nitrate was purchased from Fisher Chemical and used as received. Milli-Q grade water was used unless otherwise specified.
Characterization
Nuclear magnetic resonance (NMR) spectroscopy was carried out using a Bruker 500 MHz spectrometer with the residual solvent signal as an internal reference. UV-visible spectra were obtained using a Jasco V-530 spectrophotometer. Gel permeation chromatography (GPC) was performed with THF as solvent, on a Waters chromatograph equipped with four 5-μm Waters columns (300 × 7.7 mm) connected in series with increasing pore size (100, 1000, 10 000, and 1 000 000 Å). Waters 410 differential refractometer index (DRI) and 996 photodiode array detectors were employed. The molecular weights of the polymers were calculated relative to linear polystyrene standards.
Nanoparticle samples for transmission electron microscopy (TEM) were diluted by 1/100 with water and 5 μL was deposited on a carbon-coated TEM grid. TEM investigations were conducted using an FEI-T20 instrument, operating at 200 kV, with Scion Image software used for image analysis.
Dynamic light scattering (DLS) measurements were carried out on a Brookhaven BI-9000AT Digital Autocorrelator (Holtsville, NY, USA) equipped with an Avalanche photodiode detector and a MG vertically polarized 35 mV Helium-Neon 633 nm laser, and operated by the 9KDLSW control program. Nanoparticle samples were diluted by 1/100 with water and analyzed at a temperature of 25 °C and a fixed scattering angle of 90°.
Thermogravimetric analysis (TGA) was performed using a Mettler TGA 851e instrument under air over a temperature range of 25–800 °C at a heating rate of 10 °C per minute.
Gold nanoparticle synthesis
Gold nanoparticles were prepared in aqueous solution by citrate reduction.47 HAuCl4·3H2O (0.277 g, 0.82 mmol) was dissolved in 800 mL of boiling water, followed by addition of a solution of sodium citrate dihydrate (1.36 g, 4.6 mmol, dissolved in 68 mL of water). The resulting mixture was boiled for 30 minutes, over which time an intense ruby red color developed. The resulting gold nanoparticles were found to be 13 nm in average diameter by TEM analysis.
Hydrophobic gold nanoparticles were prepared as described in the literature.25, 48 HAuCl4·3H2O (0.25 g, 0.74 mmol) and oleylamine (2.0 g, 7.4 mmol) were dissolved in 25 mL benzene in a 50 mL round-bottom flask charged with a stirring bar. The flask was sealed with a rubber septum and the mixture sparged with argon for 10 minutes, before heating at 100°C for 80 minutes during which time the solution developed a dark purple color.
Silver nanoparticle synthesis
Silver nanoparticles were prepared in aqueous solution by borohydride reduction, using a slight modification of the method of Hao et al.49 Silver nitrate aqueous solution (0.1 M, 1.2 mL) and sodium citrate aqueous solution (0.2 M, 1.5 mL) were added to 600 mL of water and heated to 60°C. Sodium borohydride aqueous solution (1 M, 0.6 mL) was added, immediately resulting in an intense yellow-orange color, and the solution was heated at 60°C for two additional hours. The formation of silver nanoparticles with an average diameter of 15 nm was confirmed by TEM.
Gold nanorod synthesis
Gold nanorods were prepared using the method of Zijlstra et al.50 Briefly, hydrogen tetrachloroaurate aqueous solution (25 mM, 4 mL) and silver nitrate aqueous solution (6 mM, 4 mL) were added to 200 mL of 0.1 M cetyltrimethylammonium bromide (CTAB) aqueous solution. Ascorbic acid solution (0.1 M, 1.2 mL) was added, followed 10 seconds later by the addition of sodium borohydride solution (1.6 mM, 80 μL) with vigorous stirring. TEM confirmed the formation of gold nanorods with average length 33 nm and aspect ratio 4–5, together with a minor population (∼5%) of spherical gold nanoparticles.
Polymer ligand synthesis
Polymeric thiol ligands were prepared by RAFT polymerization followed by aminolysis to convert the dithioester end group to a thiol. The RAFT agent, methyl 2-phenyl-2-(phenylcarbonothioylthio)acetate, was prepared as described in the literature.51 For polymerization, RAFT agent (0.40 g, 1.3 mmol), styrene (7.0 g, 67 mmol), AIBN (0.022 g, 0.13 mmol) and a magnetic stir-bar were added to a 25 mL round-bottom flask, which was then sealed with a rubber septum. The reaction mixture was sparged with argon for 25 minutes and then heated, with stirring, at 110 °C for 80 hours. The resulting polystyrene was precipitated into cold methanol and dried under vacuum to give 4.9 g (82% recovered yield) of pink powder. Number average molecular weight of Mn = 3,900 kDa, and polydispersity index of 1.11, were determined by GPC.
The dithioester end group of the RAFT polymer was converted to a thiol by aminolysis.52 The polymer (4.5 g, 1.3 mmol) was dissolved in 50 mL THF in a 250 mL three-necked round-bottom flask. The polymer solution was subjected to three freeze-pump-thaw cycles. Hexylamine (0.39 g, 3.9 mmol) was mixed with 1 mL THF and injected into the polymer solution via an argon-purged syringe, and the solution was stirred under an argon atmosphere at room temperature for 20 hours. The aminolyzed polymer was collected by precipitation into cold methanol and dried under vacuum to give 4.1 g (92% recovered yield) of white powder. GPC analysis confirmed the absence of any disulfide formation.
Polymer grafting to gold nanoparticles
Grafting of polystyrene thiol ligands to aqueous gold nanoparticles was carried out in order to render dispersibility in organic media. The method used was a modification of the procedure reported by Merican et al.53 In a typical preparation, 100 mL of aqueous gold nanoparticles were concentrated into approximately 3 mL of aqueous solution by centrifugation (20 minutes at 13,000 × g) followed by removal of supernatant liquid. Polystyrene-thiol (5 mg) was dissolved in 3 mL of THF and mixed with aqueous gold nanoparticle solution in a 20 mL glass vial. Chloroform (3 mL) was then added to induce phase-separation of organic and aqueous solvents. The purple colored organic layer containing the polymer-grafted gold nanoparticles was extracted, and the nanoparticles isolated from ungrafted polymer by centrifugation (20 minutes at 15,000 × g). Residual solvent was evaporated under vacuum to give polymer-grafted gold nanoparticles (determined to be approximately 17 wt% polymer by TGA) in the form of a thin solid film.
The procedure for grafting polystyrene-thiol to silver nanoparticles was identical to that for gold nanoparticles, with the exception that the THF/water mixture containing nanoparticles and polystyrene thiol was adjusted to pH ∼ 2 by dropwise addition of 1 M HCl.
For polymer grafting to gold nanorods, the as-prepared nanorod solution (200 mL) was first cooled in an ice bath and centrifuged (10 minutes at 2,000 × g) to remove crystallized CTAB surfactant. Polystyrene-thiol (6 mg) was dissolved in 200 mL of inhibitor-free THF and mixed with the aqueous gold nanorod solution in a 500 mL separatory funnel. Chloroform (50 mL) and brine (10 mL) were then added to induce phase-separation of organic and aqueous solvents. The purple colored organic layer containing polymer-grafted gold nanorods was isolated and solvent removed under vacuum. Finally, the polymer-grafted nanorods were redispersed in 9 mL of THF, isolated from ungrafted polymer by centrifugation (10 minutes at 15,000 × g), and dried under vacuum.
Composite nanoparticle synthesis
Composite polymer-gold nanoparticles were synthesized by miniemulsion polymerization. In a typical experiment, 7.5 mg of 2,2′-azobis-(2-amidinopropane)dihydrochloride (V-50) and 1.3 mg of CTAB were dissolved in 2 mL of water in a 10 mL round-bottom flask charged with a stirring bar. Polymer-grafted gold nanoparticles (9 mg) were dispersed in 55 mg of divinylbenzene and this mixture was emulsified by stirring with the aqueous solution for approximately 30 minutes. The emulsion was then sonicated for 15 minutes over an ice-water bath, using a Fisher Scientific Model 500 Ultrasonic Dismembrator at 25% output power. Finally, the reaction vessel was sealed with a rubber septum, and purged with argon for 10 minutes, before heating at 50°C, with stirring, for approximately four hours. Typically, a small proportion of composite nanoparticles (<2% of the total solid content) were observed to form aggregates on the magnetic stir-bar and reaction vessel surface. Such aggregates were identified to be coagulated composite polymer-gold particles (as opposed to unencapsulated gold particle aggregates—an important distinction), and were readily removed by mild centrifugation (2 minutes at 50 × g) if necessary. Final conversion of divinylbenzene monomer was determined by gravimetry to be approximately 100%. Composite nanoparticles were stored as prepared (dispersed in aqueous solution) and were found to be stable—with minimal coagulation or sedimentation—for a period of at least several months.
Cyanide etching experiments
Citrate-stabilized gold nanoparticles prepared as above were diluted with water to a concentration of 57 mg (0.29 mmol) Au per liter, and 1 mL of this solution was added to a plastic cuvette. Potassium cyanide aqueous solution (0.29 M, 10 μL) was added to give ten molar equivalents of cyanide to gold. The intensity of the UV-visible absorbance maximum at 520 nm (due to the gold nanoparticle surface plasmon resonance) was monitored as a function of time.
Composite nanoparticles prepared as above were diluted by a factor of approximately 1/300 with water to give a gold concentration of 31 mg (0.16 mmol) Au per liter. A 1 mL volume of diluted nanoparticle solution was added to a plastic cuvette, followed by 55 μL of 0.29 M potassium cyanide solution to give 100 molar equivalents of cyanide to gold. In a separate experiment, 1 mL of diluted nanoparticle solution was mixed with 1 mL of THF in a quartz cuvette, followed by addition of 55 μL of 0.29 M potassium cyanide solution to give 100 equivalents of cyanide to gold. In both cases the intensity of the UV-visible absorbance maximum at approximately 550 nm was monitored as a function of time.
Surface functionalization of composite particles
Poly(ethylene glycol) (PEG) chains were grafted to the surface of the composite nanoparticles using thiol-ene chemistry as follows. Poly(divinylbenzene)-gold composite nanoparticles as described above (0.5 mL) was combined with 20 mg of thiol-terminated PEG (Mn = 2,000 kDa) and 5 mL of V-50 aqueous solution (2 mM) in a 25 mL round-bottom flask. The reaction mixture was purged with an argon stream for 20 minutes, before heating at 50°C, with stirring, for approximately 24 hours. The resulting 5.5 mL nanoparticle dispersion was concentrated to 0.5 mL using membrane filtration, then 5 mL of D2O was added, and the dispersion concentrated to 0.5 mL once again. This process was repeated several times in order to remove ungrafted PEG and replace the water with D2O for NMR analysis.
Results and Discussion
The desire to develop a broad composite nanoparticle platform for a variety of applications suggested miniemulsion polymerization as a viable strategy for nanoparticle formation due to its robustness and synthetic simplicity. Initially, it was necessary to address the challenges observed in previous miniemulsion polymer encapsulation procedures—in particular, the issues of low incorporation of inorganic material and inhomogeneous distribution of the inorganic nanoparticles within the polymer matrix. Gold nanoparticles (average diameter 13 nm) were chosen as a model inorganic for this work, in view of their broad potential for use in device applications and simplified visualization by TEM.2 Previous composite miniemulsion polymerizations reported in the literature have typically been carried out on a scale that required an inorganic component of the order of ∼1 g. In contrast to the inorganic nanoparticles used in these prior studies—e.g., titania,30, 31 magnetite, 34-37 silica,40, 41 etc.—which are readily purchased or prepared in multi-gram amounts, gold nanoparticles are typically prepared on a scale of ∼10 mg per 100 mL of solvent.47 This is a likely reason why miniemulsion composite polymers encapsulating gold nanoparticles have not yet been reported.
In the present work, preliminary experiments employed a miniemulsion polymerization system similar to established literature procedures,32, 54 containing a cationic surfactant (CTAB) and initiator (V-50) with styrene as monomer (see Supporting Information). In order to facilitate the synthesis of composite nanoparticles using a minimized amount of gold nanoparticles (∼10 mg), the miniemulsion system was scaled down to a total volume of approximately 2 mL (cf. typical volumes of ∼100 mL30, 34, 43). It is noted, however, that through the course of this work miniemulsion synthesis was successfully carried out on various scales from 1–20 mL in total volume. In traditional miniemulsion formulations, the monomer content is typically in the range of 10–20 wt% of the total emulsion. However, even after significantly scaling down the volume of the present emulsion system, it was found that the amount of gold relative to polymer in the resulting composite particles was unacceptably low, unless the monomer content in the formulation was reduced to the range of 1–5 wt%. Under these conditions of extremely low monomer content, a new challenge was encountered in that the styrene monomer was sufficiently water-soluble that the miniemulsion droplets were degraded by monomer dissolution before the completion of polymerization (see Supporting Information). An obvious solution to this problem was to employ a monomer with lower water solubility than styrene. In this case, divinylbenzene (∼1/10th the water solubility of styrene55) was chosen with an added benefit being the cross-linking leading to improved particle morphology/stability and allowed post-polymerization thiol-ene nanoparticle functionalization via unreacted alkene units (see below and Supporting Information).
Importance of Polymer-Grafting
A critical issue in composite miniemulsion polymerization is the successful dispersion of the inorganic material in the monomer emulsion droplets, as the quality of dispersion affects both the maximum obtainable inorganic content and the desired homogeneous distribution of inorganic material among the resulting particles. Typically, the dispersibility of inorganic nanoparticles is enhanced through the attachment of hydrophobic ligands,34, 36, 37, 39, 41 and for the present case of gold, a variety of procedures have been developed for synthesis of hydrophobic nanoparticles,5, 9, 11, 25, 48, 56 a small number of which give particularly well-defined particles, with low size polydispersity.25, 48, 57 These procedures typically employ a non-polymeric surface stabilizing group—typically a long-chain alkyl amine or thiol. Initial attempts were made to incorporate gold nanoparticles of this type (with average diameter ∼13 nm, stabilized by oleylamine25,48) into divinylbenzene miniemulsion polymerizations. Here, it was found that only a relatively small amount of gold could be successfully solubilized in divinylbenzene (∼1 mg of gold in 55 mg of divinylbenzene) and, while some composite nanoparticles were successfully formed, the gold content was minimal, and the incorporated gold nanoparticles were poorly distributed—resulting in gold aggregates and a majority of empty polymeric nanoparticles (see Figure 2). Furthermore, Figure 2 shows that in some cases the gold nanoparticles were expelled towards the surface of the composite nanoparticles, leading to only partial encapsulation.
Figure 2.

Representative transmission electron micrograph from synthesis of composite polymer-gold nanoparticles using oleylamine-stabilized gold nanoparticles. Inset at top right shows both gold nanoparticle aggregation and incomplete encapsulation by polymer.
The above results are consistent with literature reports describing poor dispersion of gold nanoparticles in polymer matrices.58 To overcome this difficulty, the small molecule hydrophobic units were replaced with polymer chains that were attached to the surface of nanoparticles58, 59 via a polymer “grafting-to” approach, using thiol-terminated polystyrene chains. This strategy was found to yield gold nanoparticles that could be conveniently isolated as a dry film and easily redispersed in a range of organic solvents, including divinylbenzene for miniemulsion polymerization. Compared with the oleylamine-stabilized gold nanoparticles described above, polystyrene-grafted nanoparticles were soluble in divinylbenzene up to considerably higher concentrations (as much as 20–30 mg gold in 55 mg of divinylbenzene). More importantly, the grafted gold particles were also found to be significantly more compatible with the polymer matrix (see Figure 3 below). It should be noted that the present gold nanoparticle grafting and encapsulation methods were found to be successful using polystyrene thiols of various molecular weights, between Mn = 1,900 and 50,000 Da. However, for consistency, all results presented here were obtained using the same polystyrene thiol, with Mn = 3,900 Da.
Figure 3.

Representative transmission electron micrograph of composite polymer-gold nanoparticles, showing 13 nm gold nanoparticles (black domains) encapsulated within larger, spherical poly(divinylbenzene) nanoparticles (gray domains). Inset at top left shows enlarged image of a single composite particle.
To investigate the external nanoenvironment and related surface chemistry of the gold nanoparticles within the composite structure, UV-visible spectroscopy was used to monitor the nature of the surface plasmon resonance of the gold nanoparticles (see Supporting Information for full spectra). The absorption spectrum of the starting citrate-stabilized gold nanoparticles in water exhibited a maximum at 520 nm, characteristic of gold nanoparticles in this size range (13 nm diameter). Grafting of thiol-terminated polystyrene chains to the gold nanoparticle surface resulted in a slight red-shift in the surface plasmon resonance to approximately 530 nm, and a further small red-shift (to ∼540 nm) was observed after encapsulation of the gold nanoparticles by miniemulsion polymerization, consistent with the changes in the local environment at the metal nanoparticle surface. Importantly, the absence of any considerable red-shift or broadening of the surface plasmon resonance confirmed that the gold nanoparticles did not undergo aggregation upon phase-transfer to organic solvent or during attachment of the thiol-terminated polystyrene ligands (also verified by TEM), and remained well stabilized within the miniemulsion droplets/polymerized composite nanoparticles.
Composite Nanoparticle Product
The size and morphology of the composite nanoparticles were investigated in detail by TEM. A representative image is presented in Figure 3 and shows the as-prepared composite nanoparticles, which have an average diameter of approximately 102 nm and a size polydispersity index of 1.2 (determined from analysis of 1000+ nanostructures)—in good agreement with results from dynamic light scattering (DLS) measurements. The encapsulated gold nanoparticles are clearly evident as well-defined spherical domains of high TEM contrast (black), distributed within the lower contrast poly(divinylbenzene) domains (gray). These findings are similar to the published results for the encapsulation of TiO2 using miniemulsion where the larger the size of composite particles the higher the number of encapsulated Au nanoparticles.29,31
TEM showed that the number of gold nanoparticles in each composite nanostructure varied considerably and was approximately proportional to the overall composite nanoparticle size. Importantly, for 13 nm gold nanoparticles, multiple gold domains could be readily incorporated into a single composite nanoparticle, and the average number of encapsulated gold nanoparticles per latex particle was determined from TEM analysis to be approximately 4.5 (with a range of 0–20 Au nanoparticles per composite nanoparticle—see Figure 4).
Figure 4.

Histogram showing the relative populations of composite nanoparticles containing different numbers of encapsulated 13 nm gold nanoparticles (compiled from TEM analysis of several hundred composite nanoparticles).
Further experiments were carried out to investigate the effect that changing the gold nanoparticle size had on the composite particles formed. Here it was observed that, for a given mass fraction of encapsulated gold nanoparticles, the average diameter of the resulting composite nanoparticles remained consistent (approx. 100 nm). However, increasing the diameter of the gold nanoparticles (between 13 and 46 nm) resulted in a decrease in the average number of encapsulated gold nanoparticles per composite particle—see Supporting Information for further details.
It is worth noting that in traditional miniemulsion formulations it is customary to include a highly water-insoluble co-stabilizer (or “hydrophobe”)—e.g., hexadecane, hexadecyl alcohol, or a polymeric species—which partitions into the monomer droplets of the emulsion and minimizes their destabilization by Ostwald ripening.27 It was found that no such additive was necessary in the present composite emulsion system: the hydrophobic polymer-grafted gold nanoparticles appeared to fulfill the function of a co-stabilizer, though theory suggests that the performance of hexadecane may be superior.28
Two features of critical interest in any new particle encapsulation methodology are the incidence of “empty” particles (in the present context, those encapsulating zero gold domains), as well as the existence of unencapsulated inorganic nanoparticles. As mentioned earlier, these two problems have been prevalent in many previous literature examples of composite nanoparticles formed by miniemulsion polymerization.30, 32, 37, 38, 41, 43
Firstly, for the present system, TEM imaging of as-prepared composite nanoparticles—i.e., without purification of any sort (Figure 3)—confirmed that the fraction of empty nanoparticles was approximately 10%. Not only is this an unusually low population of empty nanoparticles for a composite miniemulsion system, but it compares favorably with the results from various other reported nanoparticle encapsulation methodologies, in which centrifugation procedures to remove empty particles are often a mandatory requirement.23, 43 Secondly, TEM imaging (Figure 3) showed no evidence of unencapsulated gold nanoparticles with all gold nanoparticles were strictly located in the interior of latex particles, such that these composite nanoparticles had a clearly visible shell of poly(divinylbenzene), approximately 10–20 nm in thickness surrounding a composite core.
Cyanide Etching Experiments
Additional proof that gold nanoparticles were completely encapsulated by the polymer matrix was provided by treatment with a cyanide etching agent.23, 60 Particles were mixed with a large excess of potassium cyanide solution, and the UV-visible absorbance maximum corresponding to the gold nanoparticle surface plasmon resonance was monitored over time. Control experiments revealed that exposure of unmodified citrate-stabilized gold nanoparticles to ten molar equivalents of cyanide resulted in immediate and complete etching of gold, as shown in Figure 5. In contrast, the addition of 100 molar equivalents of cyanide to composite nanoparticles in aqueous solution resulted in no significant etching of gold over a period of several hours, confirming that the gold domains were not exposed at the composite nanoparticle surface.
Figure 5.

Change in the relative UV-visible absorbance maximum after exposure to excess potassium cyanide for citrate-stabilized gold nanoparticles (——●——; 10 equiv. of cyanide; λmax = 520 nm) and composite polymer-gold nanoparticles (— –■– —; 100 equiv. of cyanide; λmax = 550 nm) in water, and for composite nanoparticles in 50/50 THF/water (- - ♦ - -; 100 equiv. of cyanide; λmax = 550 nm)
Further etching experiments were conducted in the presence of organic solvent, as previous reports have shown that solvent-swelling of composite nanoparticles can render them permeable to small molecule etchants.23, 26 As shown in Figure 5, even in a 50/50 mixture of THF and water, the etching of gold from the composite nanoparticles by 100 molar equivalents of cyanide is an extremely slow process. These results strongly suggest that the non-polar cross-linked poly(divinylbenzene) matrix retains a dense network structure (despite swelling with organic solvent) that is highly impervious to the diffusion of small ions (such as CN-)—in contrast to other encapsulation methods, such as silica-coating.19
Redispersion in Organic Solvent
In addition to etch-resistance, it was found that the highly cross-linked polymer shell of the particles was able to withstand changes in the nature of the solvent. Composite nanoparticles were isolated by centrifugation (10 min at 9,000 × g) and redispersed in THF and chloroform (both good solvents for a variety of organic polymers, which readily dissolve composite nanoparticles consisting of linear polymer chains). Examination of these samples by TEM revealed composite nanoparticles that were indistinguishable from the starting particles (shown previously in Figure 3), suggesting that while these cross-linked particles may be swollen by organic solvents, the highly robust core-shell structure is not deformed or degraded. It is noted that the as-prepared composite nanoparticles did not remain stably dispersed in organic solvent (due to desorption of surfactant molecules)—aggregating and sedimenting out of solution over a period of minutes or hours. This problem is readily addressed by surface-grafting of polymer chains for steric stabilization, as discussed later.
Varying Gold Content of Composite Nanoparticles
The effect of increasing the gold loading of the composite latex particles on particle size and morphology was also examined. The formulation described above gives an expected gold content of approximately 12 wt% in the final composite nanoparticles. Two further miniemulsion formulations were prepared, where the mass of added gold nanoparticles was increased two- and three-fold, to give composite nanoparticles with expected gold content of 23 wt% and 33 wt% respectively.
The composite nanoparticles, prepared with three different gold loadings, were freeze-dried to isolate the composite materials, and subjected to thermogravimetric analysis (TGA). Figure 6 compares TGA data for the composite nanoparticles with those for a control sample: poly(divinylbenzene) nanoparticles prepared using the same miniemulsion procedure, but without added gold nanoparticles. Upon heating from 25 °C to 800 °C in air, it was observed that the majority of polymer mass loss occurs between 400 °C and 600 °C (the small mass loss at lower temperatures is attributed to residual initiator and surfactant molecules in the dried sample), leading to complete mass loss in the case of the control sample. For the composite nanoparticle samples the mass fraction remaining at the end of the measurement corresponds to the gold content of the composite nanoparticles, giving measured values of 12, 24 and 33 wt% gold—in good agreement with the values expected based on the mass of added gold nanoparticles.
Figure 6.

Thermogravimetric analysis (TGA) of pure poly(divinylbenzene) particles (————), and composite poly(divinylbenzene)-gold nanoparticles with predicted gold loadings of approximately 12 wt% (— — —), 23 wt% (—● — ●) and 33 wt% (● ● ● ●).
Representative electron micrographs of the composite nanoparticles prepared with gold content of 23 wt% and 33 wt% are presented in Figure 7. From TEM, it was clear that increasing the gold content in the miniemulsion formulation resulted in the formation of composite nanoparticles containing a greater number of gold nanoparticles (in the range of 20–100 nanoparticles encapsulated in a single latex particle) than had been previously observed. Furthermore, the number of empty latex particles was drastically decreased. Interestingly, the increase in gold loading also caused a small but appreciable increase in the size of composite latex particles, giving an average particle diameter of 121 nm for 23 wt% gold, and 144 nm for 33 wt% gold. Figure 7 also shows that when the gold content was increased to 33 wt%, the shape of some larger latex particles began to deviate from the strictly spherical shape observed at lower gold loading, resulting in some irregular geometries. Nevertheless, it is noteworthy that even for gold content in excess of 30 wt%, the present methodology consistently generates well-defined core-shell composite nanostructures.
Figure 7.

Transmission electron micrographs of composite polymer-gold nanoparticles prepared with gold content of (a) 23 wt%, and (b) 33 wt%.
Controlling Composite Nanoparticle Size
Control over nanoparticle size in miniemulsion polymerization systems is conveniently afforded by changing the amount of added surfactant, with decreasing surfactant concentration giving rise to increasing particle diameter.32 The ability to tune the size of composite polymer-gold nanoparticles was probed by varying the amount of added surfactant (CTAB) used in the miniemulsion preparation with the gold content being held constant at 12 wt%. The results are illustrated by the representative TEM images in Figures 8 and 9. As shown in Figure 8a, decreasing the surfactant concentration from 1.7 mM of CTAB to 0.57 mM (a factor of 1/3) gave an appreciable increase in nanoparticle average diameter to 134 nm (compared with the average diameter of 102 nm observed earlier). This was accompanied by a clear increase in the number of encapsulated gold nanoparticles per composite nanostructure, which is explained by the fact that an increase in the size of composite particles means a decrease in their number, so that, on average, each composite particle accommodates a greater number of gold nanoparticles.
Figure 8.

Transmission electron micrographs illustrating the effect on composite nanoparticle formation of decreasing the CTAB surfactant concentration to (a) 0.57 mM, and (b) 0.17 mM.
Figure 9.

Transmission electron micrographs illustrating the effect on composite nanoparticle formation of increasing the CTAB surfactant concentration to 17 mM, both before (a) and after (b) centrifugation to remove empty polymeric nanoparticles.
Decreasing the surfactant concentration to 0.17 mM (a factor of 1/10) (Figure 8b) gave a further increase in particle size (average diameter of 215 nm), along with the appearance of a small population of empty nanoparticles. It is not surprising to observe the formation of empty particles at very low surfactant concentrations, where the number of emulsion droplets becomes too low to capture all the radicals formed in the aqueous phase—permitting the formation of new particles by homogeneous nucleation.28
Of particular interest in these experiments with varying surfactant, was the core-shell nature of the composite particles, which became increasingly pronounced at lower surfactant concentrations. This is most evident in the case of 0.17 mM CTAB, where the gold nanoparticles are seen to be concentrated in the core region and surrounded by a ∼60 nm thick shell of poly(divinylbenzene). This well-defined core-shell structure is thought to be the result of preferential growth of poly(divinylbenzene) chains at the particle-water interface—an effect which has previously been exploited for the formation of hollow poly(divinylbenzene) particles by emulsion and suspension polymerization.61
The preparation of smaller composite particles was then investigated by increasing the surfactant concentration to 17 mM (a factor of 10). TEM imaging (see Figure 9a) reveals that composite particle formation was overwhelmed by the formation of smaller, empty poly(divinylbenzene) nanoparticles with a degree of Au nanoparticle aggregation as well. At high surfactant concentration, the presence of micelles supports the nucleation of new polymer particles outside the starting monomer droplets, and excess surfactant stabilizes the large surface area generated by the newly-formed particles.
As shown in Figure 9b, the minor population of gold-containing latex particles was readily isolated from the small empty particles by a simple centrifugation (5 min at 9,000 × g) with the average diameter of 67 nm for the composite nanoparticles generated under these conditions being significantly smaller than in earlier cases. Additionally, the removal of a significant amount of poly(divinylbenzene) in the form of empty particles resulted in composite nanoparticles with a thinner polymer shell and a higher gold to polymer ratio. Clearly, high surfactant concentration offers composite nanoparticles with significantly different size and composition—albeit with the addition of a purification step for higher surfactant concentrations.
Surface-Modification of Composite Nanoparticles using Thiol-ene Chemistry
In order to provide a synthetic platform for a diverse range of nanoparticle applications, it is crucial to develop a strategy for tailoring particle surface functionality—preferably one that is simple and efficient. In this case, thiol-ene chemistry is an ideal choice, as poly(divinylbenzene) particles are known to contain a significant fraction of unreacted vinyl “ene” groups62 (estimated at 50%63). A thiol-terminated poly(ethylene glycol) (PEG, Mn = 2,000) was chosen to demonstrate the thiol-ene reaction with composite nanoparticles, as it was anticipated that surface-grafted PEG chains would act as a steric stabilizer for the composite nanoparticles, enabling the materials to be dispersed in a variety of different solvents.
NMR analysis of the composite nanoparticles after thiol-ene reaction confirmed the successful attachment of PEG by the appearance of a signal from the methylene protons of PEG (see Supporting Information) and as noted in our previous work,44 successful PEG grafting was clearly apparent in their changed solvent properties. PEG-functionalized composite nanoparticles were isolated by centrifugation (10 min at 9,000 × g) and redispersed in THF, as shown in Figure 10, and the same process was also carried out to replace THF with a variety of other organic solvents such as chloroform, N,N-dimethylformamide and dimethylsulfoxide. PEG-functionalized composite nanoparticles could also be isolated from organic solvents and successfully redispersed in water. In contrast to the original composite particles without PEG-grafting (i.e., surfactant-stabilized), which underwent rapid aggregation in organic solvents (see Figure 10), the PEG-functionalized particles remained stable and well-dispersed in a variety of organic solvents for a period of at least several months. The dispersibility properties of the composite nanoparticles after grafting appear to be dictated by the solubility of the PEG chains.
Figure 10.

Photographs of purified composite nanoparticles redispersed in THF and left to stand for 24 hours (left), and following attachment of PEG surface groups by thiol-ene chemistry, redispersion in THF and left to stand for 24 hours (right).
Similar to earlier observations for unfunctionalized composite nanoparticles, TEM revealed no dissolution or degradation of PEG-grafted composite nanostructures, either during the thiol-ene reaction or after redispersing in organic solvents (due to their robust cross-linked nature)—TEM images of redispersed composite nanoparticles were indistinguishable from those of the original aqueous composite nanoparticles (see Figure 11b).
Figure 11.

Transmission electron micrographs of composite nanoparticles after redispersion in tetrahydrofuran (a), and after thiol-ene attachment of PEG and redispersion in chloroform (b).
Finally, while the thiol-ene approach demonstrated here for surface-modification makes use of unreacted vinyl groups inherent to the poly(divinylbenzene) matrix, the present miniemulsion polymerization methodology is also well-suited for installing a variety of other reactive groups at the particle surface through the simple addition of comonomers. This strategy has been investigated for incorporation of styrenic monomers such as vinylbenzyl chloride and vinylbenzyl azide—intended to provide pendant reactive sites for surface-modification by nucleophilic substitution and 1,3-dipolar addition (alkyne-azide “click” chemistry), respectively. In these examples, the comonomer is mixed with divinylbenzene prior to the dissolution of inorganic nanoparticles, and it has been found that up to 50 wt% of comonomer (relative to total monomer mass) may be incorporated without causing significant changes in the nature of the composite nanoparticles produced. Further experiments to confirm the efficacy of surface modification using these comonomer-functionalized particles are currently underway.64
Variability in the Shape and Nature of the Inorganic Component
A notable advantage of the present synthetic methodology over previous composite miniemulsion polymerization formulations is that it is modular in nature, therefore opening up the possibility of incorporating a variety of different inorganic nanostructures. This opens up the scope of the procedure to encapsulating other metals/inorganics, non-spherical nanostructures and combinations thereof. In demonstrating this capability, initial experiments were performed with a mixture of gold and MnFe2O4 nanoparticles (see Figures 12a and 12b, and full details in reference44), with successful encapsulation being observed for 10 wt% MnFe2O4 (11 nm diameter) and a variety of Au nanoparticles with sizes ranging from 13 to 46 nm (19 wt%).
Figure 12.
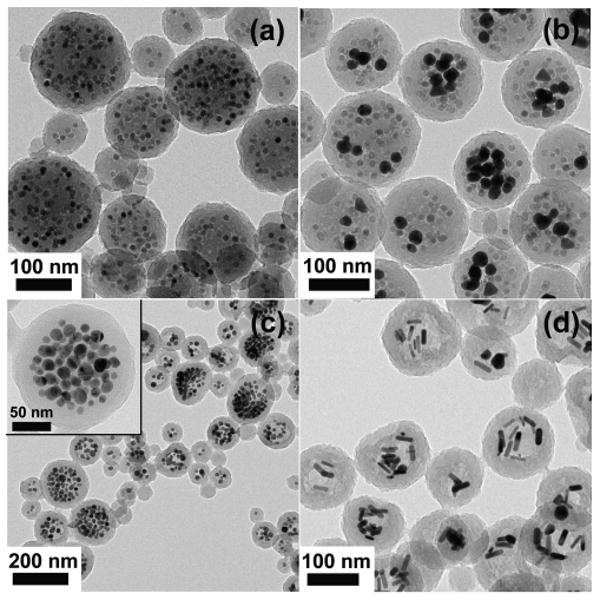
Transmission electron micrograph of composite polymer-MnFe2O4 nanoparticles (a), hybrid polymer-gold-MnFe2O4 nanoparticles (b), composite polymer-silver nanoparticles (c), and composite polymer-gold nanorods (d).
Broadening the scope of the procedure, silver nanoparticles and gold nanorods were then investigated. For the silver nanoparticles (average diameter ∼15 nm), it was found that polymer-grafting and miniemulsion polymerization procedures identical to those described previously for gold nanoparticles could be utilized, although it was observed that grafting of thiol-terminated polystyrene to silver nanoparticles was most successful when conducted at pH ∼ 2. The miniemulsion procedure produced a composite nanoparticle dispersion that was yellow-brown in color, arising from the surface plasmon resonance of the silver nanoparticles (with a UV-visible absorbance maximum at 390 nm). TEM examination, represented in Figure 12c, revealed composite poly(divinylbenzene)-silver nanoparticles whose structure was analogous to that seen earlier for gold nanoparticles: an outer shell of poly(divinylbenzene), ∼10–20 nm in thickness, with silver nanoparticles located in the composite nanoparticle interior. As in previous cases, the metal nanoparticles were well distributed, with very few empty latex particles and no observation of unencapsulated silver nanoparticles.
In changing the shape of the inorganic component, the same method developed in this work for grafting of polystyrene thiol to gold nanoparticles, using a 50/50 THF/water medium, was found to be equally effective for grafting to CTAB-stabilized gold nanorods. Encapsulation of polymer-grafted gold nanorods resulted in the familiar core-shell composite nanoparticle structure, as shown by TEM in Figure 12d. Importantly, these results for nanoparticles of different shapes and compositions, coupled with the ability to combine different inorganic nanostructures in a single composite nanoparticle, suggests that the success of the present synthetic approach is independent of the size, shape and chemical composition of the encapsulated nanoparticle and can be successfully applied to mixtures of different nanostructures. This demonstrates the platform nature of the procedure and the potential for applying this simple modular approach to the preparation of a wide range of useful composite nanostructures.
CONCLUSION
This work represents a robust and versatile platform technology for the synthesis of composite polymer-inorganic nanoparticles by miniemulsion polymerization. This method is modular in nature, yielding cross-linked composite nanoparticles that are resilient to dissolution in organic solvent and which fully encapsulate the inorganic domains within the nanoparticle interior. Significantly, this approach permits the incorporation of multiple inorganic domains of the same or different structure within a single composite nanoparticle and at the same time achieve high loadings. The combination of high loadings and localization may be of particular advantage in applications where the proximity of the inorganic domains is critical, e.g., the fabrication of “hot-spots” for surface enhanced Raman spectroscopy. To further broaden the applicability of these systems, thiol-ene chemistry was exploited for facile surface functionalization of the composite nanoparticles with the attachment of PEG chains providing steric stabilization and enabling these materials to be successfully dispersed in a wide variety of solvents. It is envisioned that the procedure developed here will provide a general platform for creating a host of new functional polymer-inorganic nanostructures with the simple nature of the procedures enabling a wide range of applications.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the National Science Foundation, through the MRSEC program under Award No. DMR05-20415 and the National Heart Lung and Blood Institute of the National Institutes of Health, as a Program of Excellence in Nanotechnology (HL080729). KVB gratefully acknowledges the support of a Postdoctoral Fellowship from the New Zealand Foundation for Research, Science & Technology. Helpful discussions with Eric Pressly and Tara Schiller are greatly appreciated, as is the advice of Dr Luis Campos regarding thiol-ene chemistry.
Footnotes
Supporting Information Available. Experimental details and representative results obtained from composite miniemulsion polymerizations with styrene as monomer; UV-vis absorption spectra for gold nanoparticles and composite polymer-gold nanoparticles; 1H-NMR spectrum of PEG-grafted composite polymer-gold nanoparticles.
References
- 1.Daniel MC, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]; Ishida T, Haruta M. Angew Chem Int Ed. 2007;46:7154–7156. doi: 10.1002/anie.200701622. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Salgueiriño-Maceira V, Liz-Marzán LM, Caruso F. Adv Mater. 2002;14:908–912. [Google Scholar]; Ouyang J, Chu CW, Szmanda CR, Ma L, Yang Y. Nature Mater. 2004;3:918–922. doi: 10.1038/nmat1269. [DOI] [PubMed] [Google Scholar]; Correa-Duarte MA, Salgueirino-Maceira V, Rinaldi A, Giersig M, Liz-Marzan LM. Gold Bull. 2007;40:6–14. [Google Scholar]; Beissenhirtz MK, Elnathan R, Weizmann Y, Willner I. Small. 2007;3:375–379. doi: 10.1002/smll.200600450. [DOI] [PubMed] [Google Scholar]
- 3.Cao YC, Jin R, Mirkin CA. Science. 2002;297:1536–1540. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]; Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]; Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]; Lee JS, Han MS, Mirkin CA. Angew Chem Int Ed. 2007;46:4093–4096. doi: 10.1002/anie.200700269. [DOI] [PubMed] [Google Scholar]; Bhattacharya R, Patra CR, Verma R, Kumar S, Greipp PR, Mukherjee P. Adv Mater. 2007;19:711–716. [Google Scholar]
- 4.Shan J, Tenhu H. Chem Commun. 2007:4580–4598. doi: 10.1039/b707740h. [DOI] [PubMed] [Google Scholar]
- 5.Leff DV, Brandt L, Heath JR. Langmuir. 1996;12:4723–4730. [Google Scholar]; Newman JDS, Blanchard GJ. Langmuir. 2006;22:5882–5887. doi: 10.1021/la060045z. [DOI] [PubMed] [Google Scholar]
- 6.Sastry M, Kumar A, Mukherjee P. Colloids Surf A Physicochem Eng Aspects. 2001;181:255–259. [Google Scholar]
- 7.Lowe AB, Sumerlin BS, Donovan MS, McCormick CL. J Am Chem Soc. 2002;124:11562–11563. doi: 10.1021/ja020556h. [DOI] [PubMed] [Google Scholar]; Zubarev ER, Xu J, Sayyad A, Gibson JD. J Am Chem Soc. 2006;128:4958–4959. doi: 10.1021/ja060782h. [DOI] [PubMed] [Google Scholar]; Weisbecker CS, Merritt MV, Whitesides GM. Langmuir. 1996;12:3763–3772. [Google Scholar]
- 8.Porter LA, Ji D, Westcott SL, Graupe M, Czernuszewicz RS, Halas NJ, Lee TR. Langmuir. 1998;14:7378–7386. doi: 10.1021/la980870i. [DOI] [PubMed] [Google Scholar]; Langry KC, Ratto TV, Rudd RE, McElfresh MW. Langmuir. 2005;21:12064–12067. doi: 10.1021/la0513555. [DOI] [PubMed] [Google Scholar]; Azzam T, Eisenberg A. Langmuir. 2007;23:2126–2132. doi: 10.1021/la0627563. [DOI] [PubMed] [Google Scholar]; Yonezawa T, Yasui K, Kimizuka N. Langmuir. 2001;17:271–273. [Google Scholar]
- 9.Manna A, Chen PL, Akiyama H, Wei TX, Tamada K, Knoll W. Chem Mater. 2003;15:20–28. [Google Scholar]
- 10.Li XM, de Jong MR, Inoue K, Shinkai S, Huskens J, Reinhoudt DN. J Mater Chem. 2001;11:1919–1923. [Google Scholar]; Maye MM, Chun SC, Han L, Rabinovich D, Zhong CJ. J Am Chem Soc. 2002;124:4958–4959. doi: 10.1021/ja025724k. [DOI] [PubMed] [Google Scholar]; Hussain I, Graham S, Wang Z, Tan B, Sherrington DC, Rannard SP, Cooper AI, Brust M. J Am Chem Soc. 2005;127:16398–16399. doi: 10.1021/ja055321v. [DOI] [PubMed] [Google Scholar]; Wang Z, Tan B, Hussain I, Schaeffer N, Wyatt MF, Brust M, Cooper AI. Langmuir. 2007;23:885–895. doi: 10.1021/la062623h. [DOI] [PubMed] [Google Scholar]
- 11.Shelley EJ, Ryan D, Johnson SR, Couillard M, Fitzmaurice D, Nellist PD, Chen Y, Palmer RE, Preece JA. Langmuir. 2002;18:1791–1795. [Google Scholar]
- 12.Duwez AS, Guillet P, Colard C, Gohy JF, Fustin CA. Macromolecules. 2006;39:2729–2731. [Google Scholar]
- 13.Zhao Y, Perez-Segarra W, Shi Q, Wei A. J Am Chem Soc. 2005;127:7328–7329. doi: 10.1021/ja050432f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gittins DI, Caruso F. ChemPhysChem. 2002;3:110–113. doi: 10.1002/1439-7641(20020118)3:1<110::AID-CPHC110>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; Latham AH, Williams ME. Langmuir. 2006;22:4319–4326. doi: 10.1021/la053523z. [DOI] [PubMed] [Google Scholar]; Yockell-Lelievre H, Desbiens J, Ritcey AM. Langmuir. 2007;23:2843–2850. doi: 10.1021/la062886b. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh SK, Pal T. Chem Rev. 2007;107:4797–4862. doi: 10.1021/cr0680282. [DOI] [PubMed] [Google Scholar]
- 16.Liz-Marzan LM, Giersig M, Mulvaney P. Langmuir. 1996;12:4329–4335. [Google Scholar]; Doering WE, Nie SM. Anal Chem. 2003;75:6171–6176. doi: 10.1021/ac034672u. [DOI] [PubMed] [Google Scholar]; Graf C, Vossen DLJ, Imhof A, van Blaaderen A. Langmuir. 2003;19:6693–6700. [Google Scholar]
- 17.Li T, Moon J, Morrone AA, Mecholsky JJ, Talham DR, Adair JH. Langmuir. 1999;15:4328–4334. [Google Scholar]; Vestal CR, Zhang ZJ. Nano Lett. 2003;3:1739–1743. [Google Scholar]; Darbandi M, Thomann R, Nann T. Chem Mater. 2005;17:5720–5725. [Google Scholar]; Gong JL, Jiang JH, Liang Y, Shen GL, Yu RQ. J Colloid Interface Sci. 2006;298:752–756. doi: 10.1016/j.jcis.2006.01.024. [DOI] [PubMed] [Google Scholar]; Stjerndahl M, Andersson M, Hall HE, Pajerowski DM, Meisel MW, Duran RS. Langmuir. 2008;24:3532–3536. doi: 10.1021/la7035604. [DOI] [PubMed] [Google Scholar]
- 18.Nystrom D, Antoni P, Malmstrom E, Johansson M, Whittaker M, Hult A. Macromol Rapid Comm. 2005;26:524–528. [Google Scholar]; Ohno K, Morinaga T, Koh K, Tsujii Y, Fukuda T. Macromolecules. 2005;38:2137–2142. [Google Scholar]; Liu SH, Han MY. Adv Funct Mater. 2005;15:961–967. [Google Scholar]; Zhao YL, Perrier S. Macromolecules. 2006;39:8603–8608. [Google Scholar]; Pastoriza-Santos I, Perez-Juste J, Liz-Marzan LM. Chem Mater. 2006;18:2465–2467. [Google Scholar]; Morinaga T, Ohkura M, Ohno K, Tsujii Y, Fukuda T. Macromolecules. 2007;40:1159–1164. [Google Scholar]
- 19.Giersig M, Ung T, LizMarzan LM, Mulvaney P. Adv Mater. 1997;9:570–576. [Google Scholar]; Poovarodom S, Bass JD, Hwang SJ, Katz A. Langmuir. 2005;21:12348–12356. doi: 10.1021/la052006d. [DOI] [PubMed] [Google Scholar]; Cavaliere-Jaricot S, Darbandi M, Nann T. Chem Commun. 2007:2031–2033. doi: 10.1039/b703811a. [DOI] [PubMed] [Google Scholar]; Burns A, Ow H, Wiesner U. Chem Soc Rev. 2006;35:1028–1042. doi: 10.1039/b600562b. [DOI] [PubMed] [Google Scholar]
- 20.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243–247. [Google Scholar]; Westcott SL, Oldenburg SJ, Lee TR, Halas NJ. Langmuir. 1998;14:5396–5401. [Google Scholar]
- 21.Chen CW, Serizawa T, Akashi M. Langmuir. 1999;15:7998–8006. [Google Scholar]; Roos C, Schmidt M, Ebenhoch J, Baumann F, Deubzer B, Weis J. Adv Mater. 1999;11:761–766. [Google Scholar]; Wang PH, Pan CY. Eur Polym J. 2000;36:2297–2300. [Google Scholar]; Suzuki D, Kawaguchi H. Langmuir. 2005;21:12016–12024. doi: 10.1021/la0516882. [DOI] [PubMed] [Google Scholar]; Kim JH, Kim JS, Choi H, Lee SM, Jun BH, Yu KN, Kuk E, Kim YK, Jeong DH, Cho MH, Lee YS. Anal Chem. 2006;78:6967–6973. doi: 10.1021/ac0607663. [DOI] [PubMed] [Google Scholar]
- 22.Ye H, Crooks RM. J Am Chem Soc. 2005;127:4930–4934. doi: 10.1021/ja0435900. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Smith AE, Lokitz BS, McCormick CL. Macromolecules. 2007;40:8524–8526. [Google Scholar]; Yusuf H, Kim WG, Lee DH, Guo Y, Moffitt MG. Langmuir. 2007;23:868–878. doi: 10.1021/la0623634. [DOI] [PubMed] [Google Scholar]; Sanchez-Gaytan BL, Cui W, Kim Y, Mendez-Polanco MA, Duncan TV, Fryd M, Wayland BB, Park SJ. Angew Chem Int Ed. 2007;46:9235–9238. doi: 10.1002/anie.200703032. [DOI] [PubMed] [Google Scholar]
- 24.Kang YJ, Taton TA. Angew Chem Int Ed. 2005;44:409–412. doi: 10.1002/anie.200461119. [DOI] [PubMed] [Google Scholar]
- 25.Kim BS, Qiu JM, Wang JP, Taton TA. Nano Lett. 2005;5:1987–1991. doi: 10.1021/nl0513939. [DOI] [PubMed] [Google Scholar]
- 26.Kim BS, Taton TA. Langmuir. 2007;23:2198–2202. doi: 10.1021/la062692w. [DOI] [PubMed] [Google Scholar]; Chen Y, Cho J, Young A, Taton TA. Langmuir. 2007;23:7491–7497. doi: 10.1021/la700494e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landfester K. Macromol Rapid Comm. 2001;22:896–936. [Google Scholar]; Schork FJ, Luo YW, Smulders W, Russum JP, Butte A, Fontenot K. Miniemulsion polymerization. In: Okubo M, editor. Polymer Particles. Vol. 175. 2005. pp. 129–255. [Google Scholar]; Asua JM. Prog Polym Sci. 2002;27:1283–1346. [Google Scholar]
- 28.El-Aasser MS, Sudol ED. J Coat Tech Res. 2004;1:1. [Google Scholar]
- 29.Erdem B, Sudol ED, Dimonie VL, El-Aasser MS. J Polym Sci Part A: Polym Chem. 2000;38:4419–4430. [Google Scholar]; Erdem B, Sudol ED, Dimonie VL, El-Aasser MS. J Polym Sci Part A: Polym Chem. 2000;38:4431–4440. [Google Scholar]
- 30.Song X, Yin G, Zhao Y, Wang H, Du Q. J Polym Sci Part A: Polym Chem. 2009;47:5728–5736. [Google Scholar]
- 31.Al-Ghamdi GH, Sudol ED, Dimonie VL, El-Aasser MS. J Appl Polym Sci. 2006;101:3479–3486. [Google Scholar]
- 32.Bechthold N, Tiarks F, Willert M, Landfester K, Antonietti M. Macromol Symp. 2000;151:549–555. [Google Scholar]
- 33.Ramirez LP, Landfester K. Macromol Chem Phys. 2003;204:22–31. [Google Scholar]; Csetneki I, Faix MK, Szilagyi A, Kovacs AL, Nemeth Z, Zrinyi M. J Polym Sci Part A: Polym Chem. 2004;42:4802–4808. [Google Scholar]; Luo YD, Dai CA, Chiu WY. J Polym Sci Part A: Polym Chem. 2008;46:1014–1024. [Google Scholar]; Liu X, Guan Y, Ma Z, Liu H. Langmuir. 2004;20:10278–10282. doi: 10.1021/la0491908. [DOI] [PubMed] [Google Scholar]; Qiu GH, Wang Q, Wang C, Lau W, Gu YL. Polym Int. 2006;55:265–272. [Google Scholar]
- 34.Xu H, Cui L, Tong N, Gu H. J Am Chem Soc. 2006;128:15582–15583. doi: 10.1021/ja066165a. [DOI] [PubMed] [Google Scholar]
- 35.Lu S, Ramos J, Forcada J. Langmuir. 2007;23:12893–12900. doi: 10.1021/la702281k. [DOI] [PubMed] [Google Scholar]
- 36.Faridi-Majidi R, Sharifi-Sanjani N. J Magn Magn Mater. 2007;311:55–58. [Google Scholar]
- 37.Joumaa N, Toussay P, Lansalot M, Elaissari A. J Polym Sci Part A: Polym Chem. 2008;46:327–340. [Google Scholar]
- 38.Tiarks F, Landfester K, Anonietti M. Macromol Chem Phys. 2001;202:51–60. [Google Scholar]
- 39.Zeng Z, Yu J, Guo ZX. Macromol Chem Phys. 2005;206:1558–1567. [Google Scholar]
- 40.Costoyas A, Ramos J, Forcada J. J Polym Sci Part A: Polym Chem. 2009;47:935–948. [Google Scholar]; Zhang SW, Zhou SX, Weng YM, Wu LM. Langmuir. 2005;21:2124–2128. doi: 10.1021/la047652b. [DOI] [PubMed] [Google Scholar]
- 41.Qi DM, Bao YZ, Weng ZX, Huang ZM. Polymer. 2006;47:4622–4629. [Google Scholar]
- 42.Lu HF, Fei B, Xin JH, Wang RH, Li L. J Colloid Interface Sci. 2006;300:111–116. doi: 10.1016/j.jcis.2006.03.059. [DOI] [PubMed] [Google Scholar]; Luo YD, Dai CA, Chiu WY. J Polym Sci Part A: Polym Chem. 2008;46:8081–8090. [Google Scholar]
- 43.Kim H, Daniels ES, Li S, Mokkapati VK, Kardos K. J Polym Sci Part A: Polym Chem. 2007;45:1038–1054. [Google Scholar]
- 44.van Berkel KY, Piekarski AM, Kierstead PH, Pressly ED, Ray PC, Hawker CJ. Macromolecules. 2009;42:1425–1427. [Google Scholar]
- 45.Wooley KL, Hawker CJ, Fréchet JMJ. Angew Chem Int Ed Engl. 1994;33:82–85. [Google Scholar]; Harth EM, Hecht S, Helms B, Malmstrom EE, Fréchet JMJ, Hawker CJ. J Am Chem Soc. 2002;124:3926–3938. doi: 10.1021/ja025536u. [DOI] [PubMed] [Google Scholar]; Hawker CJ, Fréchet JMJ. Polymer. 1992;33:1507–1511. [Google Scholar]
- 46.Gress A, Volkel A, Schlaad H. Macromolecules. 2007;40:7928–7933. [Google Scholar]; David RLA, Kornfield JA. Macromolecules. 2008;41:1151–1161. [Google Scholar]; Killops KL, Campos LM, Hawker CJ. J Am Chem Soc. 2008;130:5062–5064. doi: 10.1021/ja8006325. [DOI] [PubMed] [Google Scholar]; Nilsson C, Malmström E, Johansson M, Trey SM. J Polym Sci, Part A: Polym Chem. 2009;47:589–601. [Google Scholar]; Nilsson C, Simpson N, Malkoch M, Johansson M, Malmström E. J Polym Sci, Part A: Polym Chem. 2008;46:1339–1348. [Google Scholar]; a) Campos LM, Killops KL, Sakai R, Paulusse JMJ, Damiron D, Drockenmuller E, Messmore BW, Hawker C. J Macromolecules. 2008;41:7063–7070. [Google Scholar]; b) Boyer C, Granville A, Davis TP, Bulmus V. J Polym Sci, Part A: Polym Chem. 2009;47:3773–3794. [Google Scholar]; Vestberg R, Piekarski AM, Pressly ED, Van Berkel KY, Malkoch M, Gerbac J, Ueno N, Hawker CJ. J Polym Sci, Polym Chem. 2009;47:1237–1258. [Google Scholar]; Johnson PM, Stansbury JW, Bowman CN. J Polym Sci, Polym Chem. 2008;46:1502–1509. [Google Scholar]; Yu B, Chan JW, Hoyle CE, Lowe AB. J Polym Sci, Polym Chem. 2009;47:3544–3557. [Google Scholar]; Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Chem Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951:55–75. [Google Scholar]
- 48.Yu H, Chen M, Rice PM, Wang SX, White RL, Sun S. Nano Lett. 2005;5:379–382. doi: 10.1021/nl047955q. [DOI] [PubMed] [Google Scholar]
- 49.Hao E, Li S, Bailey RC, Zou S, Schatz GC, Hupp JT. J Phys Chem B. 2004;108:1224–1229. [Google Scholar]
- 50.Zijlstra P, Bullen C, Chon JWM, Gu M. J Phys Chem B. 2006;110:19315–19318. doi: 10.1021/jp0635866. [DOI] [PubMed] [Google Scholar]
- 51.Perrier S, Takolpuckdee P, Westwood J, Lewis DM. Macromolecules. 2004;37:2709–2717. [Google Scholar]
- 52.Lima V, Jiang XL, Brokken-Zijp J, Schoenmakers PJ, Klumperman B, Van Der Linde R. J Polym Sci Part A: Polym Chem. 2005;43:959–973. [Google Scholar]
- 53.Merican Z, Schiller TL, Hawker CJ, Fredericks PM, Blakey I. Langmuir. 2007;23:10539–10545. doi: 10.1021/la702218b. [DOI] [PubMed] [Google Scholar]
- 54.Landfester K, Bechthold N, Tiarks F, Antonietti M. Macromolecules. 1999;32:2679–2683. [Google Scholar]
- 55.Chai XS, Schork FJ, DeCinque A, Wilson K. Ind Eng Chem Res. 2005;44:5256–5258. [Google Scholar]
- 56.Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. Chem Commun. 1994:801–802. [Google Scholar]; Brust M, Fink J, Bethell D, Schiffrin DJ, Kiely C. Chem Commun. 1995:1655–1656. [Google Scholar]; Yee CK, Jordan R, Ulman A, White H, King A, Rafailovich M, Sokolov J. Langmuir. 1999;15:3486–3491. [Google Scholar]; Weare WW, Reed SM, Warner MG, Hutchison JE. J Am Chem Soc. 2000;122:12890–12891. [Google Scholar]; Cushing BL, Kolesnichenko VL, O'Connor CJ. Chem Rev. 2004;104:3893–3946. doi: 10.1021/cr030027b. [DOI] [PubMed] [Google Scholar]; Mirkhalaf F, Paprotny J, Schiffrin DJ. J Am Chem Soc. 2006;128:7400–7401. doi: 10.1021/ja058687g. [DOI] [PubMed] [Google Scholar]
- 57.Stoeva S, Klabunde KJ, Sorensen CM, Dragieva I. J Am Chem Soc. 2002;124:2305–2311. doi: 10.1021/ja012076g. [DOI] [PubMed] [Google Scholar]; Zheng N, Fan J, Stucky GD. J Am Chem Soc. 2006;128:6550–6551. doi: 10.1021/ja0604717. [DOI] [PubMed] [Google Scholar]
- 58.Corbierre MK, Cameron NS, Sutton M, Mochrie SGJ, Lurio LB, Ruhm A, Lennox RB. J Am Chem Soc. 2001;123:10411–10412. doi: 10.1021/ja0166287. [DOI] [PubMed] [Google Scholar]
- 59.Corbierre MK, Cameron NS, Sutton M, Laaziri K, Lennox RB. Langmuir. 2005;21:6063–6072. doi: 10.1021/la047193e. [DOI] [PubMed] [Google Scholar]; Balazs AC, Emrick T, Russell TP. Science. 2006;314:1107–1110. doi: 10.1126/science.1130557. [DOI] [PubMed] [Google Scholar]; Chiu JJ, Kim BJ, Yi GR, Bang J, Kramer EJ, Pine DJ. Macromolecules. 2007;40:3361–3365. [Google Scholar]; Kim BJ, Fredrickson GH, Hawker CJ, Kramer EJ. Langmuir. 2007;14:7804–7809. doi: 10.1021/la700507j. [DOI] [PubMed] [Google Scholar]
- 60.Zhu T, Vasilev K, Kreiter M, Mittler S, Knoll W. Langmuir. 2003;19:9518–9525. [Google Scholar]
- 61.Konishi Y, Okubo M, Minami H. Colloid Polym Sci. 2003;281:123–129. [Google Scholar]; Okubo M, Minami H. Colloid Polym Sci. 1997;275:992–997. [Google Scholar]; Okubo M, Konishi Y, Minami H. Colloid Polym Sci. 1998;276:638–642. [Google Scholar]; Okubo M, Konishi Y, Minami H. Colloid Polym Sci. 2000;278:659–664. [Google Scholar]
- 62.Zheng G, Stover HDH. Macromolecules. 2002;35:6828–6834. [Google Scholar]; Downey JS, Frank RS, Li WH, Stover HDH. Macromolecules. 1999;32:2838–2844. [Google Scholar]; Alam MN, Zetterlund PB, Okubo M. Macromol Chem Phys. 2006;207:1732–1741. [Google Scholar]
- 63.Law RV, Sherrington DC, Snape CE. Macromolecules. 1997;30:2868–2875. [Google Scholar]
- 64.Piekarski AM, van Berkel KY, Hawker CJ. Manuscript in preparation [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.