

Abstract
Although a variety of possible functions have been proposed for inverted repeat sequences (IRs), it is not known which of them might occur in vivo. We investigate this question by assessing the distributions and properties of IRs in the Saccharomyces cerevisiae (SC) genome. Using the IRFinder algorithm we detect 100,514 IRs having copy length greater than 6 bp and spacer length less than 77 bp. To assess statistical significance we also determine the IR distributions in two types of randomization of the S. cerevisiae genome. We find that the S. cerevisiae genome is significantly enriched in IRs relative to random. The S. cerevisiae IRs are significantly longer and contain fewer imperfections than those from the randomized genomes, suggesting that processes to lengthen and/or correct errors in IRs may be operative in vivo. The S. cerevisiae IRs are highly clustered in intergenic regions, while their occurrence in coding sequences is consistent with random. Clustering is stronger in the 3′ flanks of genes than in their 5′ flanks. However, the S. cerevisiae genome is not enriched in those IRs that would extrude cruciforms, suggesting that this is not a common event. Various explanations for these results are considered.
Electronic supplementary material
The online version of this article (doi:10.1007/s00294-010-0302-6) contains supplementary material, which is available to authorized users.
Keywords: Palindromes, Imperfect inverted repeats, Hairpins, Yeast
Introduction
An inverted repeat (IR) occurs when two exact or approximate copies of a particular DNA sequence are present in reverse complement orientation. An IR is called a palindrome if the two copies directly abut each other, so their separation distance (here called the spacer length) is zero. If the repeats are exact matches the IR is said to be perfect, otherwise it is imperfect.
Both perfect and imperfect inverted repeats are known to occur at sites involved in a variety of normal and pathological events. For example, they are found at viral origins of replication (Pearson et al. 1996; Chew et al. 2005; Leung et al. 2005). Poxviruses, which replicate by a rolling circle mechanism, have long imperfect IRs centered at the location the cutting of which resolves the linear multimer into unit length genomes. If the imperfections are removed, leaving a perfect IR, viral infectivity decreases to 5% of its original value (Costello et al. 1995).
Bacterial genomes contain IRs the properties or locations of which suggest either genomic function or specific mutational mechanisms. For example, IRs are found in leader sequences and/or terminal flanks of many genes. The latter sites are involved in alternative termination through the process of attenuation (Li et al. 1997). In E. coli, the lac operator binds lac repressor at two sites in inverted orientation. Here, imperfections in the IR sequence decrease binding affinity, and hence are important for precise regulatory control (Sadler et al. 1983).
Inverted repeats are sources of genetic instability both in prokaryotes and in eukaryotes (Gordenin et al. 1993; Bi and Liu 1996; Nag and Kurst 1997; Samadashwily et al. 1997; Lobachev et al. 1998; Moore et al. 1999; Butler et al. 2002; Lee et al. 2002; Achaz et al. 2003; Lobachev et al. 2007; Eykelenboom et al. 2008). In particular, large palindromes are associated with amplified genes (Butler et al. 1996; Rattray et al. 2005; Tanaka et al. 2005, 2007; Lewis and Cote 2006; Inagaki et al. 2007), and have been suggested to be important in late tumor progression. Inverted repeats are also known to occur at meiotic recombination hotspots (Moore et al. 1999; Butler et al. 2002).
Although IRs are present at regulatory regions governing a variety of physiological processes, it is not usually known what role, if any, the repeat itself may play in the regulatory mechanism. A variety of possibilities have been suggested. A bidirectional process involving sequence-specific protein binding could require two binding sites arranged in inverted orientation. This happens, for example, at the origin of replication of the SV40 virus (Pearson et al. 1996). Similarly, any transcriptional regulatory activity that requires either antiparallel binding of two copies of a transcription factor, or binding of a homodimeric factor with antiparallel binding sites, would require an IR.
Hairpins, the most common RNA secondary structural elements, are produced by intramolecular Watson–Crick binding. The DNA sequence encoding an RNA hairpin must be an inverted repeat. (In eukaryotes, the encoding IR copies could span multiple exons). This suggests that IRs within transcribed DNA sequences could be present in order to serve structural, and possibly regulatory, roles within the RNA transcript.
Other proposed mechanisms by which IRs might participate in regulatory processes involve the formation of alternative DNA structures in which the two repeat copies on the same DNA strand pair together to form a hairpin. (An example is shown in Fig. 1.) If both DNA strands of an IR form hairpins simultaneously, the result is a cruciform.
Fig. 1.
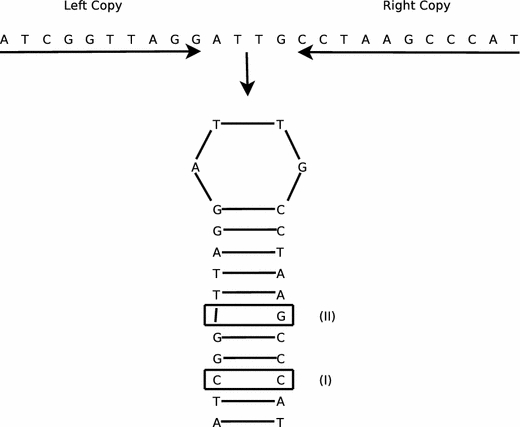
An imperfect inverted repeat and its hairpin structure. The hairpin has nine matched base pairs, one mismatch at site I, and one indel (an insertion in the right copy or a deletion in the left copy) at site II
Physiological levels of negative superhelicity have been shown to drive transitions to alternative DNA conformations, and cruciforms in particular, at susceptible sites in vitro (Gellert et al. 1983; Greaves et al. 1985; Bauer and Benham 1993; Dai et al. 1997; Benham et al. 2002; Kouzine et al. 2004; Kouzine and Levens 2007). However, while superhelical cruciformation in DNA has been documented to occur in vivo (Sinden et al. 1991; Dayn et al. 1992), to date few regulatory roles for this transition have been experimentally demonstrated. One example occurs at the leuV operon of E. coli, where the superhelically driven extrusion of a short cruciform within the transcribed DNA was shown to down-regulate transcription (Opel et al. 2004). Cruciform-binding proteins have been proposed to be associated with origins of replication (Pearson et al. 1996; Alvarez et al. 2002; Novac et al. 2002; Zannis-Hadjopoulos et al. 2008). Despite the paucity of evidence that cruciforms extrude in genomic DNA in vivo, cruciformation has been hypothesized to be involved in a variety of normal and pathological regulatory mechanisms (Pearson et al. 1996; Dai et al. 1997; Spiro and McMurray 1997; Kim et al. 1998; Shlyakhtenko et al. 2000).
Mutational events have been suggested that involve cruciforms or hairpins either directly (Ripley 1982; Akgun et al. 1997; Nasar et al. 2000; van Noort et al. 2003; Rattray et al. 2005) or indirectly (Ripley 1982; van Noort et al. 2003). Hairpin formation has been proposed to occur on the DNA lagging strand when the replication fork stalls (Voineagu et al. 2008). Extension of such hairpins has been suggested as a possible mechanism for lengthening short IRs (Dutra and Lovett 2006). It has also been proposed that imperfections in IR symmetry could be recognized and corrected, either by DNA repair mechanisms or by methyl-directed mismatch repair during replication (Ripley 1982; van Noort et al. 2003; Dutra and Lovett 2006). This could occur either on a hairpin arm or on the incorrectly templated DNA arising from intermolecular switching. However, methyl-directed mismatch repair is not expected to occur in Saccharomyces cerevisiae, the subject of this paper, since DNA methylation does not occur in this organism.
Other proposed mechanisms would extend short IRs to form longer ones. These have been associated with gene amplification (Butler et al. 1996, 2002; Rattray et al. 2005). For example, Rattray et al. (2005) implicated small IRs (4–6 base pairs (bp) in each copy, with 2–9 bp spacers) as primers for DNA synthesis via intramolecular fold-back at their 3′ ends. This would lead to the formation of large palindromes, and possibly to gene amplification. Over time, these processes would increase either the degree of perfection (Ripley 1982; Dutra and Lovett 2006) or the length (Lin et al. 1997; Dutra and Lovett 2006) of genomic IRs.
While many mutagenic or functional roles have been proposed for DNA inverted repeat sequences, it is often not known which of these activities actually occur in vivo, and with what frequencies. We investigate this question by performing a careful assessment of the distributions of both perfect and imperfect IRs in the S. cerevisiae genome. Many of the proposed roles for IRs, if they were to occur with any frequency in vivo, would result in different genomic IR distributions than would occur if IRs were evolutionarily neutral. The basic premise is that specific types of IRs that serve regulatory functions would be evolutionarily conserved, while those that are deleterious, perhaps because they are sources of genetic instability, would be preferentially eliminated. If an IR was needed for some other purpose (e.g. to form an RNA secondary structure), yet DNA cruciform extrusion would be deleterious, evolution might enrich for arrangements in which this transition is disfavored. This could involve either increased numbers of imperfections or longer spacer regions. Mechanisms that increase the length or degree of perfection of IRs would lead to corresponding enrichments of these sequence types.
In this study we use the Inverted Repeat Finder (IRF) algorithm to search for both perfect and imperfect IRs with spacer lengths ranging from 0 to 76 bp in the yeast genome. IRF is a heuristic algorithm that locates IRs with much higher reliability and efficiency than its predecessors. It has previously been used to search for long IRs in the human genome (Warburton et al. 2004). We first examine the genomic distribution of IRs in the 16 chromosomes of the S. cerevisiae (SC) genome (accession numbers NC_001133-NC_001148). We then evaluate two classes of IR features. Their inherent features are copy length (or right and left copy length for imperfect IRs), spacer length, degree of perfection, percent A+T composition, dinucleotide repeat content, percent match, percent insertion–deletion, and IRF score. The IR distributional properties studied here include the number N IR of other IRs overlapping a given IR, the number N bp of IRs overlapping a given base pair, the distributions of IRs in coding versus intergenic regions as well as in the 5′ and 3′ gene flanks of experimentally verified CDS regions, and, for each isolated IR, the distance to its nearest IR neighbor. To enable the statistical significance of our findings to be determined we randomize the S. cerevisiae genomic sequence in two ways, and compare IR properties and distributions in the actual S. cerevisiae genome with those from the randomized genomes.
The rationale for this approach is that the presence of distributional anomalies in IR numbers or features might suggest or support proposed functional or mutational roles of genomic IRs. Statistically significant enrichments at specific classes of sites could suggest evolutionary preservation. Conversely, paucities of IRs at particular positions relative to random would suggest that deleterious consequences may favor their removal.
Previous studies have found substantial numbers of perfect IRs with short spacers, and of very long imperfect IRs, in prokaryotic and in eukaryotic genomes (Lilley 1980; Schroth and Ho 1995; Cox and Mirkin 1997; Achez et al. 2000, 2003; LeBlanc et al. 2000; Lillo et al. 2002; van Noort et al. 2003; Lisnic et al. 2005; Wang and Leung 2006; Inagaki et al. 2007; Lu et al. 2007). Van Noort et al. report findings that perfect IRs occur with greater frequency than expected at random. This supports the proposal that IRs tend toward perfection, perhaps either through spontaneous mutations or by error-correcting mechanisms (van Noort et al. 2003). Other studies have shown that IRs tend to be both longer and more densely located in intergenic regions than in coding regions (Cox and Mirkin 1997; Achez et al. 2000; Lisnic et al. 2005).
This work independently confirms and greatly extends results of previous investigations of the genomic distributions of IRs in yeast. However, it is much more complete, largely because of the power of the IRF algorithm. Previous investigations of IRs in the yeast genome found at most a few hundred examples (Cox and Mirkin 1997; Achez et al. 2000; Wang and Leung 2006). The highest density found previously was 58 IRs in a study of chromosome III (Schroth and Ho 1995), which would be scale to about 2,300 IRs in the entire yeast genome. In contrast, in the present work IRF finds 100,514 IRs. We also examine the properties of these IRs in much greater depth than has previously been done, including both their inherent features and their genomic distributions.
Methods
Copy and spacer lengths
In this analysis we limit consideration to IRs with spacer lengths between 0 and 76 bp, and copy lengths larger than 6 bp. IRs with spacers longer than this are not expected to participate in either fold-back mechanisms or DNA cruciformation. Whether superhelical cruciform formation is favored or not depends on the relative magnitudes of the energy cost of the transition and the energy benefit consequent on the fractional superhelical relaxation that the transition provides (Benham 1982). To be favored for extrusion, an inverted repeat must have arm length sufficient to relax enough superhelicity to offset the energy costs associated with nucleation. The nucleation energy increases with the length of the spacer and the presence of imperfections. These energetics determine the propensity of a given IR, considered in isolation, to extrude at a given level of superhelical stress. As shown below, IRs in the S. cerevisiae genome occur on average once every 120 bp, hence many would be present within any given local region. Although each of these in principle could extrude a cruciform, in practice they compete. The IRs likely to extrude will be those the cruciformation energetics if which are most favorable. These will have long copies, short spacers and few imperfections. Using methods presented elsewhere (Benham et al. 2002), we calculate that a perfect IR with spacer length greater than 76 bp has an insignificant probability of extrusion, even in isolation (data not shown). We do not expect IRs with large spacers to be susceptible to extrusion in vivo, due to both competition and kinetic barriers involved in denaturing the large spacers (Courey and Wang 1983; Murchie and Lilley 1987).
Inverted repeat finder
In this study IRs were detected using the Inverted Repeat Finder (IRF) algorithm, the basic structure of which has been presented elsewhere (Warburton et al. 2004). The IRF does not require perfect symmetry, but rather scores complementary pairs positively and assesses a penalty for violations of symmetry, either by insertions or deletions (indels) or by mismatched bases. The scoring function used here scores Watson–Crick base pairs +2, while each indel scores −5, and each mismatch scores −3. There is no penalty for spacer length, so the algorithm can find repeat copies with wide separations, although here we limit consideration to IRs with spacer lengths less than 77 bp. All scores above 14 are reported, so the shortest perfect IR detected by IRF has copy length 7. It is important to note that with this scoring scheme the number of imperfections that can be included in a reported IR increases with copy length.
Figure 1 illustrates a simple IR with two imperfections, a mismatch and an insertion–deletion. Here, the lengths of the left and right copies are 10 and 11, respectively, and the spacer length is 4. The hairpin this IR forms is 11 bp long, with 9 matches, 1 mismatch, and 1 indel, resulting in a total score of 10.
This scoring scheme may be oversimplified, since different bases involved in either indels or mismatches need not have uniform effects on either an extruded DNA cruciform or on a hairpin arm. However, this may not matter for IRs that occur at random or are present for other purposes than cruciformation. Incorporation of these factors is not feasible in the current incarnation of the IRF (Warburton et al. 2004).
Construction of randomized genomes
To enable assessments of statistical significance, we generated two types of randomizations of the S. cerevisiae genome. The first type, the R genomes, was created by computationally implementing a Fisher–Yates shuffle on each S. cerevisiae chromosome (Fisher and Yates 1938; Knuth 1997), which randomly shuffles all the base pairs within individual chromosomes. A similar procedure was previously used by others (Lisnic et al. 2005).
In yeast the base composition of coding (i.e. genic) regions is known to differ from that of intergenic regions. As this could affect IR frequency (we show below that it does), we account for it in our second type of randomization. Here, each chromosome was partitioned into genic and intergenic regions according to their GenBank annotations. The bases in the genic regions were collected together and then distributed randomly to all the genic regions of the chromosome. The intergenic regions were randomized in a similar manner. The final result is a chromosome in which the genic and intergenic regions were separately shuffled in aggregate, while preserving the positions and lengths of these regions. This was done for each chromosome independently. The resulting randomized genomes are called the RA (randomized as annotated) genomes.
As much as 50 randomizations were made of each type (R and RA) and the IRF was applied to each. Comparison of the IR properties of the actual genome with their distributions in randomized sequences allows determinations of statistical significance for each inherent and distributional attribute, as described below.
The RA genomes are expected to generate more biologically informative null hypotheses than are the R genomes regarding the genomic distributions of IRs and their associated features. However, results for R genomes are still included for purposes of comparison with earlier work. Specifically, previous investigators have used only uniformly (i.e. R-) randomized sequences for comparing with actual IR distributions (Achez et al. 2000; LeBlanc et al. 2000; Lisnic et al. 2005).
Calculations of theoretical perfect IR frequencies
Other authors (Lillo et al. 2002; Lillo and Spano 2007) have presented formulas to compute the expected number of perfect IRs as a function of copy length in a sequence. We verified these formulas and also used them for the same purpose.
IR clustering
We examined the clustering of IRs by determining for each IR the number of other IRs that overlap it. This is called the IR overlap number, N IR. This was done separately for each genome type. We also assessed the extent of positional clustering by determining the base pair overlap number, N bp, for each base pair.
Comparing IR distributions around gene boundaries
The S. cerevisiae genome contains 6,155 annotated genes. Of these, 4,665 have experimentally verified CDS (conserved domain sequence) regions. Among these, there are 3,832 genes that have both flanking intergenic regions longer than 150 bp. We selected 450 bp regions around the gene start site for each of these 3,832 genes, and aligned them with their start positions at 150, oriented so that transcription proceeds to the right. We determined the base pair overlap number, N bp, for each of these 450 positions in each aligned sequence. This produced a distribution over the 3,832 genes of the N bp values at each location. We also did this for the terminal regions, aligned with their stop sites at position 300. This procedure was performed for the S. cerevisiae genome, and for each of the R and RA randomizations. In the latter cases we pooled together the values from the 50 randomizations of each type and then determined the average value at each position for each of the 3,832 genes.
Similar analyses were done separately for the 5′ gene flanks in divergent versus tandem intergenic regions, and for the 3′ gene flanks in convergent versus tandem regions. (Divergent, tandem and convergent refer to the directions of transcription of the abutting genes.) We also considered the distributions of IRs having short spacers, which are most susceptible to cruciformation. Here, we performed the same analysis, but limited consideration to IRs with spacer lengths in the ranges [0, 10], [11, 20], and [21, 30].
Statistical comparisons of distributions
Two types of data were developed in this study. In the first type a single number is determined for each sequence (genome or chromosome). Examples are total number of IRs, or numbers of perfect or imperfect IRs. In such cases the data for each of the randomizations also consist of a single number. As we have 50 randomizations of each type, we generate a distribution of values for each randomization type, the mean and standard deviation of which we determine. We assess the statistical significance of the number from the S. cerevisiae genome by determining how many standard deviations it is away from the mean of the randomized genomes, which is also called its z-score. This is done separately for the R and the RA randomizations.
The second type of data consist of a distribution of values for each genome. Examples include inherent IR features, such as spacer length, copy length, percent A–T (%AT), percent match (%M), or IRF score, and distributional features, such as the base pair overlap number N bp, IR overlap number N IR, and the distance d NN from an isolated IR to its nearest IR neighbor. To determine statistical significance in these cases we generate the distribution for the S. cerevisiae genome, and average together the corresponding distributions for each of the 50 RA (resp. R) genomes. We then use the KS test to determine whether these two distributions are statistically significantly different (Massey 1951; Marsaglia et al. 2003). The KS test is non-parametric, and makes no assumptions regarding the nature of the distributions being compared. It is regarded as a conservative test, so situations where it finds significant differences can be viewed as reliable.
For inter-chromosomal comparisons as well as comparisons involving gene boundary regions, significance levels were made more stringent by applying the Bonferroni correction to account for the fact that multiple questions were being asked of what was arguably the same dataset. This is not necessary when testing a single base pair position, but if we wish to impose a p value cutoff at all positions simultaneously, we must take into account that we are doing multiple testing, where adjacent base pair tests are directly drawing on the same (or very nearly the same) IR dataset. Unless otherwise stated, all references to “statistically significant” results from KS tests, refer to p values smaller than 0.01, adjusted with the appropriate Bonferroni correction if required.
In many of the analyses reported here the data from all chromosomes in the S. cerevisiae genome were placed into a single set, the entire genome set, and analyzed as a single entity. This was done for two reasons. First, the inherent features of IRs were found to show no statistically significant differences between chromosomes. And second, larger sets give more informative statistical comparisons. When we compared the left and right copy length distributions of imperfect IRs by these procedures we also found that they are statistically indistinguishable in all genomes. So in later analyses involving imperfect IRs we consider only the lengths of the left copies.
Results
IR numbers
We used the Inverted Repeat Finder (IRF) program to search for perfect and imperfect IRs in the 16 chromosomes of the S. cerevisiae genome (NC_001133-NC_001148), and in the R and RA randomizations of that genome. We considered only IRs with spacer lengths between 0 and 76 and copy lengths greater than 6. With the above constraints we found 100,514 IRs in the S. cerevisiae genome, an average frequency of approximately one every 120 bp. The randomized genomes contain, on average, fewer IRs than the S. cerevisiae genome, with means and standard deviations given by μ RA = 89,109, σ RA = 337, and μ R = 91,008, σ R = 298. So the total number of IRs in the S. cerevisiae genome is z RA = 34 standard deviations above the RA genome mean, and z R = 32 standard deviations above the R genome mean. There are approximately 11,400 more IRs in the S. cerevisiae genome than would be expected at (RA) random, based on composition. Although this is only a 12.7% enrichment, it is statistically highly significant.
The total numbers of IRs in each chromosome of the S. cerevisiae, RA, and R genomes were found to vary linearly with chromosome length, with values of r 2 in all cases on the order of 0.99 (data not shown). (Here r 2 is the fraction of the observed variation that can be ascribed to the deterministic linear relationship, and 1 − r 2 is the fraction due to random variations about this linear function).
Short-spacer, long-copy IRs
The subset of IRs that would be most competitive for cruciform extrusion are those with long copy lengths and short spacers. This subset would also be expected to be most susceptible to both fold-back mechanisms and intermolecular switching. Indeed, it has been shown that in yeast, the degree that an IR stimulates deletion and/or recombination events is directly related to its copy length and inversely related to its spacer length (Lobachev et al. 1998). IR-mediated recombination events in E. coli were found to be dramatically reduced with increased spacer length (Bi and Liu 1996). Similar results were also found by others (Chalker et al. 1993; Kogo et al. 2007; Lisnic et al. 2009). Thus, short-spacer, long-copy IRs are likely to be the most structurally and/or mutationally susceptible in the genome.
To investigate these questions, we constructed the long-copy, short-spacer set, consisting of all IRs with spacers less than 11 and copy lengths longer than 9. This is done separately for the IRs in the S. cerevisiae genome, and for those in its RA randomizations. The S. cerevisiae genome contained 9,130 long-copy, short-spacer IRs, approximately 9.1% of the total. Interestingly, none of the perfect IRs in this set had copy and spacer lengths that would render them susceptible to cruciform extrusion. While there are IRs in the S. cerevisiae genome that have cruciform-susceptible spacer and copy lengths in this region, none is perfect. As imperfections increase the extrusion energy of a cruciform, they drastically diminish its competitiveness.
We examined four inherent IR properties, spacer length, copy length, percent match, and percent A+T, in the full set of IRs and in the long-copy, short-spacer subset. The results are shown in Fig. 2 for the full set (data not shown for the long-copy, short-spacer set). As shown in Fig. 2a, the number of IRs that have a given spacer length remain relatively constant for spacers greater than 1. This holds true in both the S. cerevisiae genome and its RA randomizations, in both the full sets and the long-copy, short-spacer subsets. For palindromes, which have spacer lengths 0, and for those with spacer lengths 1, IR counts in all cases were somewhat larger. Figure 2b shows that the number of IRs having a given copy length decreases approximately exponentially within both the S. cerevisiae and RA genomes.
Fig. 2.
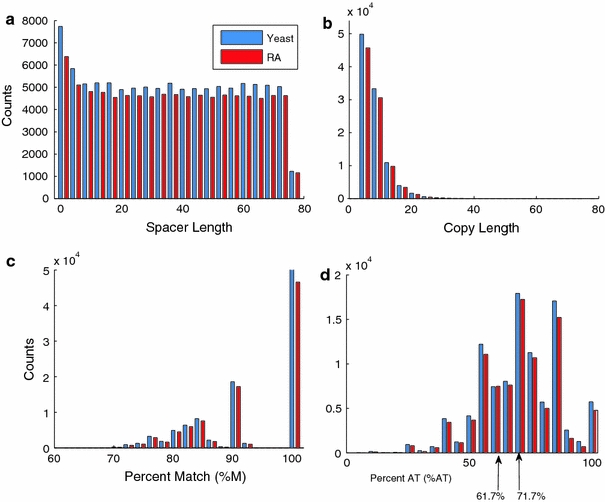
The distribution of four inherent features—spacer length (a), copy length (b), percent match (c), and percent A+T (d)—of the full set of IRs found in this study. Values for the 100,514 IRs found in the S. cerevisiae genome are indicated in blue, while those for a representative RA genome are in red. In part (d) we indicate by arrows the S. cerevisiae genomic A+T content (61.7%) and the average A+T content of the S. cerevisiae IR set (71.7%)
The long-copy, short-spacer subset has a smaller proportion of perfect IRs relative to the full genome. Interestingly, this trend is also observed in the RA random sequences. The distribution of A+T content is somewhat broader, more even and with a higher average value in the long-copy, short-spacer subset (72.8%) than in the full S. cerevisiae genomic set (71.7%) (Fig. 2d). (The genome average A+T-richness is 61.7%.) The long-copy, short-spacer sets did not show any other significant distributional differences between the S. cerevisiae and RA or R IRs sets. So analyzing the full IR dataset will not mask other interesting inherent or distributional features of the long-copy, short-spacer subset that is most susceptible to cruciformation and mutational process.
These results differ slightly from those observed in yeast by others (Schroth and Ho 1995). That reference finds that attributes which are expected to favor cruciform extrusion (high A+T content and short spacer) are overrepresented in their set of IRs. However, those authors identified only 58 perfect IRs in yeast chromosome III, which is a much smaller sample than the 100,514 IRs examined here.
Inherent IR features
Next, we determined the pairwise correlations between the inherent IR features. The results are shown in Fig. 3 for the full IR set. (Data not shown for the long-copy, short-spacer subset). We find no correlation between the copy length and spacer length (Fig. 3a), or between the percent match and spacer length (Fig. 3b), in both the full genomic IR set and the long-copy, short-spacer subset. That is, IRs do not tend to be either more or less perfect as the size of the spacer region increases. The percent match and copy length are negatively correlated both in the S. cerevisiae and in the RA genomes, for the full set of IRs and for the long-copy, short-spacer subset, as shown in Fig. 3c. This means that longer IRs are less perfect on average than shorter ones. This is expected from the IRF scoring scheme, which allows more imperfections at longer copy lengths. The weakness of this correlation in the S. cerevisiae genome relative to that found in the RA randomizations could be due to in vivo error correction and/or possibly as a spontaneous generation of IRs from some repair process that need not act on an existing, shorter IR. At present, there is too little data to conclude to what degree either proposed mechanism may or may not contribute with any certainty. However, if this weakness is the result of in vivo error correction, our finding of insignificant correlation between spacer length and the degree of perfection (%M) in the S. cerevisiae genome suggests that its mechanisms are not strongly influenced by spacer length.
Fig. 3.
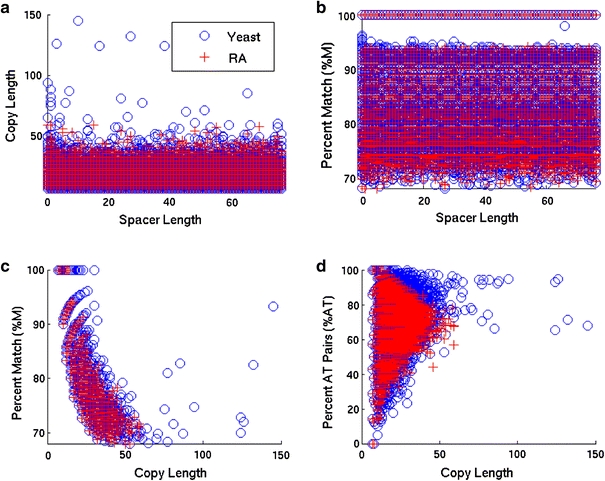
The correlations are shown between pairs of inherent IR features in the S. cerevisiae genome (blue) and in a representative RA (randomized) genome (red). No correlation is found between spacer length and either copy length (a) or percent match (b) in either case. The strong negative correlation between percent match and copy length seen in (c) results from the scoring scheme of the IRFinder algorithm, which allows more imperfections in longer IRs. A very weak correlation exists between percent AT and copy length (d)
The A+T content of IRs
The IRs in S. cerevisiae genome are significantly more A+T-rich than is the genome itself (71.7% vs. 61.7%). This trend toward A+T-richer IRs is also observed in the RA randomizations (average of 71.3% A+T), and hence is probably due largely to base composition effects. However, there is essentially no correlation between copy length and percent A+T for either the S. cerevisiae or RA genomes. That is, although there are a few hundred more A+T-rich genomic IRs than would be expected at random, there is no copy length dependence to this trend.
Palindromic dinucleotides in IRs
Next, we consider whether IRs in the S. cerevisiae genome are enriched specifically for palindromic dinucleotide repeats. We consider ApT and TpA repeats separately, but we denote them collectively as AT dinucleotide repeats. Similarly, we denote either CpG or GpC repeats as CG dinucleotide repeats. These are the only dinucleotides that have inverted repeat symmetry, and hence the only ones which can form IRs when repeated.
We first compared the numbers and lengths of AT and of GC dinucleotide repeats in the S. cerevisiae genome with those in its RA randomizations, as shown in Fig. 4. Because the yeast genome is relatively A+T-rich, GC dinucleotide repeats are both much fewer in number and shorter. (Its genomic base composition is 30.9% A, 30.8% T, 19.1% G, and 19.2% C.) This data show the S. cerevisiae genome to be significantly enriched in AT dinucleotide repeats up to lengths of n = 12–13. For example, there are 116 (AT)12 repeats in the S. cerevisiae genome, where none is expected at RA random. Figure 4b shows corresponding data for GC dinucleotides. Here, enrichment is not observed past repeat number n = 5, where the enrichment averages less than 1 repeat per chromosome. So GC repeats clearly can have no relevance for IR formation.
Fig. 4.
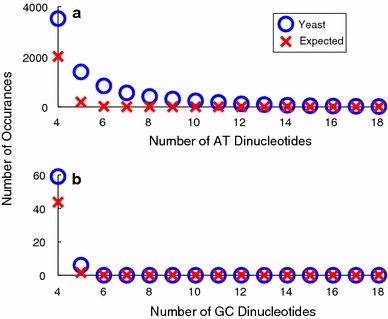
The numbers of ApT and TpA dinucleotide repeats in the S. cerevisiae genome are shown in part a as a function of repeat length. Part b shows the corresponding data for GpC and CpG dinucleotides. Here, the S. cerevisiae genome is represented by blue circles while the average numbers in the RA randomizations are shown as red x’s
We next considered whether this dinucleotide enrichment has an effect on the IR numbers. Figure 5 shows the percentage of IRs in the S. cerevisiae genome, and in a representative RA genome, that contain either n consecutive ApT repeats, or n consecutive TpA repeats. One sees that only small fractions of the IRs in the S. cerevisiae genome contain AT dinucleotide repeats having n > 2, and that these numbers are entirely consistent with random. We note that less than 1% of IRs in either the S. cerevisiae or RA genomes possess dinucleotide repeats with n > 3. This data show that the 12.7% genomic enrichment of IRs is not significant due to expansion of AT dinucleotide repeats, nor are long IRs the result of closely occurring independent dinucleotide repeats (shown in the supplementary materials, SFigure 1). For repeat numbers n = 1 or 2, the IR count is weakly positively correlated with copy length. This correlation is weaker in the S. cerevisiae genome than in the RA randomized genomes, and drops steadily beyond n = 2. This means that longer IRs do not contain more instances of (ApT)n or (TpA)n for a given n than do short IRs. These results show that neither A+T content nor the presence or extension of AT dinucleotide repeats is significantly responsible for the observed enrichment of the S. cerevisiae genome in either total IR numbers or in long IRs.
Fig. 5.
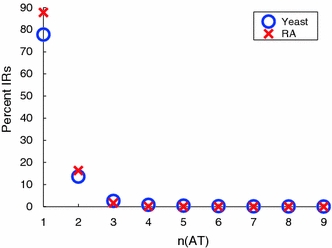
The percentage of IRs in the S. cerevisiae genome (blue circles) and in a representative RA genome (red x’s) that contain one or more ApT or TpA dinucleotide repeat of length n, for n ≥ 1
Perfect IRs
The S. cerevisiae genome is significantly enriched in perfect inverted repeats relative to random. Figure 6 plots in semilog coordinates the number of perfect IRs as a function of copy length for the S. cerevisiae, R, and RA genomes, together with expected numbers calculated using a theoretical method presented elsewhere (Lillo and Spano 2007). At every copy length the perfect IR number is statistically significantly higher in the S. cerevisiae genome than in either the R or the RA genomes. Moreover, the proportional overrepresentation of perfect IRs becomes more pronounced at longer copy lengths. Indeed, the S. cerevisiae genome contains longer perfect IRs than were found in any of the 100 randomized genomes. The longest perfect IR found in the S. cerevisiae genome had copy length 30, while the average number of perfect IRs with copy length larger than 15 was less than one both in the R and in the RA genomes. The R and the RA genomes are both seen to fit the theoretical line quite well, while the S. cerevisiae genome shows significant deviation. This observation supports the possibility that biological processes increase the degree of perfection of genomic IRs.
Fig. 6.
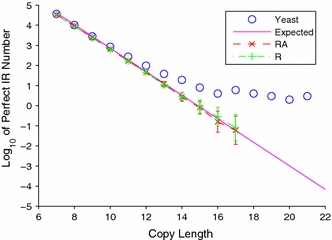
The number of perfect inverted repeats is plotted as a function of copy length in semilog coordinates. The data for the S. cerevisiae genome are shown in blue, while the means for the R- and the RA-randomizations are shown in green and red, respectively, with error bars corresponding to one standard deviation. The theoretically calculated expected numbers for a genome the base composition of which is 40% C+G are shown in magenta. The S. cerevisiae genome is significantly enriched in perfect inverted repeats for every copy length
Figure 7 shows the fractions of IRs that are perfect as a function of copy length, together with the means and error bars (representing two standard deviations on either side of the mean) for this fraction in the R and in the RA genomes. (The latter lines are difficult to distinguish because they closely overlie each other.) Because the number of imperfections allowed by the IRF search algorithm increases with copy length, one expects the proportion of perfect IRs to decrease. This is indeed the behavior seen in the R and RA randomized genomes. But in the S. cerevisiae genome a statistically highly significant enrichment in the proportion of perfect IRs is seen at all copy lengths. This increase in perfect IR numbers is larger than their fractional share of the 12.7% enrichment of all IRs documented above. Thus, while the S. cerevisiae genome is enriched both in perfect and in imperfect IRs relative to random, the enrichment is greater for perfect IRs. This further supports the view that in vivo processes may induce a trend toward perfection.
Fig. 7.
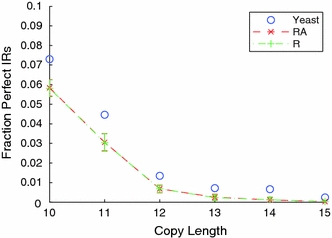
The fraction of perfect inverted repeats are shown for the S. cerevisiae (blue), RA (red), and R (green) genomes, respectively. The error bars here represent two standard deviations about the mean. The S. cerevisiae genome has a significantly greater proportion of perfect inverted repeats than either randomization. Because the scoring scheme of the IRF search algorithm only allows imperfections in IRs longer than 9 bp, the data are confined to this region
The genomic distribution of IRs
Next, we examined the distribution of IRs within the S. cerevisiae genome and its randomizations. We wished to determine whether there is any significant clustering (or anti-clustering) of IRs, if so whether clustering happens at biologically significant locations, and whether the set of clustered IRs have different properties than the non-clustered ones. To this end we first partitioned all IRs in each genome into the overlapping subset, which is all IRs that overlap at least one other IR, and the non-overlapping subset consisting of all isolated IRs. (Here, we regard each IR as comprising the region between the start of its left copy and the end of its right copy, including the spacer region.) We then examined three important distributional parameters—the number, N bp, of IRs that overlap a given base pair, the number, N IR, of other IRs that overlap a given IR, and, for each isolated IR, the distance, d NN, to its nearest IR neighbor.
We find that 36.4% of the base pairs in the S. cerevisiae genome are overlapped by at least one IR. (When the maximum allowed spacer length is increased from 76 to 130 this number becomes approximately 70.0%.) Further, 66,117 of the IRs in the S. cerevisiae genome are overlapping while 34,397 (34.2%) are isolated.
Numbers of overlapping and nonoverlapping IRs
The S. cerevisiae genome is significantly enriched in overlapping IRs, and has fewer non-overlapping IRs, relative to both the R and RA genomes. The total number of overlapping IRs exceed the mean for the R genomes by z R = 39 standard deviations, and exceeds the RA-mean by z RA = 35 standard deviations. The number of non-overlapping IRs is z R = −16 standard deviations smaller than the R-mean, and z RA = −13 standard deviations smaller than the RA-mean. This high degree of overlap shows that IRs in the yeast genome have a significant tendency to cluster.
Interchromosomal comparisons in the S. cerevisiae genome
We used the KS test to compare the distributions of inherent IR features as well as N IR, N bp, and d NN between the chromosomes of the S. cerevisiae genome. Here, the overlapping and non-overlapping sets were considered separately. Our results indicate that the distributions of IR features are not significantly different between chromosomes when restricted to the overlapping or the non-overlapping sets, just as was shown previously for the full sets. For this reason, subsequent analyses of these features were performed with all chromosome sets pooled together.
Comparison of overlapping with non-overlapping IRs
Next, we used the KS test to compare the distributions of each inherent IR feature (copy and spacer length, percent match, percent indel, percent A+T, and IRF Score) between the non-overlapping and the overlapping IR sets of the S. cerevisiae genome. In all cases the null hypothesis was rejected with p values less than 10−4, indicating that the IRs comprising these two sets are statistically significantly different in every inherent attribute. In particular, overlapping IRs have larger average spacer lengths than non-overlapping IRs (~38.5 bp vs. ~33.2 bp), and slightly longer copy lengths (~11.2 bp vs. ~10.1 bp). This is not surprising, as longer IRs would have a higher chance of overlapping other IRs. The non-overlapping IRs also tend to be slightly (but significantly) more perfect.
Analysis of overlapping IRs
We next compared the distributions of the positional and inherent IR features of the overlapping IR set in the S. cerevisiae genome with those from the overlapping sets in its R and RA randomizations. We performed KS tests to compare the distribution of each attribute in the S. cerevisiae genome to its distribution in either of the randomized genomes. In every case a p value of <10−4 was found, as shown in Table 1 (top right).
Table 1.
Statistically significant differences in detected IR subsets
| Non-overlapping IRs | Overlapping IRs | |||
|---|---|---|---|---|
| S. cerevisiae versus R | S. cerevisiae versus RA | S. cerevisiae versus R | S. cerevisiae versus RA | |
| Number | + (fewer) | + (fewer) | + (more) | + (more) |
| Spacer | − | − | + | + |
| Copy | + | + | + | + |
| Score | − | − | + | + |
| %AT | + | + | + | + |
| %Match | + | + | + | + |
| %Indel | + | + | + | + |
| N IR | NA | NA | + | + |
| d NN | + | + | NA | NA |
| S. cerevisiae non-overlapping genic versus non-overlapping intergenic | S. cerevisiae overlapping genic versus overlapping intergenic | |
|---|---|---|
| Spacer | + | + |
| Copy | − | + |
| Score | − | + |
| %AT | + | + |
| %Match | − | + |
| %Indel | − | + |
| N IR | NA | + |
| d NN | + | NA |
Statistically significant differences are defined as returning a KS test p value smaller than 0.01 when the null hypothesis of identity is rejected. The symbol + represents a statistical difference (p value less than 0.01) while − indicates that no difference was detected
Figure 8 shows semilog plots of the number of IRs that are overlapped by N IR others (Fig. 8a), and the number of base pairs that are overlapped by N bp IRs (Fig. 8b), for each of the three genome types. The data for each genome type fit a straight line to high precision. This shows that both N bp and N IR decrease exponentially with overlap number. It is clear from the figure that the exponential decay rates for the R and RA genomes are essentially the same, and steeper than that for the S. cerevisiae genome, again indicative of IR clustering.
Fig. 8.
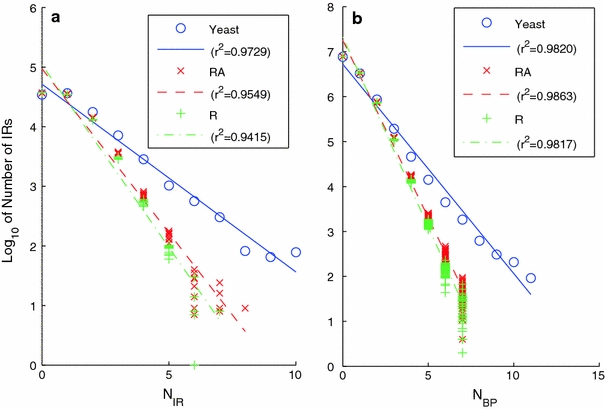
Part a shows the number of inverted repeats that are overlapped by N IR others, together with regression lines, plotted for the S. cerevisiae (blue), RA (red), and R (green) genomes. Part b shows the number N bp of distinct inverted repeats that overlap a given base pair. Both plots are presented in semilog coordinates. There is significant clustering of IRs in the S. cerevisiae genome relative to random, as shown by the larger overlap numbers it attains
Parameters reflective of clustering also have very high extreme values. In the S. cerevisiae genome there is a base pair that is overlapped by N bp = 11 different IRs, while the maximum achieved in any of the 50 RA genomes was N bp = 7. The largest number attained in any of the 50 R randomizations was N bp = 6. The distribution of IRs in six representative highly overlapped regions are shown in Fig. 9. The number of IRs in each of these regions is quite high. Each has at least one base pair with N bp ≥ 11 and contains at least one IR with N IR ≥ 10. In every case the region of highest overlap occurs in the 3′ flank of a gene. These examples suggest a preference for clustering to occur in intergenic regions, and specifically at the 3′ ends of genes, within either tandem or convergent intergenic regions.
Fig. 9.
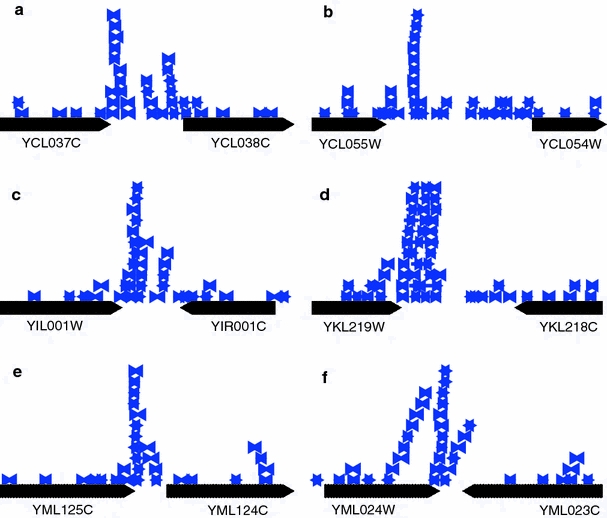
The six regions of the S. cerevisiae genome are shown where the highest levels of IR overlap are attained. Here IRs are represented by blue arrows, one arrow for each copy, pointing toward the center of symmetry. The flanking genes are shown in black with the arrow indicating transcriptional direction. Regions (a–b), (c), (d), (e–f) are located on chromosomes 3, 9, 11, and 13 respectively
It is possible that the observed higher degree of overlap in the S. cerevisiae genome than in the randomized ones could be due in part to the existence of a small number of long IRs. The longest IR seen in this survey is comprised of two near neighbor Ty elements, YGRCTy1-3 and YGRWTy2-2, on chromosome 7, and is over 5,900 bp long. This is the only case where Ty elements or LTRs were found in this study, because it is the only instance where a pair in reverse orientation is located within 76 bp of each other. Excluding this case, the S. cerevisiae genome contains several overlapping IRs with copy lengths in the range of 300–400 bp. In contrast, the longest overlapping IR within any of the 100 randomized genomes (either R or RA) has copy length of 92 bp. However, the number of IRs that are overlapped by N IR others remains effectively exponential even at the high values, as shown in Fig. 8a. So whatever excess there may be of long IRs in the S. cerevisiae genome relative to its randomizations, the overall effect produces a consistently higher distribution at all overlap numbers. Moreover, the average copy lengths of overlapping IRs do not differ greatly between the S. cerevisiae genome and its randomizations. The mean copy length for the former is μ S. cerevisiae ≈ 14.7 bp, while for the RA randomized genomes it is μ RA ≈ 14.0 bp.
Analysis of non-overlapping IRs
Next, we compared the features of the non-overlapping IR set from the S. cerevisiae genome with those of the non-overlapping sets from the R and RA genomes. Here, the KS test found statistically significant differences at the p < 10−2 level in copy length, percent match, percent indels, d NN, and percent A+T (see Table 1). However, the null hypothesis could not be rejected at this significance level for spacer length or for IRF score.
Overall, the non-overlapping IRs in the S. cerevisiae genome tend to have marginally shorter copy lengths, and slightly fewer indels than would be expected at random. The mean percent indels was about 9% smaller in the S. cerevisiae non-overlapping IR set than in those from either the R or the RA genomes. This effect may be due in part to the scoring scheme used by the IRF algorithm, in which the allowed number of imperfections increases with arm length. So a portion of the observed slightly higher frequency of perfection in the non-overlapping set may be a consequence of its shorter average copy length.
We next examined the genome-wide distribution of the distance d NN from an isolated IR to its nearest neighbor. IRs with d NN > 300 were excluded from this analysis as outliers because they are sufficiently infrequent as to give poor statistics beyond this length. This excluded less than 1% of the non-overlapping IRs in any genome. The distribution of nearest neighbor distances in the S. cerevisiae genome is not significantly different from its distributions in its R and RA randomizations. In all cases there is an excellent fit to the same exponential distribution (data not shown). Exponential waiting times are characteristic of memoryless processes (Feller 1968). This means that the position along a chromosome of an isolated IR has no influence on the location of the sequentially next isolated IR. So, in contrast to the overlapping IRs, the non-overlapping IRs are randomly distributed within the S. cerevisiae genome. There is no tendency for isolated IRs either to cluster or to anti-cluster; the distribution of isolated IRs in the S. cerevisiae genome is entirely consistent with random expectations.
Comparisons of IRs in coding versus non-coding genomic regions
Here, we investigate whether the observed clustering of IRs seen above in the S. cerevisiae genome is related in any way to its transcriptional structure. The release of the S. cerevisiae genome sequence analyzed here (version 7) contains 6,155 annotated genes, comprising 72.8% of the genome. We partitioned this genome into coding (genic) and non-coding (intergenic) regions according to its annotation, and looked at the IR distributions within each category. We also subdivided the intergenic regions according to the orientations of their abutting genes into convergent, tandem, and divergent sets, and examined these separately.
We found that 66.5% of the IRs identified by IRF are located either wholly inside or overlapping coding regions, while 39.2% are within or overlapping intergenic regions. (Here, we define overlap as having at least one base pair within a region of each annotation). Of these, 33.5% are wholly within intergenic regions, while 5.7% overlap the boundaries between genic and intergenic regions. This indicates an enrichment of IRs within intergenic regions and/or overlapping the start and/or end positions of genes relative to genic regions. Anecdotal evidence of this is seen in Fig. 9, which shows that the six regions in the S. cerevisiae genome having the highest overlap numbers are all intergenic. IRs that serve regulatory functions in transcriptional initiation and termination would be expected to be located largely in intergenic regions.
We also observed that short IRs are located with lower frequencies in genic regions than within or overlapping intergenic regions (data not shown). When the intergenic regions are subdivided further into tandem, convergent, and divergent, a higher percentage of the S. cerevisiae IRs are found to overlap either convergent or tandem regions than is observed for the RA genome, while the percentage overlapping divergent intergenic regions is slightly lower in the S. cerevisiae genome than in the RA genomes. (Data also not shown.) This indicates that the most significant deviations from random occur at those intergenic regions located in the 3′ flanks of genes, where transcripts terminate. This matter is examined further below.
Next, we performed separate investigations of the non-overlapping and the overlapping IR sets to determine whether their IR features are significantly different when they are located in coding versus non-coding regions. As shown in Table 1, the distributions of all features of the overlapping IR sets are statistically significantly different between coding and non-coding regions. The average copy length of overlapping IRs is longer in intergenic regions than in coding regions, while their average spacer length is shorter and their percent match is slightly lower. Thus, IRs within genes tend to be shorter, to have longer spacers, and to be more perfect than those in intergenic regions. The average overlap number N IR was larger for intergenic overlapping IRs than for those in coding regions. The maximum number of overlaps for an IR was 10 within intergenic regions, but only 8 for those within or overlapping genes. This again suggests that the increase in clustering observed in the S. cerevisiae genome relative to random is concentrated in intergenic regions.
Similar comparisons made using the non-overlapping IR set show that non-overlapping IRs have relatively uniform feature distributions, with few significant differences between coding and non-coding regions (see Table 1).
Comparing IRs in gene flanks
We aligned the annotated genes of the S. cerevisiae genome, orienting them so transcription proceeds to the right. Translation stop locations were aligned in a similar manner. This was done for each of the 3,832 genes that are annotated as experimentally verified in the S. cerevisiae genome and that have flanking intergenic regions longer than 150 bp. The aligned regions were 450 bp long, with the gene boundary placed so it contains 150 bp of intergenic region and 300 bp of coding region. We determined N bp, the number of IRs that overlap the base pair at each location in each sequence. We then computed the mean of this value at each position, which gives a measure of the IR density at each site relative to the gene start (or stop) location. This was also done for the R and the RA genomes.
The results of this analysis are shown in Fig. 10a for 5′ flanks, and in Fig. 10b for 3′ flanks. We see that in the S. cerevisiae genome the IR density substantially increases both just before gene starts and just after gene stops, the latter being the larger. Comparison with the RA randomizations shows that approximately half of the upstream enrichment and a quarter of the downstream enrichment can be attributed to the difference in base composition between intergenic and genic regions. This is consistent with the findings of others (Lillo et al. 2002; Lisnic et al. 2005; Lu et al. 2007).
Fig. 10.
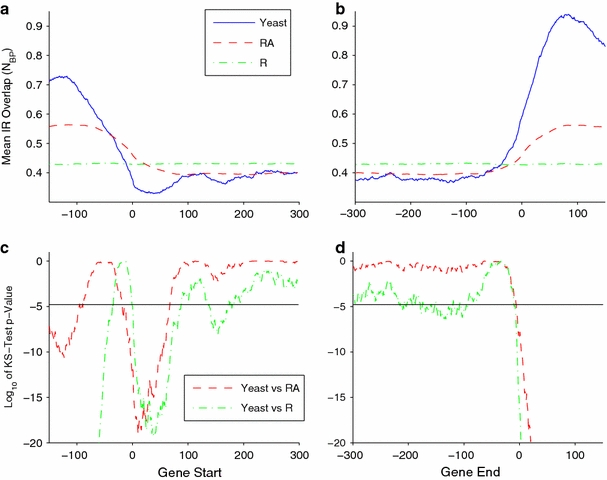
The mean overlap number N bp is shown for 3,832 genes aligned at their start (a), and stop positions (b). The results for the S. cerevisiae genome are shown in blue, while those for the RA- and R-genomes are in red and green, respectively. We compared the distributions at each position using the Kolmogorov–Smirnov test. The p values assessing statistical significance found this way at each position are plotted logarithmically in parts (c) and (d). The threshold for significance is shown as a horizontal line in each case. We note that significance can occur through either enrichment or paucity relative to random. This shows a significant enrichment of IRs in the 5′ and 3′ flanks of genes, with greater enrichment in the downstream, 3′ flanks. Within coding regions IRs occur at rates consistent with random, given their base composition. Within the first 80 bp after the gene start there are significantly fewer IRs than expected at random
These results show that the statistically significant clustering of IRs in the S. cerevisiae genome that was documented above does not occur at random, but instead is concentrated at the gene start and stop positions. The increase in IR density is larger in the downstream 3′ flanks of genes than in their upstream 5′ flanks. Figure 10a also shows a significant paucity of IRs in the S. cerevisiae genome just after the gene start position. This is not observed in either the RA or R genomes.
Within coding regions, the mean base pair overlap number in the S. cerevisiae genome is approximately constant at all positions 100 bp or more into the gene, and approaches the mean for the RA randomizations. This indicates neither a paucity nor an enrichment of IRs relative to what would be expected at random for regions with the genic base composition.
We performed the KS test to determine the statistical significance of these observations. The results are shown in Fig. 10c, d, plotted as the base 10 logarithms of the p values found for each position. We use a cutoff value for statistical significance of –4.65, indicated by a horizontal line. This includes a Bonferroni correction to account for the fact that we are testing a separate hypothesis at each of the 450 bp. As the figure shows, the increase in the IR density is statistically significant relative to the RA randomizations (red curves) in the regions ending approximately 100 bp upstream from gene start positions, and in regions extending at least 150 bp downstream from gene stop positions. The dip in IR density within the first 75 bp of genes is also statistically significant.
We note that the same procedure executed on all 6,155 annotated genes in the S. cerevisiae genome produces a qualitatively similar result with the sites of statistical significance occurring at the same locations (data not shown).
IRs in the three types of intergenic regions
To further examine IR enrichment within gene flanks, we subdivided the set of intergenic regions into the three disjoint subsets of convergent, divergent, and tandem, according to the directions of transcription of their abutting genes. The S. cerevisiae genome was annotated to have 1561 convergent, 1569 divergent, and 2906 tandem intergenic regions whose ends are located more than 400 bases from chromosome telomeres. We found that 8.0% of the IRs in yeast either overlap or are contained entirely within convergent intergenic regions. For divergent intergenic regions this number is 11.9%, while for tandem regions it is 19.5%. As convergent, divergent, and tandem regions comprise 4.1, 9.4, and 13.3% of the genome respectively, the overlap of intergenic regions is not simply proportional to the number of base pairs in each region type.
To assess the relationships, if any, between IR densities and specific types of intergenic region boundaries (i.e. 3′ or 5′ gene flanks), we compared start positions in divergent regions to those in tandem regions, and stop positions in convergent regions with those it tandem regions. (Data not shown.) Qualitatively, these results are similar to those of Fig. 10. However, the IR density dip immediately after the gene start is now seen to be strongest for divergently oriented genes and is essentially absent for genes starting at tandem regions. The IR density in the upstream, 5′ flank is somewhat less in the divergent than in the tandem case. The density peak in the 3′ flank that occurs immediately after the gene stop position is higher for convergently oriented genes than for tandemly oriented ones. Each of these differences is statistically significant. Additionally, the density of IRs as measured by N bp increases monotonically from divergent, to 5′ tandem, to 3′ tandem, to convergent intergenic regions. This agrees with our earlier finding that the IR density is higher at 3′ flanks than at 5′ flanks. In the discussion we consider possible explanations for these findings.
IR spacer length distributions in gene flanks
The above results document a significant concentration of IRs in the 5′ flanks of genes. In eukaryotes, these sites are known to experience significant negative supercoiling due to transcription (Kouzine et al. 2008). This raises the possibility that some of the IRs located there might serve regulatory functions involving superhelical cruciform extrusion. So we assessed whether the distributions of IRs located at these sites are enriched for those that would be susceptible to cruciformation.
Spacer length is the IR feature that most directly influences hairpin fold-back or cruciform extrusion, with short-spacer lengths being favored over long ones. As there is high density of IRs in the S. cerevisiae genome, and as cruciformation would also have to compete with other structural transitions that can be driven by superhelicity, only the IRs with the shortest spacers would have a chance to extrude cruciforms. So we separated the IRs into three ranges of spacer lengths: [0, 10], [11, 20], and [21, 30]. The overlap number N bp was calculated at each position for each set separately, and the KS test was used to compare the distribution in the S. cerevisiae genome with that for the RA genomes. A similar procedure was done for the 3′ flanks.
The 5′-gene flanks show no differences from random at the 5% significance level at any location. Indeed, there are slightly fewer IRs with short-spacer lengths than expected at random in 5′-gene flanks. However, there is a strong statistical enrichment of those IRs that have the shortest spacers in the 3′ flanks of genes. (Data not shown.) The 3′-flank IRs with spacers in [0, 10] have more significant enrichment than those with spacers in [11, 20], while those with spacers in [21, 30] show no statistically significant difference from RA random.
Discussion
We have examined the distributions and properties of those IRs in the S. cerevisiae genome that have copy length at least 7 bp, and spacer length no greater than 76 bp. Comparisons with distributions of the corresponding properties in randomizations of this genome enabled the assessment of statistical significance of variations from random. The basic results of our analyses are these.
IRs in yeast are more numerous, longer, and more perfect than expected
We found 100,514 IRs having the above attributes in the S. cerevisiae genome, one every 120 bp on average. This is a 12.7% enrichment above the mean for the RA randomized genomes, which is statistically highly significant. The average IR density is also quite high, with 36% of the base pairs being overlapped by one or more IR. Longer IRs are overrepresented in this genome, and the number and fraction of perfect IRs also exceeds random, with this enrichment increasing with copy length.
The observed excess of IRs does not result from the presence of long dinucleotide repeats, specifically (ApT)n or (TpA)n. Only a statistically insignificant proportion of IRs in the S. cerevisiae genome contain long dinucleotide repeats, and their dinucleotide repeat contents do not correlate with copy length. Longer IRs do not tend to be either more or less A–T rich than shorter ones.
IR clustering
Only 34% of the IRs in the S. cerevisiae genome are isolated; the other 66% overlap at least one other IR. This is a significant excess over random expectations. Moreover, their degrees of overlapping are also higher than expected. While the set of overlapping IRs have significant distributional anomalies, the set of non-overlapping IRs is distributed in a way that is consistent with random.
We document a significantly higher IR density in the 5′ and 3′ flanks of genes, as measured by the base pair overlap number N bp, while this density within coding regions is consistent with random. This result is consistent with the findings of earlier palindrome and perfect IR searches both in prokaryotes and in eukaryotes (Lillo et al. 2002; Lisnic et al. 2005; Lu et al. 2007). The degree of enrichment in IR density is least for 5′ flanks in divergent intergenic regions, greater for 5′ tandem regions, still greater for 3′ flanks in tandem regions, and greatest for 3′ flanks in convergent regions. This is opposite to the amount of transcriptionally driven negative supercoiling that would be present in these domains.
Possible functional roles of IRs
Several possible functional roles have been suggested for inverted repeat sequences. We describe each in turn, and consider whether the statistical data presented here support it.
Mutagenesis
Several mechanisms for hairpin-directed mutagenesis have been suggested, which would increase the number of perfect IRs and/or decrease the degree of IR imperfection. Error-correction mechanisms involving either intra- or intermolecular switching would tend to reduce the number of imperfections in a given IR and/or increase its length (Ripley 1982; van Noort et al. 2003; Rattray et al. 2005). Our observation of significant enrichments of both long IRs and perfect IRs in the S. cerevisiae genome would suggest that mechanisms of these types may indeed be operative in yeast. (We note that methyl-directed mismatch repair would not occur in yeast, as the DNA of this organism is not methylated.) However, our result that spacer length is not correlated with the degree of perfection would also suggest that these mechanisms, if they are responsible for the enrichment of perfect IRs, are relatively insensitive to spacer length.
RNA secondary structure
Hairpins are important structural features of RNA that are encoded by inverted repeat DNA sequences. If hairpins were functionally important in mRNA, they would be evolutionarily conserved. One expects that this would lead to an enrichment of IRs in transcribed regions. However, we have shown that there is no observed enrichment in genic regions of the S. cerevisiae genome; the number of IRs present there is consistent with random for their base composition. This suggests that the number of IRs that are present in order to enable specific hairpins to form in mRNA may be too small to confer statistical significance. One possible explanation is that most IRs in mRNA transcripts are neutral, neither conferring advantages or disadvantages. An alternative, although perhaps less probable, explanation is that those IRs that are present in genic regions do in fact encode functionally important RNA hairpins, but that other IRs would be deleterious, perhaps interfering with the important ones. This might happen, for example, if hairpins affected the rate of translation or the number of proteins made per mRNA. So the functional IRs could be enriched while the non-functional ones are selected against. If these effects counterbalance, this could result in an overall distribution that is consistent with random in these regions.
If functional hairpins were important in mRNA, but the cruciforms their encoding DNA might form were deleterious, one would expect these IRs to have attributes that disfavor cruciformation. This could be an enrichment in imperfections or a preference for long spacers. Neither of these was observed in genic regions.
Bidirectional processes
If a bidirectional process requires the sequence-specific binding of a protein, the binding sites may be arranged in inverted repeat orientation. This is known to occur at the replication origin of SV40 and other viruses. Unfortunately, the functional replication origins in yeast are too few and (at present) too poorly characterized to enable us to assess the frequency of IRs at these sites.
The substantial clustering of IRs within intergenic regions that was documented here suggests the possibility that at least some of these may serve regulatory functions. A variety of possible roles have been suggested for IRs in transcriptional control events taking place at 5′- and 3′-gene flanks.
A transcriptional regulatory process that involves either antiparallel binding of two copies of a transcription factor, or binding of a homodimeric factor with antiparallel binding sites, would require an inverted repeat-binding site. This could account for some of the observed IR enrichment at 5’-gene flanks. There are only two documented instances of a palindromic yeast transcription factor binding site in the TRANSFAC database (Wingender et al. 1996), and no instances of an IR-binding site with the attributes to be found by the IRF search used in this study. It would be difficult to assess the prevalence of antiparallel pairs of transcription factor binding sites from available data, as this would require the accurate prediction of such sites based on their base sequences. Unfortunately, genome-wide binding site predictions based on sequence motifs commonly have such high false positive error rates as to be useless in practice.
The IRs in 3′-gene flanks could be involved in transcriptional termination, transcript release, polyadenylation or some other event occurring there. In principle this could occur through bidirectional binding, cruciform or hairpin extrusion, or some other process. (The latter situations will be considered below.) However, no such proposed processes in eukaryotes are known to us. It is also possible that IRs in general, or perfect IRs in particular, are created, lengthened or enriched through the previously discussed error-correction schemes. If IRs at deleterious sites are removed by selection, an abundance that is concentrated at the sites where they are not deleterious would result. This also could explain the clustering of IRs in gene 3′ flanks.
Regulatory cruciform extrusion
The possibility that IRs serve regulatory functions through the extrusion of DNA cruciforms has received considerable attention. In these proposals the driving force for extrusion is assumed to be negative DNA superhelicity. There is little evidence in this study to support such models.
Most of the negative superhelicity in eukaryotic DNA is stabilized by nucleosomal winding. The global average level of uncompensated superhelicity is approximately zero. However, transcription is known to drive substantial levels of negative superhelicity back toward the promoter, and the symmetric level of superhelicity forwards toward the gene terminus (Kouzine et al. 2008). So transcription can induce substantial negative superhelicity in gene 5′ flanks, and positive superhelicity in 3′ flanks.
We find that the degree of enrichment in IR density is least for 5′ flanks in divergent intergenic regions, greater for 5′ tandem regions, still greater for 3′ flanks in tandem regions, and greatest for 3′ flanks in convergent regions. This is opposite to the degree of transcriptionally driven negative supercoiling that would felt in these domains. Our results show that IRs with short spacers, and hence the greatest susceptibility to superhelical cruciformation, are significantly enriched only in 3′ gene flanks. This transition is unlikely to occur in these regions because transcription drives positive superhelicity there.
Although we do find a substantially increased IR density in the 5′ flanks of genes, these IRs are not significantly enriched—and indeed are slightly depleted—in ones having the short spacers. This evidence, though circumstantial, supports theories involving mutagenic or binding, rather than extrusional, roles for the IRs in gene 5′ and 3′ flanks.
Our findings do not preclude the possibility that some of these IRs may be involved in regulatory extrusion events, especially as transcription drives strong negative superhelicity into 5′-gene flanks. But it does suggest that the number of IRs that are evolutionarily conserved for this purpose may not be enough to leave a statistical signature in the genome. Alternatively, other IRs could be deleterious there, as they might compete with the regulatory cruciform extrusion events, and hence be evolutionarily selected against. It is possible that these trends could approximately cancel, leaving no overall statistical signature. It is hoped that targeted experimental analysis, particularly of IR copy and spacer length effects, in the 5′ flanks of genes will provide more concrete information on these questions.
Fold-back mechanisms associated with transcription would not be expected to occur in 3′ intergenic regions because they are not transcribed. However, while either fold-back or intermolecular switching could still occur in these regions, it is possible that the resulting mutations would not be as deleterious when associated with intergenic regions, or with those intergenic regions solely associated with transcription termination. As the putative in vivo mechanisms that provide the tendencies toward IR perfection or the creation or lengthening of IRs are inherently mutagenic, the altered sequences might possibly be stable only in regions not associated with protein production or with transcription initiation or control.
Finally, it is important to note that DNA sequences can serve more than one regulatory function. For example, an IR that binds one or more transcription factors could be prevented from doing so by its forming a hairpin or cruciform. A homodimer that binds to an IR in duplex form may not be able to bind to its cruciform. These could serve as mechanisms to regulate gene expression. Even if an IR were perfect, the molecular binding characteristics of the interstrand duplex and the hairpin arms may be different. Gene expression could be affected by which of these mutually exclusive types of binding occurred. There are a variety of activating or inhibitory strategies that could be based on these and similar events.
Electronic supplementary material
Below is the link to the electronic supplementary material.
SFig. 1 The numbers of occurrences of the dinucleotide repeats (ApT)n or (TpA)n within IRs are plotted as functions of (left) copy length. Parts (a) through (j) give separate plots for each repeat length 1 ≤ n ≤ 9. Data for the S. cerevisiae genome are indicated by blue circles while corresponding data from a representative RA genome are shown as red x’s (EPS 4728 kb)
Acknowledgments
ES was supported in part by grant DMS-0135345 from the National Science Foundation. GB and YG were supported in part by NSF grant IIS-0612153 and National Institutes of Health grant 1 R01 GM072084. CJB was supported in part by NSF grants DBI-0416764 and DBI-0850214, and by grant NIH RO1 HG004348 from the National Institutes of Health.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
List of abbreviations
- RA
Random genome where the regions annotated as coding are shuffled separately from the non-coding regions within a chromosome
- R
Random genome where each chromosome is shuffled uniformly
- IR
Inverted repeat
- Indel
Insertion–deletion error within a hairpin or cruciform structure
- KS
Kolmogorov–Smirnov test
- %M
Percent of the base pairs in a hairpin or cruciform arm that are Watson–Crick paired
- %I
Percent of the base pairs in a hairpin or cruciform arm that are involved in an insertion or deletion
- %AT
Percent A–T nucleotide composition of an IR, not the same as the percent AT dinucleotide pairs
- Nbp
Number of IRs overlapping a given base pair
- NIR
Number of IRs overlapping a given IR
- dNN
Distance to the next closest IR, zero for overlapping IRs
- r2
Fraction of observed variation that can be ascribed to the deterministic linear relationship; 1 − r 2 is the fraction due to random variations about this linear function
- ρ
Pearson’s correlation coefficient indicates the strength of the linear correlation; 1 in the case of an increasing linear relationship, −1 in case of a decreasing linear relationship
- μ
Mean
- σ
Standard deviation
Contributor Information
Eva M. Strawbridge, Phone: +1-773-7028035, FAX: +1-773-7029787, Email: emstrawb@math.uchicago.edu
Gary Benson, Email: gbenson@bu.edu.
Yevgeniy Gelfand, Email: ygelfand@bu.edu.
Craig J. Benham, Email: cjbenham@ucdavis.edu
References
- Achaz G, Coissac E, Netter P, Rocha EPC. Associations between inverted repeats and the structural evolution of bacterial genomes. Genetics. 2003;164:1279–1289. doi: 10.1093/genetics/164.4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achez G, Coissac E, Viari A, Netter P. Analysis of intrachromosomal duplications in yeast Saccharomyces cerevisiae: a possible model for their origin. Mol Biol Evol. 2000;17:1268–1275. doi: 10.1093/oxfordjournals.molbev.a026410. [DOI] [PubMed] [Google Scholar]
- Akgun E, Zahn J, Baumes S, Brown G, Liang F, Romanienko PJ, Lewis S, Jasin M. Palindrome resolution and recombination in the mammalian germ line. Mol Cell Biol. 1997;17:5559–5570. doi: 10.1128/mcb.17.9.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez D, Novac O, Callejo M, Ruiz MT, Price GB, Zannis-Hadjopoulos M. 14-3-3 sigma is a cruciform DNA binding protein and associates in vivo with origins of DNA replication. J Cell Biochem. 2002;87:194–207. doi: 10.1002/jcb.10294. [DOI] [PubMed] [Google Scholar]
- Bauer WR, Benham CJ. The free energy, enthalpy and entropy of native and of partially denatured closed circular DNA. J Mol Biol. 1993;234:1184–1196. doi: 10.1006/jmbi.1993.1669. [DOI] [PubMed] [Google Scholar]
- Benham CJ. Stable cruciform formation at inverted repeat sequence in supercoiled DNA. Biopolymers. 1982;21:679–696. doi: 10.1002/bip.360210314. [DOI] [PubMed] [Google Scholar]
- Benham CJ, Savitt AG, Bauer WR. Extrusion of an imperfect palindrome to a cruciform in superhelical DNA: complete determination of energetics using a statistical mechanical model. J Mol Biol. 2002;316:563–581. doi: 10.1006/jmbi.2001.5361. [DOI] [PubMed] [Google Scholar]
- Bi X, Liu LF. DNA rearrangement mediated by inverted repeats. Proc Nat Acad Sci USA. 1996;93:819–823. doi: 10.1073/pnas.93.2.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DK, Yasuda LE, Yao M-C. Induction of large DNA palindrome formation in yeast: Implications for gene amplification and genome stability in eukaryotes. Cell. 1996;87:1115–1122. doi: 10.1016/S0092-8674(00)81805-X. [DOI] [PubMed] [Google Scholar]
- Butler DK, Gillespie D, Steele B. Formation of large palindromic DNA by homologous recombination of short inverted repeat sequences in Saccharomyces cerevisiae. Genetics. 2002;161:1065–1075. doi: 10.1093/genetics/161.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker AF, Okely EA, Davison A, Leach DRF. The effects of central asymmetry on the propagation of palindromic DNA in bacteriophage-lambda are consistent with cruciform extrusion in vivo. Genetics. 1993;133:143–148. doi: 10.1093/genetics/133.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew DSH, Choi KP, Leung M-Y. Scoring schemes of palindrome clusters for more sensitive prediction of replication origins in herpesviruses. Nucleic Acids Res. 2005;33:e134. doi: 10.1093/nar/gni135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello E, Sahli R, Hirt B, Beard P. The mismatched nucleotides in the 5′-terminal hairpin of minute virus of mice are required for efficient viral DNA replication. J Virol. 1995;69:7489–7496. doi: 10.1128/jvi.69.12.7489-7496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courey AJ, Wang JC. Cruciform formation in a negatively supercoiled DNA may be kinetically forbidden under physiological conditions. Cell. 1983;33:817–828. doi: 10.1016/0092-8674(83)90024-7. [DOI] [PubMed] [Google Scholar]
- Cox R, Mirkin SM. Characteristic enrichment of DNA repeats in different genomes. Proc Natl Acad Sci USA. 1997;94:5237–5242. doi: 10.1073/pnas.94.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Greizerstein MB, Nadas-Chinni K, Rothman-Denes LB. Supercoil-induced extrusion of a regulatory DNA hairpin. Proc Natl Acad Sci USA. 1997;94:2174–2179. doi: 10.1073/pnas.94.6.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayn A, Malkhosyan S, Mirkin SM. Transcriptionally driven cruciform formation in vivo. Nucleic Acids Res. 1992;20:5991–5997. doi: 10.1093/nar/20.22.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra BE, Lovett ST. Cis and trans-acting effects on a mutational hotspot involving a replication template switch. J Mol Biol. 2006;358:300–311. doi: 10.1016/j.jmb.2005.11.071. [DOI] [PubMed] [Google Scholar]
- Eykelenboom JK, Blackwood JK, Okely E, Leach DRF. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell. 2008;29:644–651. doi: 10.1016/j.molcel.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Feller W. An introduction to probability theory and its applications. 3. New York: Wiley; 1968. [Google Scholar]
- Fisher RA, Yates F (1938) Statistical tables (Example 12). London
- Gellert M, O’Dea MH, Mizuuchi K. Slow cruciform transitions in palindromic DNA. Proc Natl Acad Sci USA. 1983;80:5545–5549. doi: 10.1073/pnas.80.18.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordenin DA, Lobachev KS, Degtyareva NP, Malkova AL, Perkins E, Resnick MA. Inverted DNA repeats: a source of eukaryotic genomic instability. Mol Cell Biol. 1993;13:53155–55322. doi: 10.1128/mcb.13.9.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves DR, Patient RK, Lilley DMJ. Facile cruciform formation by an (A-T)34 sequence from a xenopus globin gene. J Mol Biol. 1985;185:461–478. doi: 10.1016/0022-2836(85)90064-6. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Lewis SM, Wu X, Ma C, Munroe DJ, Fuess S, Storm T, Kay MA, Nakai H. DNA palindromes with a modest arm length of >=20 base pairs are a significant target for recombinant adeno-associated virus vector integration in the liver, muscles, and heart in mice. J Virol. 2007;81:11290–11308. doi: 10.1128/JVI.00963-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EL, Peng H, Exparza FM, Maltchenko SZ, Stachowiak MK. Cruciform-extruding regulatory element controls cell-specific activity of the tyrosine hydroxylase gene promoter. Nucleic Acids Res. 1998;26:1793–1800. doi: 10.1093/nar/26.7.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth DE. The art of computer programming. 3. Reading: Addison-Wesley; 1997. [Google Scholar]
- Kogo H, Inagaki H, Ohyo T, Kato T, Emanual BS, Kurahashi H. Cruciform extrusion propensity of human translocation-mediating palindromic AT-rich repeats. Nucleic Acids Res. 2007;35:1198–1208. doi: 10.1093/nar/gkm036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Levens D. Supercoil-driven DNA structures regulate genetic transactions. Front Biosci. 2007;12:4409–4423. doi: 10.2741/2398. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Liu J, Sanford S, Chung H-J, Levens D. The dynamic response of upstream DNA to transcription-generated torsional stress. Nature Struct Mol Biol. 2004;11:1092–1099. doi: 10.1038/nsmb848. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Sanford S, Elisha-Feil Z, Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat Struct Mol Biol. 2008;15:146–154. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- LeBlanc MD, Aspelagh G, Buggia NP, Dyer BD. An annotated catalog of inverted repeats of Caenorhabditis elegans chromosomes III and X, with observations concerning odd/even biases and conserved motifs. Genome Res. 2000;10:1381–1392. doi: 10.1101/gr.122700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Sinden RR, Roth DB. Targeted transposition by the V(D)J recombinase. Mol Cell Biol. 2002;22:2068–2077. doi: 10.1128/MCB.22.7.2068-2077.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M-Y, Choi KP, Xia A, Chen LHY. Nonrandom clusters of palindromes in herpesvirus genomes. J Comput Biol. 2005;12:331–354. doi: 10.1089/cmb.2005.12.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Cote AG. Palindromes and genomic stress fractures: bracing and repairing damage. DNA Repair. 2006;5:1146–1160. doi: 10.1016/j.dnarep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Li X, Lindahl L, Sha Y, Zengel JM. Analysis of the Bacillus subtilis S10 ribosomal protein gene cluster identifies two promoters that may be responsible for transcription of the entire 15-kilobase S10-spc-alpha cluster. J Bacteriol. 1997;179:7046–7054. doi: 10.1128/jb.179.22.7046-7054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley DMJ. The inverted repeat as a recognizable structural feature in supercoiled DNA molecules. Proc Natl Acad Sci USA. 1980;77:6468–6472. doi: 10.1073/pnas.77.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo F, Spano M. Inverted and mirror repeats in model nucleotide sequences. Phys Rev E. 2007;76:1914–1926. doi: 10.1103/PhysRevE.76.041914. [DOI] [PubMed] [Google Scholar]
- Lillo F, Basile S, Mantegna RN. Comparative genomics study of inverted repeats in bacteria. Bioinformatics. 2002;18:971–979. doi: 10.1093/bioinformatics/18.7.971. [DOI] [PubMed] [Google Scholar]
- Lin CT, Lyu YL, Liu LF. A cruciform-dumbbell model for inverted dimer formation mediated by inverted repeats. Nucleic Acids Res. 1997;25:3009–3016. doi: 10.1093/nar/25.15.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisnic B, Svetec I-K, Saric H, Nicolic I, Zgaga Z. Palindrome content of the yeast Saccharomyces cerevisiae genome. Curr Genet. 2005;47:289–297. doi: 10.1007/s00294-005-0573-5. [DOI] [PubMed] [Google Scholar]
- Lisnic B, Svetec IK, Stafa A, Zgaga Z. Size-dependent palindrome-induced intrachromosomal recombination in yeast. DNA Repair. 2009;8:383–389. doi: 10.1016/j.dnarep.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Lobachev KS, Shor BM, Tran HT, Taylor W, Keen JD, Resnick MA, Gordenin DA. Factors affecting inverted repeat stimulation of recombination and deletion in Saccharomyes cerevisiae. Genetics. 1998;148:1507–1524. doi: 10.1093/genetics/148.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Rattray A, Narayanan V. Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front Biosci. 2007;12:4208–4220. doi: 10.2741/2381. [DOI] [PubMed] [Google Scholar]
- Lu L, Jia H, Droge P, Li J. The human genome-wide distribution of DNA palindromes. Funct Integr Genomics. 2007;7:221–227. doi: 10.1007/s10142-007-0047-6. [DOI] [PubMed] [Google Scholar]
- Marsaglia G, Tsang W, Wang J. Evaluating Kolmogorov’s distribution. J Stat Softw. 2003;8:1–4. [Google Scholar]
- Massey FJ. The Kolmogorov–Smirnov test for goodness of fit. J Am Stat Assoc. 1951;46:68–78. doi: 10.2307/2280095. [DOI] [Google Scholar]
- Moore H, Greenwell PW, Liu C-P, Arnheim N, Petes TD. Triplet repeats form secondary structures that escape DNA repair in yeast. Proc Natl Acad Sci USA. 1999;96:1504–1509. doi: 10.1073/pnas.96.4.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchie AIH, Lilley DMJ. The mechanism of cruciform formation in supercoiled DNA: initial opening of central basepairs in salt-dependent extrusion. Nucleic Acids Res. 1987;15:9641–9654. doi: 10.1093/nar/15.23.9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag DK, Kurst A. A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics. 1997;146:835–847. doi: 10.1093/genetics/146.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasar F, Jankowski C, Nag DK. Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol Cell Biol. 2000;20:3449–3458. doi: 10.1128/MCB.20.10.3449-3458.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novac O, Alvarez D, Pearson CE, Price GB, Zannis-Hadjopoulos M. The human cruciform-binding protein, CBP, is involved in DNA replication and associates in vivo with mammalian replication origins. J Biol Chem. 2002;277:11174–11183. doi: 10.1074/jbc.M107902200. [DOI] [PubMed] [Google Scholar]
- Opel ML, Aeling KA, Holmes WM, Johnson RC, Benham CJ, Hatfield GW. Activation of transcription initiation from a stable RNA promotor by a Fis protein-mediated DNA structural transmission mechanism. Mol Microbiol. 2004;53:665–674. doi: 10.1111/j.1365-2958.2004.04147.x. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Zorbas H, Price GB, Zannis-Hadjopoulos M. Inverted repeats, stem-loops, and cruciforms: significance for initiation of DNA replication. J Cell Biochem. 1996;63:1–22. doi: 10.1002/(SICI)1097-4644(199610)63:1<1::AID-JCB1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–1399. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley LS. Model for the participation of quasi-palindromic DNA sequences in frameshift mutation. Proc Natl Acad Sci USA. 1982;79:4128–4132. doi: 10.1073/pnas.79.13.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler JR, Sasmore H, Betz JL. A perfectly symmetric lac operator binds to the lac repressor very tightly. Proc Natl Acad Sci USA. 1983;80:6785–6789. doi: 10.1073/pnas.80.22.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadashwily GM, Raca G, Mirkin SM. Trinucleotide repeats affect DNA replication in vivo. Nat Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- Schroth GP, Ho PS. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 1995;23:1977–1983. doi: 10.1093/nar/23.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko LS, Hsieh P, Grigoriev M, Potaman VN, Sinden RR, Lyubchenko YL. A cruciform structural transition provides a molecular switch for chromosome structure and dynamics. J Mol Biol. 2000;296:1169–1173. doi: 10.1006/jmbi.2000.3542. [DOI] [PubMed] [Google Scholar]
- Sinden RR, Zheng G, Brankamp RG, Allen KN. On the deletion of inverted repeated DNA in Escherichia coli: effects of length, thermal stability, and cruciform formation in vivo. Genetics. 1991;129:991–1005. doi: 10.1093/genetics/129.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro C, McMurray CT. Switching of DNA secondary structure in proenkephalin transcriptional regulation. J Biol Chem. 1997;272:33145–33152. doi: 10.1074/jbc.272.52.33145. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Bergstrom DA, Yao M-C, Tapscott SJ. Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat Genet. 2005;37:320–327. doi: 10.1038/ng1515. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Cao Y, Bergstrom DA, Kooperberg C, Tapscott SJ, Yao M-C. Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol Cell Biol. 2007;27:1993–2002. doi: 10.1128/MCB.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort V, Worming P, Ussery DW, Rosche WA, Sinden RR. Strand misalignments lead to quasipalindrome correction. Trends in Genet. 2003;19:365–369. doi: 10.1016/S0168-9525(03)00136-7. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Leung FCC. Long inverted repeats in eukaryotic genomes: recombinogenic motifs determine genomic plasticity. FEBS Lett. 2006;580:1277–1284. doi: 10.1016/j.febslet.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G. Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004;14:1861–1869. doi: 10.1101/gr.2542904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knuppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannis-Hadjopoulos M, Yahyaoui W, Callejo M. 14-3-3 cruciform-binding proteins as regulators of eukaryotic DNA replication. Trends Biochem Sci. 2008;33:44–50. doi: 10.1016/j.tibs.2007.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SFig. 1 The numbers of occurrences of the dinucleotide repeats (ApT)n or (TpA)n within IRs are plotted as functions of (left) copy length. Parts (a) through (j) give separate plots for each repeat length 1 ≤ n ≤ 9. Data for the S. cerevisiae genome are indicated by blue circles while corresponding data from a representative RA genome are shown as red x’s (EPS 4728 kb)