

Abstract
Positional cloning is the approach of choice for the identification of genetic mutations underlying the pathological development of diseases with simple Mendelian inheritance. It consists of different consecutive steps, starting with recruitment of patients and DNA collection, that are critical to the overall process. A genetic analysis of the enrolled patients and their families is performed, based on genetic recombination frequencies generated by meiotic cross-overs and on genome-wide molecular studies, to define a critical DNA region of interest. This analysis culminates in a statistical estimate of the probability that disease features may segregate in the families independently or in association with specific molecular markers located in known regions. In this latter case, a marker can be defined as being linked to the disease manifestations. The genetic markers define an interval that is a function of their recombination frequencies with the disease, in which the disease gene is localised. The identification and characterisation of chromosome abnormalities as translocations, deletions and duplications by classical cytogenetic methods or by the newly developed microarray-based comparative genomic hybridisation (array CGH) technique may define extensions and borders of the genomic regions involved. The step following the definition of a critical genomic region is the identification of candidate genes that is based on the analysis of available databases from genome browsers. Positional cloning culminates in the identification of the causative gene mutation, and the definition of its functional role in the pathogenesis of the disorder, by the use of cell-based or animal-based experiments. More often, positional cloning ends with the generation of mice with homologous mutations reproducing the human clinical phenotype. Altogether, positional cloning has represented a fundamental step in the research on genetic renal disorders, leading to the definition of several disease mechanisms and allowing a proper diagnostic approach to many conditions.
Keywords: Positional cloning, Linkage analysis, Mutation screening, Gene functional analysis, Mendelian renal disease
Introduction
Positional cloning is one of the most important molecular approaches to genetic disorders. Since its introduction in research practice in the early 1980s, it has been widely utilised for defining the molecular origin of genetic diseases with Mendelian traits and still represents a necessary research step in this area. The term identifies a series of molecular techniques (not a single one) that usually lead, during successive steps, to the definition of causative genes. Overall, the strategy of positional cloning starts with the identification of a candidate gene, according to chromosomal location, and is followed by mutation analysis in affected individuals. The second phase, which follows the identification of relevant mutations, consists of experiments aimed at defining the causative relationship between a gene mutation and an aberrant phenotype, very often including in vitro tests and/or experiments on animal models (Fig. 1). There are, however, several alternative strategies that allow the identification of a causative gene, starting from animal models or from proteomic approaches and that are nowadays considered as an integral part of positional cloning. They constitute the so-called inverse genetics, a term that indicates how genotype identification is followed by phenotype characterisation, in contrast to “forward genetics”, in which the study of phenotype leads to genotype identification [1]. Inverse genetics is gaining increasing popularity, thanks to the completion of genome sequencing, the improvement of databases, and the use of modern proteomic techniques that allow the detection of even very tiny differences in the proteomes of patients with respect to control samples. These strategies, already fully described in the report by Groenen and van den Heuvel [2], are out of the scope of this teaching section.
Fig. 1.
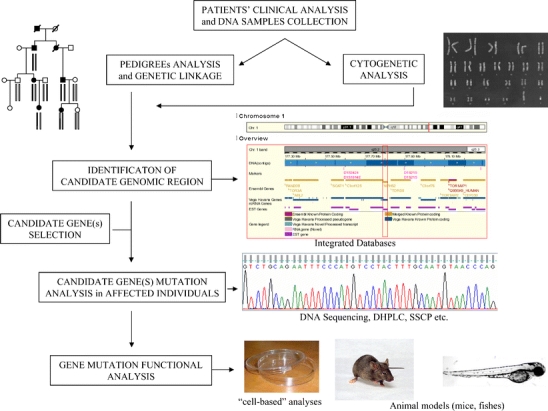
Main steps in positional cloning of genetic disorders (DHPLC denaturing high-performance liquid chromatography, SSCP single-strand conformation polymorphism)
There is a long list of renal diseases characterised, over years, by traditional positional cloning, including the identification of causative genes of cystic diseases with dominant (ADPKD1, ADPKD2) and recessive traits, Alport’s syndrome, nephronophthisis and medullary cystic diseases. These are only examples of disorders with relatively frequent occurrence, but the list of rarer conditions is obviously quite long.
As already pointed out, traditional positional cloning consists of different consecutive steps that will be reviewed separately.
Patient recruitment and DNA sample collection
An accurate clinical analysis, with proper classification of patients according to established criteria, is an essential aspect during this preliminary phase, since a mistake in interpreting clinical data would cause an inappropriate classification. In this phase, the disease of interest should be clearly differentiated from an already characterised condition, to avoid unnecessary duplication of analysis. The use of public databases such as GeneClinics (http://www.geneclinics.org) and OMIM (Online Mendelian Inheritance in Man, a catalogue of human genes and genetic disorders, http://www.ncbi.nlm.nih.gov/) facilitates the search for both clinical aspects and gene mutations associated with a specific disease. In addition, these databases indicate available molecular diagnostic tests that integrate clinical data for a fine classification of patients. Only conditions that are of new description or for which responsible genetic mutations are still to be identified are to be considered for positional cloning. A basic point in this respect is to consider the clinical variability usually associated with specific gene mutations that suggests the extension of the clinical study to family members possibly sharing one or more clinical signs. The type of inheritance that is inferred from pedigree analysis indicates segregation modes that are a key aspect of the problem. It must be clearly distinguished between simple Mendelian and complex traits, the latter being characterised by mutations/variants in different genes with often additive effects and environmental interactions.
DNA samples are then collected from all the members of interest after informed consent had been obtained.
Positional cloning starting from cytogenetic evidence
A major contribution to the identification of disease genes comes from studies in patients with chromosome anomalies that, when present, suggest a direct causative link with the disease. Balanced translocations or inversions are of particular interest, since, in principle, they are not associated with loss or gain of DNA material. Therefore, the association of an “apparently” balanced translocation/inversion with an abnormal phenotype may indicate that the aberration is not really balanced and that a small quantity of DNA, undetectable by traditional cytogenetic analysis, has been lost during rearrangement. Alternatively, a balanced translocation may have interrupted the disease gene or separated it from an expression regulatory region. In these cases, cloning of chromosome breakpoints leads to the identification of the disease-associated gene. The identification of the gene for polycystic kidney disease (PKD1) is a good example of this approach [3]. In 1994, in a Portuguese family with polycystic kidneys, the European Polycystic Kidney Disease Consortium isolated a gene encoding a 14-kb transcript that was disrupted by a chromosome translocation. In this family the mother had a balanced translocation, 46,XX t(16;22)(p13.3;q11.21), which had been inherited by her daughter. The mother and the daughter with the balanced translocation had the clinical features of PKD1, while the parents of the mother were cytogenetically normal, with no finding of renal cysts on ultrasound examination. The consortium isolated a gene spanning the breakpoint which was found mutated in other patients with PKD1.
Deletions are other chromosome aberrations that may favour positional cloning in some conditions. Depending on the extent of a deletion, few or several genes may be deleted, which results in multiple-organ effects that identify a condition also known as “contiguous gene syndrome”. Mental retardation is often associated with large chromosomal deletions.
Interstitial or telomeric chromosome deletions extending over small regions may be of interest. In general, unlike translocations, deletion breakpoints may be located at some distance from the disease gene, confining the search for a candidate gene to small genomic regions flanked by the deletion breakpoints. Several patients with de novo mutations are, in fact, carriers of chromosome alterations, implying that classical cytogenetic G-banded metaphase chromosome analyses must be performed in the case of de novo mutations.
Microarray-based comparative genomic hybridisation (array CGH) is a new method allowing the detection of subliminal chromosome anomalies throughout the genome. Basically, the method consists of the use of DNA from matched control and patient cells in competitive fluorescence in situ hybridisation on oligonucleotide-array probes designed in silico. Image-processing software reveal genomic regions where the ratio between two fluorescence signals from control and patient DNA deviates from expectation, indicating gain or loss of DNA material. Prospective studies have demonstrated that array CGH has the potential to detect all types of genomic imbalance, including deletions, duplications, aneuploidies and amplifications, with detection rates ranging from 5% to 17% in individuals with normal results from prior routine cytogenetic testing [4]. CGH has already achieved great results in the vast area of the search for genes involved in tumour development. The recent paper by Rivera et al. [5], who identified a new gene, WTX (a Wilms’ tumour gene on the X chromosome), inactivated in sporadic Wilms’ tumours, is only an example of this application. The authors searched for DNA copy-number changes in 51 primary tumour specimens by performing a genome-wide scan with array CGH. Results indicated small deletions at chromosome Xq11.1 overlapping the WTX gene in five out of 26 male patients. By using the more classical fluorescence in situ hybridization (FISH) cytogenetic analysis, they also detected heterozygous deletions in six out of 25 female patients, affecting the active copy of the X chromosomes. Finally, point mutations were also identified in additional patients. Notably, no tumour with a deletion or point mutation in WTX contained mutations in any of the other two genes, namely WT1 and β-catenin, known to be involved in Wilms’ tumours, which suggests that inactivation of the WTX gene may account for a distinct subset of nephroblastomas. The importance of the results obtained by the use of array CGH in identifying disease genes will encourage its application to the search for genes involved in renal hereditary diseases.
Positional cloning starting from genetic linkage analysis
A positional approach, based on linkage analysis in families with multiple affected individuals, is still the method of choice for identifying rare mutations implicated in simple Mendelian diseases. For complex traits, which involve several interacting genes and/or multifactorial mechanisms, new statistical methods have been set up, based on the analysis of smaller family units. Transmission analysis of single or multiple molecular markers in affected sibling pairs and in trios (simple families composed of parents and one affected child) is an emerging technique in complex traits, in addition to comparison of unrelated patients and controls for population-based association studies. So far, only limited application of these techniques has been reported (for reviews see Forabosco et al. [6]).
The logical basis for genetic mapping lies in the occurrence of genetic recombination of maternal and paternal homologues, which results from crossing over during meiosis. Linkage analysis tests whether specific disease characteristics segregate at meiosis independently or in association with genetic markers widespread in the different segments of the human genome. DNA single nucleotide polymorphisms (SNPs) and short tandem repeats, also called DNA microsatellites, are utilised as markers of the segment of DNA of interest. SNPs are the most common form of DNA sequence variation, occurring when a single nucleotide—A, T, C, or G—in the genome differs among individuals or between paired chromosomes in an individual. Changes can occur in both coding (gene) and non-coding regions of the genome, most of them showing no effect on cell function. DNA microsatellite markers consist of short sequences, typically from one to four nucleotides, repeated in tandem several times and therefore forming highly informative multiple alleles. On the other hand, SNPs are less informative and less useful for human genetic mapping (Fig. 2). In spite of this, SNPs have the advantage of being more diffusely present than microsatellites in the human genome (one every 100 to 300 bases compared with one every 1,000–3,000). In addition, while microsatellite analysis requires DNA sequencing, SNPs may be rapidly characterised by using DNA arrays. It is clear, however, that the choice of using SNP or microsatellites in linkage analysis depends on their availability in the genomic region of interest and on the variability among the members of families under study [1].
Fig. 2.
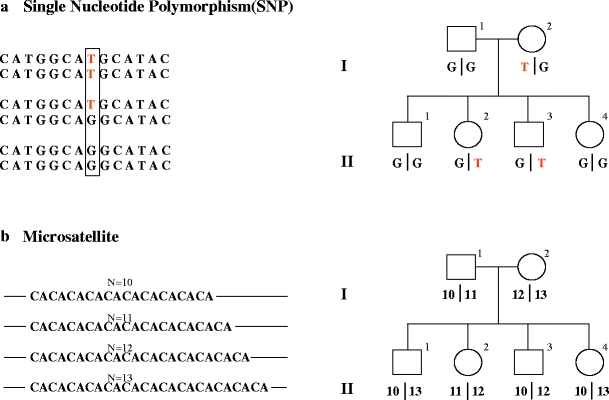
Genetic markers: DNA single nucleotide polymorphisms and microsatellites. a Example of SNP. Left Fragments of DNA with the same sequence containing a difference only in a single nucleotide. The two alleles (G and T) give rise to three possible genotypes: TT homozygous, T/G heterozygous and GG homozygous. On the right-hand side: example of SNP hypothetical segregation in a pedigree. b Example of DNA microsatellite. Left Fragments of DNA with the same sequence containing a (CA) microsatellite. The polymorphism consists of different numbers of the dinucleotide (CA) present in each of the four possible alleles represented (N = 10 to N = 13). Right Example of DNA microsatellite polymorphism hypothetical segregation in a pedigree
When an association is observed, linkage analysis defines by statistical probability whether the association occurs by chance or because the disease gene and the tested genetic markers are closely located in the same genomic segment [1, 6].
The principal method of linkage analysis is the calculation of the logarithm of the odds (lod) score, which is based on the genetic recombination frequencies generated by meiotic cross-overs. Since the probability of a cross-over occurring between two loci is a function of the distance between them, it is possible to estimate this latter by means of the frequency of recombination. If this frequency, known as the recombination fraction (θ), is small, two loci should be closely located in the genome. Conversely, when two loci are distant they will segregate independently and θ will be equal to 50%.
The lod score (Z value) is represented by the logarithm of the odds ratio between the likelihood that one or more markers are linked (recombination fraction = θ) or not linked (recombination fraction = 50%) to the disease phenotype. In other words, the lod score analysis tests the hypothesis that the recombination fractions between the genetic markers and the clinical trait observed are significantly different from 50%, which is the value predicted by the hypothesis of random association. Non-random association is defined by values of the lod score equal to or higher than 3.3, a value actually considered to be the threshold for accepting linkage with 5% probability of false positive results [6].
In general, linkage analysis indicates the interval in which the disease gene is localised in close proximity with the associated markers. The genetic interval is measured in centimorgans (cM), 1 cM corresponding to a 1% chance of a recombination event. In terms of physical distance, 1 cM corresponds, on average, to 1 million DNA base pairs (1 megabase, 1 Mbp). The number of candidate genes that should be considered for mutation analysis depends on the size of the candidate region, so that a strong effort is required to define the smallest candidate region. Usually, after initial genetic mapping that defines a candidate region of 10 Mbp, the region is narrowed by increasing the number of polymorphic markers and, possibly, by looking at other informative families. In principle, a family with 100 informative meioses might restrict the locus of a Mendelian disease to a candidate region of approximately 1 cM, that is, 1 million DNA base-pairs that correspond to very few genes.
Identification of a candidate gene
The step that follows the definition of a critical region for a given disease is the identification of candidate genes. It is based on the analysis of available databases from genome browsers such as Ensembl (http://www.ensembl.org) or Santa Cruz Genome Browser UCSC (http://genome.ucsc.edu) that contain large numbers of data covering the whole genome but few areas that are still to be characterised. In such cases, alternative strategies should be designed. Databases contain information on gene maps and specific expression profiles and, in some cases, provide relevant hints on functions. For genes not yet cloned, or in the case of limited expression data in specific tissues, hypothetical transcripts are defined, as deduced from bio-informatic tools. Genome browsers provide a considerable number of data on expression and functions of paralogous and orthologous genes in different animal species, including data on mouse mutants that may be essential in the strategy. Once the list is available, it is necessary to rank genes endowed in the critical zone on the basis of expression and/or function consistent with disease development. One practical criterion is to consider those genes that are expressed in organs and tissues involved in the disease phenotype, either during adulthood or embryonic development. When not available in databases, expression data should be generated by new laboratory investigations using classical and innovative strategies based on reverse transcriptase-polymerase chain reaction (RT-PCR), Northern blot, in situ hybridisation and expression arrays [1]. Since it is accepted that humans and mice have similar expression patterns, many experiments can be carried out on mouse tissues, especially if the use of embryonic tissues is required. Other practical approaches for identifying candidate genes are to consider functions of specific genes and/or, in the case of similar diseases, functional homologues that are involved in similar diseases. For example, an ion channel may be a good candidate for diseases of the renal tubule. Again, molecules of the cilia may be good candidates for nephronophthisis and/or other renal cyst diseases.
Search for mutations and functional analysis
The search for functional mutations in a candidate gene is the step following positional cloning. The term mutation defines a change in DNA sequence in a given gene that is not present in the general population. The presence of the mutation is not, however, ‘per se’, a direct demonstration of a causative relationship with a clinical phenotype. In fact, causative mutations should be clearly related to a functional impairment that is caused by the change itself. Stop codons, frame-shifts, nonsense mutations and micro-deletions are, in this sense, most probative of a structural change, since they lead to the generation of short unstable transcripts and/or short proteins lacking functional domains. Another piece of evidence proving a direct involvement in the pathogenesis of the disease is the demonstration of an aberrant effect in animals.
Among different methods for detecting new mutations [i.e. single strand conformational polymorphism (SSCP); heteroduplex analysis (HA)], the most widely utilised are denaturing high-performance liquid chromatography (DHPLC) and direct sequencing after PCR amplification of exon fragments of genomic DNA. DHPLC gives information about the specific elution profile of variants, which must then be demonstrated by direct sequencing. It is a rapid and completely automatic approach and is, for these reasons, the technique of choice to utilise in screening studies. Deletions, inversions and/or mutations involving non-exonic regions (i.e. intronic and promoter regions) are difficult to detect. In the former case, the use of more sophisticated techniques is required. Pulsed-field gel electrophoresis and Southern blot, FISH on chromosome metaphases and DNA microsatellite analysis with reconstruction of inherited haplotypes are the techniques of choice for detecting large molecular deletions. Array CGH for smaller deletions has been described above. Intronic mutations may be involved in the splicing of the mature transcripts and, therefore, affect expression. Splicing errors of RNA molecules, as well as mutations affecting more, in general, transcription regulatory sequences, may be detected by RT-PCR in tissues in which the candidate gene is expressed (see [7] for an overview on the RNA maturation process and RT-PCR technique). Briefly, the method consists of a first step that includes reverse transcription, followed by PCR amplification, of an expressed gene using its RNA, instead of genomic DNA, as starting material. By using this technique one can directly and quite rapidly test a gene of interest for mutations possibly impairing its qualitative or quantitative expression, as mutations in transcription regulatory sequences or in sites regulating RNA splicing may do. Unfortunately, this approach is often confined to genes expressed in blood cells or fibroblasts, because of their accessibility, while the definition of splicing mutations in target organs is limited, due to tissue sampling. A key point of positional cloning that should always be kept in mind is that there are mutations with no clear functional effects that cannot be readily related to the pathogenesis of a disease. In general, they are missense mutations, causing only minor changes in DNA sequence without relevant effects on gene expression and function. Owing to their minor impact on gene function, the distinction between missense mutations and rare neutral variants may be, in some cases, difficult and require screening studies in a general population. Functional tests carried out in cells and based on in vitro techniques are required in these cases to confirm a relationship with human pathology. Expression studies involve transfection of cell lines with a mutated cloned cDNA and are mainly based on functional tests searching for impairment of cell physiology. An alternative to the use of cDNA cloned in the laboratory is the use of cells deriving from patients presenting the basic defect. In general, cell experiments require positive and negative (i.e. wild type cells) controls. Sometimes, it is possible to rescue the abnormal cell phenotype by transfecting the mutated cells with a wild type form of the gene, obtaining a confirmation of the pathogenetic mechanisms.
As already discussed, experiments on animals are necessary for final validation. They consist of definitive suppression (knock out) of a gene and are, in some cases, based on the generation of animal strains incorporating the same mutations observed in humans (knock in) [8]. Several spontaneous, chemically-induced or transgenic mouse mutants have been generated in the past few years and are maintained in animal facilities (such as The Jackson Laboratories in the USA, http://www.jax.org/ and the EMMA archive in Europe, http://www.emma.rm.cnr.it/). Chemical mutagenesis causing random allelic point mutations in mice is, nowadays, a promising technique for creating animal models of human diseases. The most popular method to obtain chemical mutagenesis is the use of N-ethyl nitroso-urea (ENU), which has been widely utilised to create abnormal phenotypes. ENU mice have already been generated and characterised on a phenotypic basis by international consortiums and are available on request. Therefore, the advantage of ENU mice is their clinical characterisation, while their drawback is the lack of genetic information, which requires a lot of molecular work. Phenotype screenings and genetic and molecular analyses of ENU mice are now in progress, with the clear objective of building up an informative system to be utilised at any occurrence [9].
A key aspect, and probably the major drawback, of animal models, considering knock-out and knock-in mouse types, is the phenotype that often does not reflect human beings.
An emerging alternative to the use of mouse mutagenesis for functional genomics and disease modelling is represented by the system of morpholino-phosphorodiamidate-based anti-sense oligonucleotides (MPOs) for gene-specific knock-down in Zebrafish (Danio rerio) [10].
MPOs, originally developed as therapeutic agents, are oligonucleotides in which the ribose sugar has been replaced with a morpholine moiety, and the phospho-diester linkage between nucleotides has been replaced with a phospho-rodiamidate non-ionic linkage. When an MPO hybridises to its target RNA, it is hypothesised to block the 40S ribosomal subunit scanning process, thereby effectively preventing initiation of translation. MPOs seem to work in the animal systems where they can be delivered early in development by micro-injection. The Zebrafish is well suited as a model organism, due to the ease of delivery by micro-injection. Other advantages of the use of Zebrafish are the fast rate of development and the transparency of embryos, which allows easy visualisation of fundamental vertebrate developmental processes. Since this teleost, unlike simple invertebrates, maintains the biological and genomic complexity found in higher vertebrates, its use in developmental disorder studies has been increased. Recently, this system has also been successfully used to obtain models of renal disorders [11, 12]. Results obtained to date highlight the efficiency and rapidity of the method and highly encourage its use as a tool for in vivo function assessment of vertebrate genomes.
Most recent applications in nephrology
An example of the successful application of positional cloning in nephrology is the identification of a gene for a variant of nephrotic syndrome, recently reported by Hinkes et al. [12]. These authors utilised haplotype-based linkage analysis to delimit the critical region to an interval of 4.0 Mb on chromosome 10q23.32–q24.1 that contains 43 predicted genes. Candidate gene selection prior to mutational analysis was based on the hypothesis that nephrotic syndrome should rely on mutations in a gene expressed in podocytes. Glomerular DNA expression of the 43 genes of the critical region was evaluated with microarrays that demonstrated a reproducible expression profile at this site for only three genes. Mutational analysis showed different homozygous nonsense and frameshift mutations of phospholipase c epsilon (PLCE1) in affected individuals that predicted the synthesis of truncated forms of the protein, with loss of function. The confirmation of a functional implication of PLCE1 in glomeruli was obtained in Zebrafish in which the plce1 knock-down produced an altered structure of slit-diaphragm, effacement and disorganisation of podocyte foot processes, with consequent disruption of the glomerular filtration barrier and oedema. It is noteworthy that the knock-out mouse model for PLCE1 reported in 2005, before the finding of mutations in nephrotic syndrome [13], presented no renal phenotype and, most importantly, no nephrotic syndrome. The presence of modifier genes in the mouse strain utilised for the knock-out of the gene may explain this apparent contradiction and also gives an idea of the problems that can be encountered with animal models of human diseases. The evidence in Zebrafish is sufficient, in this case, to prove the direct relationship between functional mutations of PLCE1 and nephrotic syndrome.
Conclusions
Positional cloning based on genome-wide mapping and candidate gene analysis will probably remain the method of choice for defining Mendelian traits in the near future. Among basic requirements that have to be fulfilled prior to the start of the molecular approach, the availability of informative families with clear clinical phenotypes still remains a challenge for researchers. Owing to technology advancements, an increasing number of molecular tools, such as adequate DNA markers, information on gene sequences and expression profiles and functions, will allow the extension of positional cloning to those conditions still lacking molecular characterisation. Multifactorial diseases represent the new frontier that has to be rapidly faced. The definition of regulatory mechanisms for gene expression (transcriptome), a better knowledge of protein structure and functions (proteome) and new animal models of the disease are essential topics for future studies.
Acknowledgements
Data were critically discussed with Prof. Rosanna Gusmano, and we acknowledge her role. The manuscript was revised by Miss Capurro.
References
- 1.Strachan T, Read A. Human molecular genetics. 3. London: Garland; 2003. [Google Scholar]
- 2.Groenen PJ, Heuvel LP. Teaching molecular genetics: chapter 3—proteomics in nephrology. Pediatr Nephrol. 2006;21:611–618. doi: 10.1007/s00467-006-0064-z. [DOI] [PubMed] [Google Scholar]
- 3.The European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 4.Shaffer LG, Bejjani BA. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115:303–309. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- 5.Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, Kim JC, Feinberg AP, Gerald WL, Vargas SO, Chin L, Iafrate AJ, Bell DW, Haber DA. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642–645. doi: 10.1126/science.1137509. [DOI] [PubMed] [Google Scholar]
- 6.Forabosco P, Falchi M, Devoto M. Statistical tools for linkage analysis and genetic association studies. Expert Rev Mol Diagn. 2005;5:781–796. doi: 10.1586/14737159.5.5.781. [DOI] [PubMed] [Google Scholar]
- 7.Knoers NV, Monnens LA. Teaching molecular genetics: chapter 1—background principles and methods of molecular biology. Pediatr Nephrol. 2006;21:169–176. doi: 10.1007/s00467-005-2154-8. [DOI] [PubMed] [Google Scholar]
- 8.Claij N, Peters DJ. Teaching molecular genetics: chapter 2—transgenesis and gene targeting: mouse models to study gene function and expression. Pediatr Nephrol. 2006;21:318–323. doi: 10.1007/s00467-005-2110-7. [DOI] [PubMed] [Google Scholar]
- 9.Guenet JL. The mouse genome. Genome Res. 2005;15:1729–1740. doi: 10.1101/gr.3728305. [DOI] [PubMed] [Google Scholar]
- 10.Sumanas S, Larson JD. Morpholino phosphorodiamidate oligonucleotides in zebrafish: a recipe for functional genomics? Brief Funct Genomic Proteomic. 2002;1:239–256. doi: 10.1093/bfgp/1.3.239. [DOI] [PubMed] [Google Scholar]
- 11.Kramer-Zucker AG, Wiessner S, Jensen AM, Drummond IA. Organization of the pronephric filtration apparatus in zebrafish requires nephrin, podocin and the FERM domain protein mosaic eyes. Dev Biol. 2005;285:316–329. doi: 10.1016/j.ydbio.2005.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Muller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’Toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nurnberg P, Hildebrandt F. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res. 2005;97:1305–1313. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]