

Abstract
Rett syndrome is a severe neurodevelopmental disorder mainly caused by mutations in the transcriptional regulator MeCP2. Although there is no effective therapy for Rett syndrome, the recently discovered disease reversibility in mice suggests that there are therapeutic possibilities. Identification of MeCP2 targets or modifiers of the phenotype can facilitate the design of curative strategies. To identify possible novel MeCP2 interactors, we exploited a bioinformatic approach and selected Ying Yang 1 (YY1) as an interesting candidate. We demonstrate that MeCP2 interacts in vitro and in vivo with YY1, a ubiquitous zinc-finger epigenetic factor regulating the expression of several genes. We show that MeCP2 cooperates with YY1 in repressing the ANT1 gene encoding a mitochondrial adenine nucleotide translocase. Importantly, ANT1 mRNA levels are increased in human and mouse cell lines devoid of MeCP2, in Rett patient fibroblasts and in the brain of Mecp2-null mice. We further demonstrate that ANT1 protein levels are upregulated in Mecp2-null mice. Finally, the identified MeCP2–YY1 interaction, together with the well-known involvement of YY1 in the regulation of D4Z4-associated genes at 4q35, led us to discover the anomalous depression of FRG2, a subtelomeric gene of unknown function, in Rett fibroblasts. Collectively, our data indicate that mutations in MeCP2 might cause the aberrant overexpression of genes located at a specific locus, thus providing new candidates for the pathogenesis of Rett syndrome. As both ANT1 mutations and overexpression have been associated with human diseases, we consider it highly relevant to address the consequences of ANT1 deregulation in Rett syndrome.
INTRODUCTION
Epigenetic silencing is a fundamental mechanism in the prevention of inappropriate gene expression during development and differentiation. In particular, epigenetic phenomena play important roles in brain development and neuronal functions. MeCP2, which silences gene expression by binding methylated sequences of the genome via the methyl-CpG-binding domain (MBD) and by recruiting transcriptional corepressors capable of altering chromatin structure, is essential for proper brain maturation and function. Even though the precise mechanisms by which MeCP2 mediates transcriptional regulation remain elusive, MeCP2 appears to engage diverse cofactors such as the repressors Sin3A, the Histone H3 lysine 9 methyltransferase and ATRX (1–4) or the activator CREB1 (5). Thus, its association with different binding partners seems to be fundamental for the fine regulation of specific target genes (6).
MECP2 mutations are responsible for Rett syndrome (RTT), a severe neurological disorder that affects almost exclusively girls by their second year of life. Several heterogeneous symptoms characterize RTT including autistic behavior, mental retardation and reduced intellectual capacities (7). Moreover, it has been demonstrated that not only the lack of, but also a subtle increase in MeCP2 protein levels can lead to the development of an RTT-like phenotype and to severe forms of mental retardation (8). Male mice with Mecp2 deletions or over expressing MeCP2 develop severe neurological phenotypes that result in death in early adulthood, confirming the importance of a proper regulation of MeCP2 levels (9). Interestingly, it has recently been demonstrated that MeCP2 dysfunction in glia might also be involved in RTT pathogenesis (10,11).
Considering that the identification of novel MeCP2 interacting factors might contribute to a better comprehension of the molecular mechanisms by which MeCP2 acts, we exploited a bioinformatics approach to identify possible novel MeCP2 interactors. Based on the principle that functionally correlated genes tend to be expressed very similarly in space and time, we screened large repositories of published microarray data to identify genes that are consistently co-expressed with MeCP2 (12,13). Among the different genes identified by this screening, the ubiquitous zinc-finger transcription factor Yin Yang 1 (YY1) was of particular interest, as it can either activate or repress transcription of a wide range of genes (14). Its repressive functions have been suggested to play important roles in cell growth and differentiation. YY1 also plays a significant role in the establishment of epigenetic signals as it can recruit Polycomb proteins on DNA, thus leading to histone post-transcriptional modifications and stable transcriptional repression possibly through the interaction with a HDAC1/HDAC2-containing chromatin remodeling complex (14–16). Interestingly, YY1 has been associated with fascioscapulohumeral muscular dystrophy (FSHD), a common myopathy causally related to the reduction of D4Z4 tandem repeats located on chromosome region 4q35 (17,18). YY1 belongs to the multi-protein D4Z4 recognition complex (DRC), which binds these genomic repeats, leading to the formation of a chromatin structure that regulates the expression of some genes located upstream of D4Z4, including Adenine Nucleotide Translocator 1 (ANT1) and FSHD Region Gene 2 (FRG2). D4Z4 deletion results in the lack of DRC binding and the consequent aberrant overexpression of 4q35 genes (18). Here we show that MeCP2 interacts directly with YY1 and regulates the ANT1 gene, which encodes a mitochondrial protein, and FRG2, a gene of unknown function, which is specifically upregulated in FSHD. Notably, the lack of MeCP2 causes, in both humans and mice, an increase of ANT1 expression that could be relevant to some of the symptoms of RTT.
RESULTS
Identification of YY1 as a potential interactor of MeCP2 by co-expression analysis
It is well known that MeCP2 regulates the expression of several target genes through association with different interacting partners and that its role in control of gene transcription is context dependent. Thus, identification of molecules interacting with MeCP2 may provide useful information about the molecular network regulated by this protein. To provide a high-confidence list of new possible partners of MeCP2, we resorted to a co-expression-based approach (12). To this aim, we first concentrated on human cDNA microarray experiments deposited in the Stanford Microarray Database, which included data from four independent MeCP2 probes, covering partially overlapping sets of experimental conditions. For every probe, we generated a ranked list of genes showing the highest co-expression with MeCP2, as previously described (12). The final list of candidate MeCP2 partners (Supplementary Material, Table S1) was obtained by selecting only genes found in at least two independent MeCP2 co-expression lists. Interestingly, the analysis of the known function of these genes revealed that a significant fraction of them encode nuclear proteins involved in the control of gene expression (Supplementary Material, Table S2). More importantly, a well-known MeCP2 interactor, ATRX, was among the top hits of our list (4). As MeCP2 is strongly conserved between species and may regulate gene expression in a tissue-specific manner, to identify the candidates that are more likely to play a role in the context of RTT, we performed a second screening on an independent platform, looking for genes that are strongly co-expressed with MeCP2 in the central nervous system (CNS) of humans and mice. The list of these genes, obtained using the procedures described in Ala et al. (13) on a CNS-specific oligonucleotide-based microarray data set, is shown in the Supplementary Material, Table S3. Strikingly, the only gene common to both lists of candidates is the transcription factor YY1. Keeping in mind the capacity of this factor to repress transcription through its association with chromatin remodeling factors, we selected it for further validation.
YY1 associates with MeCP2 in vitro and in vivo
To verify the interaction of MeCP2 and YY1 in vitro, we performed a classical glutathione S-transferase (GST) pull-down assay. Figure 1A shows that in vitro translated MeCP2 binds immobilized GST–YY1 but not GST alone. We next defined the region of MeCP2 that interacts with YY1 by deletion analysis. The pull-down assay was repeated using deletion derivatives of the methyl-binding protein (Fig. 1A). The obtained data indicate that neither the N-terminal region (amino acids 1–162) containing the MBD nor the C-terminal region (amino acids 311–486) are able to bind YY1; in contrast, the first 53 residues of the TRD (amino acids 202–255) appear to be the minimal region of MeCP2 sufficient for the interaction with YY1. Additionally, a comparison of the auto radiographic signals indicates that the intervening domain between the MBD and the TRD also contributes significantly to the association. We then tested whether the association between the two proteins might be direct. For this purpose, we performed a far-western experiment in which GST–YY1 and GST were purified, separated by SDS–PAGE, renatured after their transfer to a nitrocellulose membrane and probed with purified recombinant MeCP2 (Fig. 1B). As shown, MeCP2 interacts with GST–YY1 but not with GST. We thus conclude that MeCP2 and YY1 interact directly in vitro and that part of the TRD and the intervening domain define the interaction surface for this association.
Figure 1.

MeCP2 and YY1 interact directly in vitro. (A) The MeCP2 region that binds YY1 was identified by GST pull-down assays in which full-length in vitro translated [35S]methionine-labeled MeCP2 or its mutated derivatives were incubated with immobilized recombinant GST and GST–YY1. The MeCP2 derivatives are shown schematically to the left (filled circles indicate the methionines present in MeCP2) and the corresponding autoradiograms to the right. + and − indicate the presence and absence, respectively, of interaction. (B) A far-western assay (FW) was performed with 1 µg of GST and GST–YY1, separated by SDS–PAGE, transferred to a nitrocellulose membrane and probed with recombinant MeCP2 after renaturation. MeCP2 was detected with anti-MeCP2 antibodies (lanes 1 and 2). Coomassie staining (Coom.) was used to visualize the recombinant GST and GST–YY1 proteins (lanes 3 and 4, respectively) used in the far-western assay.
The capability of the two proteins of interacting in vivo was analyzed by co-immunoprecipitation (Co-IP; Fig. 2). HEK293 cells were transiently transfected with Flag-YY1 and Myc-MeCP2 and the total cell extract was immunoprecipitated with anti-Flag antibodies. The subsequent immunoblotting with anti-MeCP2 and anti-YY1 antibodies demonstrated a specific co-precipitation of the methyl-binding protein with YY1 (Fig. 2A). The interaction was also confirmed with endogenous YY1 and MeCP2 in mouse brain total cell extracts; in fact, YY1 antibodies, but not unrelated mouse IgGs, were able to co-precipitate both proteins (Fig. 2B). In addition, immunofluorescence experiments of NIH3T3 cells indicated that the two proteins co-localize to some extent in the nuclear compartment (Fig. 2C). On the basis of the results shown in Figures 1 and 2, we concluded that MeCP2 and YY1 interact in vitro and in vivo.
Figure 2.

MeCP2 and YY1 interact in vivo. (A) Co-IP of exogenously expressed Myc-MeCP2 and Flag-YY1. HEK293 cells were transiently transfected with vectors expressing Myc-MeCP2 and Flag or Flag-YY1 in different combinations. A total cell extract (TCE) was immunoprecipitated with anti-Flag antibodies, and the precipitated proteins were revealed with anti-MeCP2 (monoclonal) and anti-YY1 (polyclonal) antibodies. Inputs corresponding to 10% of the TCEs were also analyzed. (B) Co-IP of endogenous MeCP2 and YY1 from a mouse brain extract. YY1 was immunoprecipitated from a P28 mouse brain TCE and the precipitated proteins revealed with anti-MeCP2 and anti-YY1. Mouse IgGs were used as negative control. Input corresponds to 10% of the TCE. (C) Endogenous YY1 and MeCP2 partially co-localize in NIH3T3 cells. Cells were immunostained with anti-MeCP2 (a, polyclonal) and anti-YY1 (b, monoclonal) antibodies and counterstained with DAPI (d). Merged images are shown in (c).
MeCP2 and YY1 regulate ANT1 expression
Interestingly YY1, HMGB2 and nucleolin constitute a multi-protein complex DRC that binds the D4Z4 module, regulates chromatin organization and mediates transcriptional repression of the 4q35 genes, FRG1, FRG2 and ANT1 (18). We thus reasoned that MeCP2 might contribute to the YY1-dependent regulation of 4q35 gene expression and that a reduction of MeCP2 levels would cause some derepression of 4q35 genes. We therefore depleted HeLa cells for MeCP2 and YY1 by small interfering RNA (siRNA) and verified by western blotting that the level of both proteins was reduced by >80% (Fig. 3A). The expression levels of some 4q35 genes were then analyzed by quantitative real-time PCR (qRT-PCR) (Fig. 3B and data not shown). MeCP2 or YY1 depletion causes a significant increase of ANT1 mRNA levels (Fig. 3B), whereas a slight effect is observed for FRG1 and FRG2 (data not shown). Importantly, silencing of both MeCP2 and YY1 leads to an additive increase in ANT1 mRNA expression (Fig. 3B), suggesting that the two proteins cooperatively contribute in controlling the activity of this gene. As mitochondrial defects have often been associated with RTT (19–21), we decided to focus our attention on the mitochondrial gene ANT1. We therefore analyzed the expression of ANT1 in cells from RTT patients carrying mutations in MECP2 (Fig. 4). ANT1 mRNA levels were evaluated in fibroblasts from a patient carrying a 705delG mutation, affecting codon 235 (bar 1), one with a pQ244X truncating mutation (bar 2) and two patients with the pT158M missense mutation in the MBD (bars 3 and 4). The results shown in Fig. 4A demonstrated that ANT1 expression was significantly upregulated in the samples carrying mutated MeCP2. Moreover, as our results raised the possibility that functional inactivating mutations of MECP2 might lead to inappropriate expression of other 4q35 genes, we also analyzed expression of FRG2. Interestingly, significantly increased FRG2 levels were observed in the three analyzed samples. We conclude that MeCP2 mutations cause a consistent increase in the expression of at least two 4q35 genes, even though the induction levels seem to depend on the specific mutation and/or genetic background.
Figure 3.
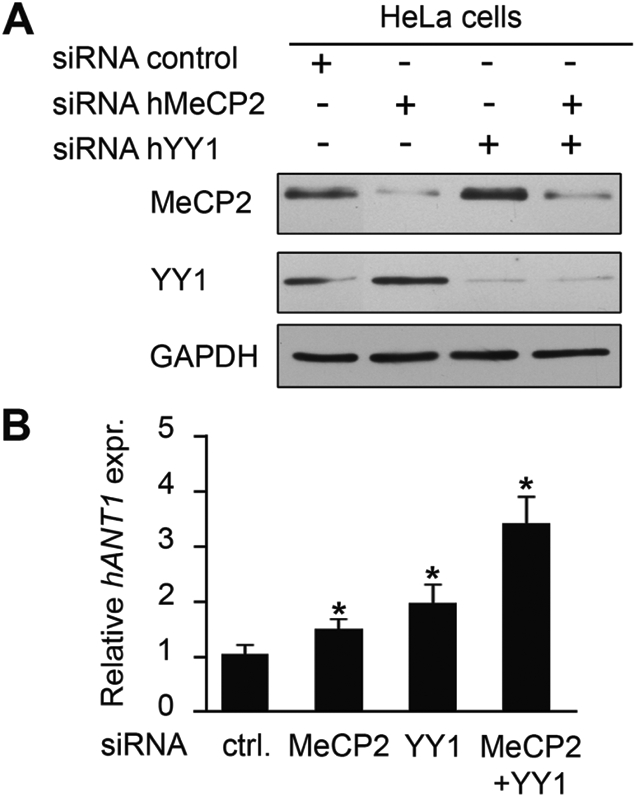
ANT1 expression is upregulated in MeCP2- and YY1-depleted cells. (A) HeLa cells were transiently transfected with the indicated siRNAs; total cell extracts were collected 24 h post-transfection and analyzed by western blotting using anti-MeCP2 and anti-YY1 antibodies. (B) qRT-PCR analysis of ANT1 mRNA expression relative to Actin performed on RNA extracted from siRNA-treated HeLa cells (A). ANT1 expression in control cells transfected with a scrambled siRNA was normalized to 1 (Ctrl). Statistical analysis was performed on samples analyzed in triplicate. Significance was tested using the t-test. *P < 0.05. Error bars indicate standard deviations. This experiment is representative of three independent experiments.
Figure 4.
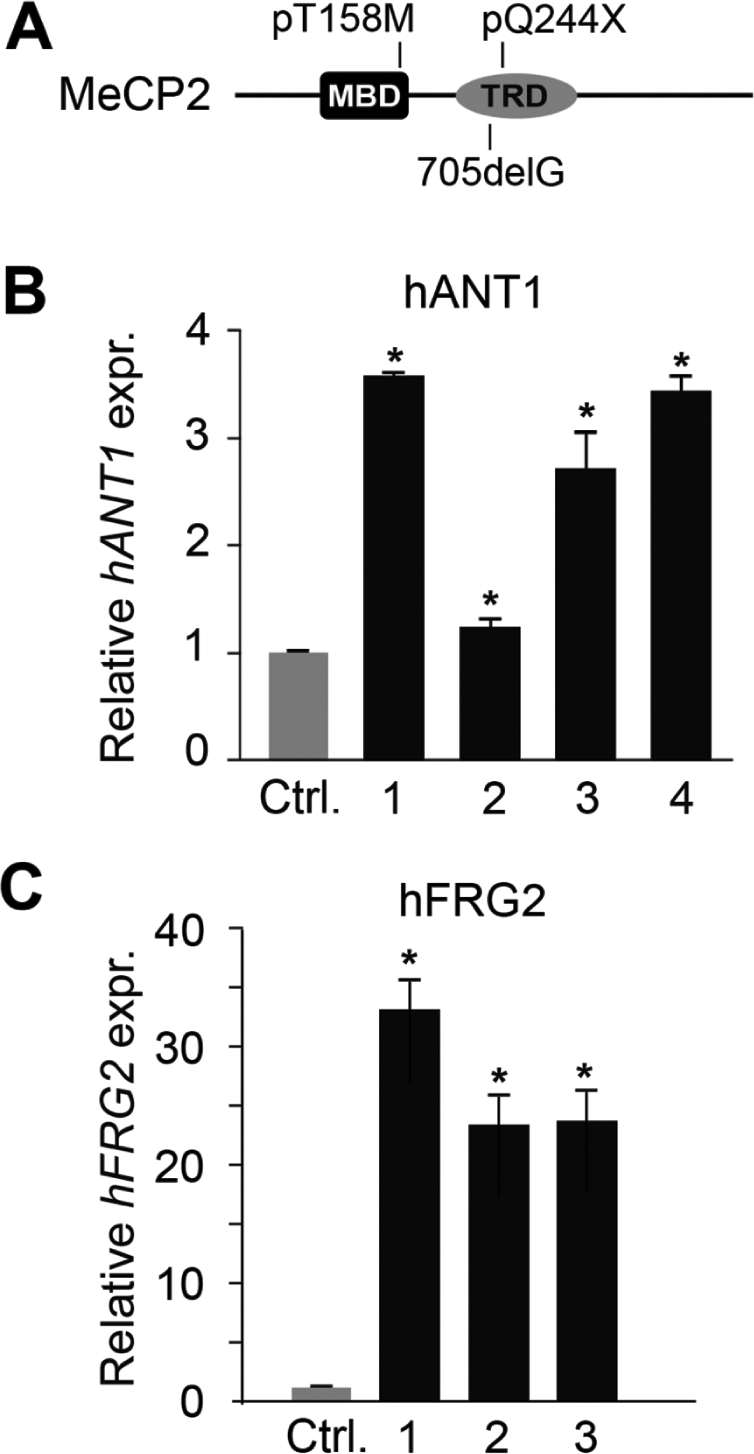
ANT1 and FRG2 mRNAs are upregulated in RTT patients' fibroblasts. (A) Schematic illustration of the MeCP2 mutations in the analyzed Rett patients' fibroblast. (B) ANT1 mRNA levels in primary fibroblasts from four RTT patients with different MECP2 mutations were measured by qRT-PCR analysis and normalized to Actin. (C) FRG2 mRNA levels were analyzed in fibroblasts from three RTT patients by qRT-PCR analysis and normalized to B2M levels. The different bars correspond to the following MECP2 mutations: bar 1, 705delG; bar 2, pQ244X; bars 3 and 4, pT158M. ANT1 and FRG2 mRNA expression in fibroblasts from three unaffected patients (Ctrl) was normalized to 1. Statistical analysis was performed on samples analyzed in triplicate. Significance was tested using the t-test. *P < 0.05. Error bars indicate standard deviations.
Based on the fact that D4Z4 repeats, as well as FRG2, are absent in the murine genome, we investigated whether MeCP2 and YY1 modulate Ant1 transcription independent of the repeats. Ant1 mRNA levels were determined by qRT-PCR in N2a cells depleted for the two proteins (Fig. 5A). Whereas the absence of MeCP2 or YY1 causes a subtle increase in Ant1 expression, the depletion of both proteins generates an additive effect, confirming that MeCP2 and YY1 also cooperate in regulating Ant1 levels in murine cells.
Figure 5.

MeCP2 and YY1 depletion increases Ant1 expression in murine cells and brain. (A) N2a cells were transiently transfected with the indicated siRNAs; total RNA was extracted 24 h post-transfection and analyzed by qRT-PCR to determine Ant1 mRNA levels relative to GAPDH. Ant1 expression in control cells transfected with a scrambled siRNA was normalized to 1 (Ctrl). Statistical analysis was performed on samples analyzed in triplicate. Significance was tested using the t-test. P < 0.05. Error bars indicate standard deviations. This experiment is representative of three independent experiments. (B) Representative qRT-PCR analysis showing Ant1 up regulation in the brain and cerebellum (Cer.) of one Mecp2-null mouse (KO). Similar results were observed with six other mutant animals in which we consistently observed a more pronounced augmention in the cerebellum. The data were evaluated using the t-test. *P < 0.05. Error bars indicate standard deviations. Ant1 mRNA levels are relative to GAPDH. Ant1 expression in the WT mouse was normalized to 1. (C) Western blot showing ANT1 protein levels in total extracts of tibial muscles from normal and Mecp2-null mice. Numbers below the western blot indicate the increase in ANT1 levels normalized to α-tubulin with respect to the wild-type controls.
Interestingly, as shown in Fig. 5B, we also found a significant increase in Ant1 mRNA levels in both total brain and cerebellum of Mecp2-null mice, supporting the role of MeCP2 in regulating Ant1 expression in mouse tissues. In contrast, no differential Ant1 expression was found in fibroblasts derived from Mecp2-null embryos and their wild-type litters (data not shown). As ANT1 deregulation was observed in fibroblasts derived from RTT patients, these data might indicate a stage-specific regulation. Of the three murine Ant genes, Ant1 is considered to be the muscle-specific adenine nucleotide translocator (22); by western blotting, we therefore compared ANT1 protein levels in muscles from Mecp2-null mice with their wild-type litters and, importantly, we found a significant increase in the mutant mice (Fig. 5C).
MeCP2 and YY1 bind to the Ant1 promoter
The regulation of Ant1 expression by MeCP2 and YY1 was further investigated to address the potential interaction of the two proteins with its promoter sequences. To achieve this, we performed chromatin immunoprecipitation (ChIP) assays on chromatin from wild-type mouse cerebella. We analyzed the region of the Ant1 promoter that extends ∼2000 bp upstream of the transcription start site and subdivided it for PCR amplification in six shorter regions schematically represented in Fig. 6A. MeCP2 is not bound to the region encompassing the transcription start site (+86 to −188) but appears particularly enriched on the region spanning nucleotides −389 to −1517; YY1 binding appears confined to the region limited by nucleotides −840 to −1709 (Fig. 6B). These data show that both proteins interact with the Ant1 promoter. As MeCP2 is generally recognized as a methyl-binding protein capable of repressing transcription of methylated genes, we investigated the presence of methyl-CpG sites in the Ant1 regions bound by MeCP2. Bisulfite sequencing of the Ant1 promoter in cerebellum revealed a high concentration of methylated CpG dinucleotides between −1205 and −1709 and a low density in the region from −840 to −1205 (Fig. 6C). Consistent with recent findings (23), the many CpG sites at the proximal promoter region (from +86 to −840) are not methylated. Overall, our data do not show a perfect coincidence between MeCP2 binding and DNA methylation. Indeed, they suggest a strong residency of MeCP2 on fragments c–e, therefore including DNA segments that lack or contain very little CpG methylation.
Figure 6.

MeCP2 and YY1 bind to a specific region of the mouse Ant1 promoter. (A) Schematic representation of the Ant1 promoter region used for PCR amplification of immunoprecipitated DNA. The sequence was amplified by PCR in six sub regions (a–f). The numbers −188, −389, −840, −1205, −1517 and −1709 indicate the relative distance upstream of the transcriptional start site (black arrow). Vertical bars in the upper part indicate CpG dinucleotides. (B) ChIP reveals binding of MeCP2 and YY1 to a common region of the Ant1 promoter. Chromatin from mouse cerebella was immunoprecipitated with anti-MeCP2 and anti-YY1 antibodies or control IgGs and analyzed by PCR. Non-precipitated chromatin was used as control (Input). (C) CpG methylation of the Ant1 promoter region. The promoter region was divided into four sub-regions (a + b, c, d and e + f) and the methylation status of CpGs monitored by bisulfite analysis. The filled and open circles represent the methylated and unmethylated CpG dinucleotides, respectively. A mean of 10 separate clones was analyzed for each region. Partial methylation was observed in d, e + f.
DISCUSSION
Mutations in the X-linked MECP2 gene are the main cause of RTT, a severe neurodevelopmental disorder, considered as the second most common cause of mental retardation in females. The disease onset is marked by a neurological decline, which appears around 6–18 months of age when a sudden deceleration in head growth associated with progressive loss of previously acquired skills, stereotypic hand movement, muscle hypotonia, breathing disturbances, autonomic dysfunctions and severe cognitive impairment appear.
Mice deficient for MeCP2 have a wide range of neurological and physiological abnormalities that mimic the human disorder (9). Although no specific treatments for RTT are available, the recently discovered disease reversibility in mice suggests that there are therapeutic possibilities for Rett patients (24). Therefore, understanding the molecular pathogenesis of RTT becomes of major importance for developing curative strategies.
Several scientific reports suggest that MeCP2 works as an epigenetic transcriptional repressor that binds methylated DNA and causes the formation of a compact, transcriptionally inert chromatin structure, mainly by recruiting repressive chromatin-modifying complexes (1,2). However, more recently, it has been proposed that MeCP2 might also function as a transcriptional activator (5). Nonetheless, there is a general consensus that MeCP2-related disorders are caused by changes in the expression levels of several genes. Furthermore, it has been proposed that the protein partners of MeCP2, and therefore its mechanisms of actions, might change in a DNA template-dependent manner (6). Here we show that YY1 interacts with MeCP2 and participates in the repression of the ANT1 gene. YY1 is a ubiquitously expressed transcription factor, belonging to the Polycomb proteins that can either activate or repress transcription depending upon cell type, stimuli and/or promoter context (14). YY1 is expressed in all major brain regions with levels that vary depending on the brain structure, but with a significantly higher expression in neurons than in glia. The relevance of YY1 in the maintenance of cell homeostasis in the adult brain is suggested by its involvement in the pathogenesis of different disorders such as schizophrenia, Parkinson's and Alzheimer's disease (25). Importantly, the conditional knockout of YY1 in the oligodendrocyte lineage determines an arrest in cell differentiation leading to tremor and ataxia (26).
Interestingly, YY1 is part of a multi-protein complex together with HMGB2 and nucleolin that regulates 4q35-associated genes (18). As we demonstrate that MeCP2 and YY1 interact in vivo and cooperate in the repression of genes associated with 4q35, we tested the relevance of MeCP2 and YY1 interaction for RTT. Remarkably, analysis of 4q35 gene expression revealed the significant upregulation of ANT1 and FRG2 in fibroblasts from RTT patients bearing MeCP2 mutations.
ANT1, which encodes an adenine nucleotide translocator, is a central component of the mitochondrial permeability transition pore complex, whereas FRG2 is a gene of unknown function, which was found uniquely expressed in FSHD (18,27). As mitochondria have long been implicated in RTT, we decided to focus our further studies on this gene. Interestingly, we found that the pQ244X mutation, which maintains most of the minimal region of interaction we identified in Figure 1, leads to a very subtle deregulation of ANT1. Vice versa, the frameshift mutation occurring at residue 235 (patient 705delG), which interferes to a larger extent with the surface of interaction, has effects comparable to that of the missense mutation, affecting the capability of MeCP2 to bind methylated DNA (28). Even though this result correlates with the importance of the interaction of MeCP2 and YY1 for a proper repression of the ANT1 promoter, we only want to highlight that ANT1 is derepressed in all samples where MeCP2 is malfunctioning. Indeed, we are aware that the genetic background and the level of X-chromosome inactivation might influence the result in addition to the specific mutation. Notably, in RTT human primary fibroblasts, we also observed a considerable activation of FRG2, the most proximal gene to the D4Z4 repeats. This observation reinforces the idea that MeCP2 contributes substantially to the chromatin organization at 4q35 and MECP2 mutations might thus affect 4q35 gene expression significantly.
Additionally, our experiments revealed that MeCP2 and YY1 control Ant1 expression even in murine cells and tissues, whose genome lacks the D4Z4 repeat, including the FRG2 gene. Expression analyses conducted in both total brain and cerebellum of Mecp2-null adult mice, a mouse model of RTT, and in embryonic fibroblasts from the same mouse model revealed the selective increase of Ant1 in neural tissues. As we observed an effect of MECP2 mutations on ANT1 mRNA levels in human fibroblasts, we hypothesize that MeCP2 might regulate ANT1 in a stage-specific manner even though we cannot exclude that a tissue-specific regulation might also exist. Ant1 has a significantly higher expression in skeletal muscles where it represents the vast majority of adenine nucleotide translocators; by western blotting we were also able to demonstrate that ANT1 protein levels were significantly increased in muscles from mice devoid of MeCP2. The same analysis was performed on brain extracts. Even though our not shown data do suggest an upregulation of the protein in neuronal samples, the results are less clear. We believe that this is mainly due to the fact that the antibody recognizes all ANT isoforms including ANT2 that is also significantly expressed in brain.
Interestingly, our ChIP results clearly indicate that the identified ANT1 regulation is a direct effect of MeCP2 binding, even though our methylation studies did not show an exact correspondence between MeCP2 occupancy and the methylation profile of the regulatory sequences analyzed. This result might represent the capability of the MBD to bind unmethylated DNA, albeit that we favor the hypothesis that the observed presence of MeCP2 on DNA fragments containing very little or no methylation is given by the TRD or the C-terminal portion of the protein. In fact, it has been demonstrated that the TRD of MeCP2 is able to bind non-specifically to DNA and to cross-link unmethylated DNA and nucleosomal arrays into oligomeric superstructures (29), whereas the C-terminal portion leads to chromatin compaction through nucleosome–MeCP2–nucleosome interactions (30).
Collectively, our results show that YY1 is a novel partner of MeCP2 and that ANT1 is a target gene of these two proteins; the lack of a functional MeCP2 protein leads to the upregulation of ANT1 in both human and mouse cells. ANT1 is a nuclear gene encoding the mitochondrial adenine nucleotide translocase-1, a bi-functional protein that transports ADP and ATP across the mitochondrial membrane, and regulates the mitochondrial permeability transition pore, involved in initiating the apoptotic response (31). Even though there are three different ANT isoforms in humans, only ANT1, which is predominantly expressed in skeletal muscles and heart, but is also present in brain, has thus far been associated with mitochondrial diseases in humans. In fact, ANT1 mutations have been linked in humans to inherited progressive external ophthalmoplegia (32,33). Moreover, Ant1−/− mice develop mitochondrial myopathy associated with ragged-red fibers and abnormal mitochondria and a hypertrophic cardiopathy (34). Importantly, ANT1 overexpression induces apoptosis and a link between the excessive apoptosis observed in tissues from DCM (dilated cardiomyopathy) patients and a dramatic upregulation of ANT1 in heart tissue of these patients has been proposed (35). It has been postulated that ANT1 derepression might correlate with the higher sensitivity of muscle cells to oxidative stress observed in FSHD, one of the most common hereditary myopathies. This disease is generally characterized by the adult onset of a progressive and spatially restricted muscle weakness; in the most severe cases, extramuscular manifestations such as mental retardation and seizures have also been described (36). Even though, so far, Rett symptoms are considered to be the result of neuronal defects, in the future it might be relevant to address what are the consequences of ANT1 deregulation in both neuronal and muscle cells deficient for MeCP2.
Finally, our results provide evidence that MeCP2 is capable of influencing expression of genes associated with the D4Z4 repeat on chromosome 4. Notably, D4Z4 is an element with heterochromatin features, belonging to a family of repetitive elements, which are present at 1q12 and on the short arm of acrocentric chromosomes in the human genome (37). It is therefore plausible to speculate that investigating the same molecular mechanism involving different chromosomes containing this repetitive element might lead to a deregulation of genes important for the onset of RTT symptoms.
MATERIALS AND METHODS
Co-expression analysis
In order to identify high-confidence putative partners of MeCP2, we performed two different co-expression analyses, conforming to very stringent criteria. In the first analysis, we obtained a list of human genes consistently co-expressed with MeCP2 in a previously described set of microarray experiments extracted from the Stanford Microarray Database (12). The analyzed data set was composed of 2803 ratiometric experiments, comprising in total 74 588 probes that contained four independent MeCP2 probes. For every MeCP2 probe, we calculated the Pearson correlation coefficient with all other probes and ranked the remaining probes according to the values of this index. The final list (Supplementary Material Table S1) was obtained considering only those genes that fell in the top 1% rank in at least two of the four lists. In the second analysis, we obtained a list of the genes displaying brain-specific co-expression with MeCP2 in both human and mouse. In this case, a large set of brain-specific oligonucleotide array experiments (618 human and 473 mouse samples, respectively) were downloaded from the Gene Expression Omnibus. A tissue-specific conserved co-expression network was then produced, using the procedure previously described (13). The list of putative MeCP2 partners (Supplementary Material, Table S3) was obtained by extracting from the network MeCP2 first-level links.
Cell cultures, fibroblast clones and animals
The human cervical epithelial carcinoma cell line, HeLa, and the human embryo-derived kidney cell line, HEK293, were maintained in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), streptomycin (100 g/ml) and 2 mm glutamine. Mouse neuroblastoma cells, N2a, were grown in MEM supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 g/ml) and 2 mm glutamine. All cells were grown at 37°C and 5% CO2. Three fibroblast strains from females with clinically diagnosed RTT and two wild-type controls were obtained from Coriell Cell Repositories. MECP2 mutations are as follows: GM16548: 730C > T (pQ244X); GM7982: 705 delG; GM17880: 473 C > T (pT158M). The fourth RTT fibroblast clone with the pT158M MeCP2 mutation and fibroblasts from one healthy control patient were a kind gift from A. Martinuzzi. The cells were grown in monolayer and were trypsinized for expansion at confluency. Growth rates and cellular morphology were closely monitored. The B6.129P2©-Mecp2tm1-Bird mice (38) from the Jackson Laboratory were maintained on a C57BL/6 background. Hemizygous mutant males were generated by crossing heterozygous knockout females to C57BL/6 males. All experiments were performed on hemizygous Mecp2-deficient males of about 4 weeks of age. Wild-type males of the same litter and age were used as controls for each hemizygous Mecp2-deficient male analyzed.
Plasmids
The following plasmids have already been described: pMyc-MeCP2 (39), pGST-hYY1 (18). pFlag-YY1 contains the hYY1 sequence cloned in- frame with the Flag-tag in p3xFlag-myc-CMV (Sigma-Aldrich).
GST pull-down assays
Expression and purification of GST and GST–YY1 fusion proteins were performed using glutathione–Sepharose 4B (Amersham Pharmacia Biotechnology) according to the manufacturer's instructions. Immobilized GST proteins (5 µg) were incubated with 20 µl of in vitro translated [35S]methionine labeled MeCP2 in 300 µl of binding buffer [PBS, 0.1% Triton X-100, 1 mm phenyl-methylsulfonyl fluoride (PMSF), 1 mg/ml bovine serum albumin (BSA), protease inhibitor mixture] for 3 h at 4°C, and the beads were subsequently washed with PBS, 0.1% Triton X-100, 0.1% NP-40, 1 mm PMSF. Bound proteins were fractionated by SDS–PAGE and detected by autoradiography of the dried gel. In vitro translated full-length MeCP2 was obtained using the TNT T7 Coupled Reticulocyte Lysate System (Promega) with MeCP2 as template. [35S]Methionine-labeled MeCP2 derivatives were obtained using the TNT T7 Quick for PCR DNA (Promega).
Far-western assay and western blotting
Purified GST and GST–YY1 (1 µg) were resolved by SDS–PAGE, transferred to a nitrocellulose membrane (Hybond; Amersham Biosciences) and renatured by overnight incubation at 4°C in binding buffer (50 mm Tris–HCl, pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.5% Triton X-100, 10% glycerol, 5% non-fat dried milk). After two washes with PBS and 0.2% Tween-20, the membrane was probed with 3 µg of recombinant hMeCP2 purified from Escherichia coli (40) in binding buffer without milk for 2 h at 4°C. MeCP2 was detected by western blotting with polyclonal or monoclonal anti-MeCP2 antibodies (Sigma-Aldrich). To detect ANT1 protein by western blotting, we used a polyclonal anti-Ant1 antibody (Santa Cruz).
Co-immunoprecipitation
HEK293 cells were transfected with pFlag-YY1 and/or pMyc-MeCP2 expression vectors. After 24 h, cells were collected, resuspended in lysis buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA) supplemented with phosphatase and protease inhibitors (1 mm Na3VO4, 1 mm DTT, 5 mm NaF, 1 mm PMSF and protease inhibitor mixture), sonicated and centrifuged for 15 min at 13 000g at 4°C. After pre-clearing the extracts for 1 h with 10 µl of 100% mouse IgG–agarose beads at 4°C, 25 µl of 100% EZviewRed Anti-Flag M2 Affinity gel (Sigma-Aldrich) was added and the mixture incubated for 2 h 30 min at 4°C. Immunocomplexes were collected by centrifugation, washed three times with the above lysis buffer and twice with the lysis buffer containing 400 mm NaCl. The immunocomplexes were detected by western blotting with monoclonal anti-MeCP2 (Sigma-Aldrich) and polyclonal anti-YY1 (Santa Cruz Biotechnology) antibodies. For immunoprecipitation of endogenous proteins, P28 mouse brains were extracted by the addition of lysis buffer as above and centrifuged for 20 min at 13 000g. The pre-cleared extract was incubated with monoclonal anti-YY1 (Santa Cruz Biotechnology) or an unrelated IgG overnight at 4°C and immunocomplexes precipitated with protein-G–agarose (Invitrogen). The immunocomplexes were analyzed by western blotting with polyclonal anti-YY1 and anti-MeCP2 (Sigma-Aldrich) antibodies.
Bisulfite treatment and sequencing analysis
Bisulfite treatment of 1 µg of mouse cerebellum genomic DNA was carried out using the MethylDetector™ kit (Active Motif). Four microliters of bisulfite-treated DNA were amplified by PCR. Primers used for this analysis were designed using the MethPrimer software (41) and the forward and reverse sequences were: (a + b) 5′-GAGTTGAGAGAGATTAGGAGTTAATAAAGGAG-3′ and 5′-CCCCTTAATACTTCAAAAAAAACTCTTTTTAACC-3′; (c) 5′-GGTTAAAAAGAGTTTTTTTTGAAGTATTAAG-3′ and 5′-CCTTAAAAAAAACTCAAAACCTAATCC-3′; (d) 5′-GTGGAAGGGGTAGTAGTATGGAAAATATGTATAG-3′ and 5′-GTGGAAGGGGTAGTAGTATGGAAAATATGTATAG-3′; (e + f) 5′-GTTTATGGAAATTGTTGTGTGAAATTGTG-3′ and 5′-CTATACATATTTTCCATACTACTACCCCTTCCAC-3′. PCR products were cloned into the pGEM-T Easy vector (Promega) and about 10 colonies for each insert were sequenced for analysis of the percentage of methylated CpGs.
Chromatin immunoprecipitation
Cerebella from adult mice (age 5 months) were fixed by incubation in 1.8% formaldehyde in PBS. After 5 min, the tissue was mechanically chopped, the fixation continued for 10 min at room temperature under gentle agitation and then quenched with 0.125 m glycine for 5 min. Pelleted cells were washed with PBS and homogenized in a Dounce homogenizer. After centrifugation, cells were resuspended in 1 ml of lysis buffer (5 mm PIPES pH 8.0; 85 mm KCl, 0.5% NP40, 1 mm PMSF and protease inhibitor mixture). Lysates were triturated through a 25-guage needle to remove lumps and incubated on ice for 15 min with an additional 1 ml of lysis buffer. Nuclei were then harvested by spinning at 4000g for 5 min at 4°C, resuspended in 700 µl of sonication buffer (1% SDS; 10 mm EDTA, pH 8.0; 50 mm Tris–HCl, pH 8; 1 mm PMSF and protease inhibitor mixture) and incubated for 10 min on ice. To obtain chromatin DNA fragments of an average of 400 bp, the extract was sonicated for 5 min using 35 s pulses (40% amplitude) with a 15 s rest between pulses with a Branson Digital Sonifier (Branson Ultrasonic) at power setting of 20–30 W. The chromatin was then cleared by centrifugation, pre-cleared with protein G–agarose (Invitrogen) and subjected to overnight immunoprecipitation with rabbit polyclonal antibodies against MeCP2 (Sigma) and YY1 (Santa Cruz Biotechnology) or with immunopurified IgG (Upstate). Antibody precipitates were bound to protein G–agarose (Invitrogen) for 1 h with agitation and washed five times with buffer A (0.1% SDS; 2 mm EDTA, pH 8.0; 20 mm Tris–HCl, pH 8; 1% Triton X-100; 150 mm NaCl; 1 mm PMSF and protease inhibitor mixture), four times with buffer B (0.1% SDS; 2 mm EDTA, pH 8.0, 20 mm Tris–HCl, pH 8; 1% Triton X-100; 500 mm NaCl; 1 mm PMSF and protease inhibitor mixture) and once with Tris–EDTA. Antibody precipitates were then extracted twice with elution solution (1% SDS; 100 mm NaHCO3), NaCl was added to a final concentration of 200 mm and cross-links were reversed by overnight incubation at 65°C. The proteins were digested with 200 µg/ml of proteinase K (Sigma) for 2 h at 44°C. The DNA was recovered by phenol–chloroform extraction and ethanol precipitated using glycogen as carrier. DNA was resuspended in 30 µl of nuclease-free water, and 1 µl was used for PCR amplifications. The forward and reverse primers used for this analysis were: (a) 5′-GCTCCCGGAAGCGTCTT-3′ and 5′-GAAAGCCGGGCGCACA-3′; (b) 5′-CAAAAAGAGCTTCCCTTGAAGC-3′ and 5′-GAAGACGCTTCCGGGAGCT-3′; (c) 5′-CTAATAGCACTCATTCTTGTGG-3′ and 5′-CTTGGGATTCTCTCGTG-3′; (d) 5′-CTCCAGCAACATGGTAACG-3′ and 5′-CCACAAGAATGAGTGCTATTAG-3′; (e) 5′-GCCAAATGGAGTAGACACAC-3′ and 5′-CAAAGAAGACACACGCATAGAC-3′; (f) 5′-GAGAGTCAGAAAGGCTGTGG-3′ and 5′-GTGTGTCTACTCCATTTGGC-3′.
Small interfering RNA assay
The oligonucleotides used for the siRNA experiments were obtained from Dharmacon RNA Technologies and were as follows: siRNAMeCP2 (sense) 5′-GGAAAGGACUGAAGACCUGUU-3′, (antisense) 5′-CAGGUCUUCAGUCCUUUCCUU-3′; siRNAYY1 (sense) 5′-CAUAAAGGCUGCACAAAGAUU-3′, (antisense) 5′-UCUUUGUGCCUUUAUGUU-3′. A scrambled siRNA was used as negative control. HeLa cells were transfected with Lipofectamine 2000 (Invitrogen) with 20 nm siRNA (final concentration). N2a cells were transfected with the same siRNA (1 nm each) using Metafectene™ Pro (Biontex) according to the manufacturer's instructions. After 24 h, the cells were harvested and used for western blotting and qRT-PCR assays. No major effects in cell cycle regulation and proliferation were observed probably because of the limited time occurring between the transfection and the harvesting of cells.
Reverse transcription and qRT-PCR analysis
Total RNA was isolated from mouse brains and cerebella using an AURUM™ Total RNA Fatty and Fibrous Tissue kit (Bio-Rad) according to the manufacturer's instructions. Total RNA from the different cell lines was extracted using the TRIzol RNA extraction Reagent (Invitrogen) and genomic DNA eliminated with DNase I (New England Biolabs). cDNAs were prepared from 1 µg of purified RNA using SuperScript II (Invitrogen), and 1 µl was PCR-amplified. qRT-PCR analysis was performed using a Chromo 4™ Detector real-time PCR machine with IQ SYBR green Supermix or IQ Supermix (Bio-Rad). Each independent sample was analyzed in triplicate from RNA collected from at least three independent siRNA assays or was confirmed with n = 7 animals. All qRT-PCR values were normalized to either Actin or GAPDH, as indicated. For expression analysis of FRG2, which requires more sensitivity and specificity, a TaqMan assay was performed. cDNAs were prepared from 2 µg of total RNA, isolated as above, and 1 µl was analyzed by RT-PCR with ABI PRISM 7500 FAST (Applied Biosystems) using B2M levels as an endogenous control for data normalization.
Forward and reverse primer sequences used are: Ant1: 5′-tcgtaggatgatgatgcagtct-3′ and 5′-gctccttcatcttttgcaatct-3′; Gapdh: 5′-ATTGTCAGCAATGCATCCTG-3′ and 5′-ATGGACTGTGGTCATGAGCC-3′; ANT1: 5′-TGGGCHAGAGCACGAACG-3′ and 5′-CACCACACAATCAATGATCCCTTTG-3′; ACTA1: 5′-TCCTTCCTGGGCATGGAG-3′ and 5′-AGGAGGAGCAATGATCTTGATCTT-3′. TAQ-MAN ASSAY: FRG2: 5′-GGAGTGCAGCTTGTCATTGAATAA-3′, 5′-CGAATTGACGGTGTTTGGACTC-3′ and probe: 5′-AGACCTATGCCGCTTGCTGTGCCC-3′; B2M: 5′-CGCTCCGTGGCCTTAGC-3′, 5′-AATCTTTGGAGTACGCTGGATAGC-3′ and probe: 5′-CTCGCGCTACTCTCTCTTTCTGGCCTG-3′.
Immunofluorescence staining
NIH3T3 cells were seeded on glass coverslips and transfected with Lipofectamine 2000 (Invitrogen). The cells were fixed with 4% paraformaldehyde and stained with monoclonal anti-YY1 antibodies (Santa Cruz Biotechnology; diluted 1:100) and polyclonal anti-MeCP2 (Sigma; diluted 1:2000) in PBS, 1% FBS, 0.1% Triton-X-100 for 1 h at room temperature. Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor 555-conjugated anti-mouse IgGs (Molecular Probes) were used as secondary antibodies. Nuclei were stained with DAPI (Sigma-Aldrich). Microscopic analysis was performed with an Olympus BX51 fluorescence microscope.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by Fondazione Cariplo (N.L.), Fondazione Comunitaria del Varesotto onlus (N.L.), Telethon (N.L.), NIH-NINDS (R.T.) FSHD Global (R.T.), EuroRETT (N.L., C.K.-N.), AIRC (N.L.) and FP7-PEOPLE-ITN-2008 (C.K.-N., R.T.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Martinuzzi for providing RTT fibroblasts, and ProRETT Ricerca for their continuous trust in the laboratory.
Conflict of Interest statement. The authors have no conflict of interest.
REFERENCES
- 1.Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. doi:10.1038/561. [DOI] [PubMed] [Google Scholar]
- 2.Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 3.Fuks F., Hurd P.J., Deplus R., Kouzarides T. The methyl-CpG binding protein MeCP2 links DNA methylation to histone methylation. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. doi:10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nan X., Hou J., Maclean A., Nasir J., Lafuente M.J., Shu X., Kriaucionis S., Bird A. Interaction between chromatic proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl Acad. Sci. USA. 2007;104:2709–2714. doi: 10.1073/pnas.0608056104. doi:10.1073/pnas.0608056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. doi:10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klose R., Bird A. MeCP2 repression goes nonglobal. Science. 2003;302:793–795. doi: 10.1126/science.1091762. doi:10.1126/science.1091762. [DOI] [PubMed] [Google Scholar]
- 7.Chahrour M., Zoghbi H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. doi:10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Van Esch H., Bauters M., Ignatius J., Jansen M., Raynaud M., Hollanders K., Lugtenberg D., Bienvenu T., Jensen L.R., Gecz J., et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005;73:422–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricceri L., De Filippis B., Laviola G. Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav. Pharmacol. 2008;19:501–517. doi: 10.1097/FBP.0b013e32830c3645. doi:10.1097/FBP.0b013e32830c3645. [DOI] [PubMed] [Google Scholar]
- 10.Ballas N., Lioy D.T., Grunseich C., Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. doi:10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maezawa I., Swanberg S., Harvey D., LaSalle J.M., Jin L.W. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. doi:10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellegrino M., Provero P., Silengo L., Di Cunto F. CLOE: identification of putative functional relationships among genes by comparison of expression profiles between two species. BMC Bioinformatics. 2004;5:179. doi: 10.1186/1471-2105-5-179. doi:10.1186/1471-2105-5-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ala U., Piro R.M., Grassi E., Damasco C., Silengo L., Oti M., Provero P., Di Cunto F. Functional annotation and identification of candidate genes by computational analysis of normal tissue gene expression data. PLoS Comput. Biol. 2008;4:e1000043. doi: 10.1371/journal.pcbi.1000043. doi:10.1371/journal.pcbi.1000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas M.J., Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. doi:10.1016/S0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson F.H., Park K., Atchison M.L. Polycomb recruitment to DNA in vivo by the YY1 REPO domain. Proc. Natl Acad. Sci. USA. 2006;103:19296–19301. doi: 10.1073/pnas.0603564103. doi:10.1073/pnas.0603564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Y.L., Yang W.M., Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. doi:10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tupler R., Gabellini D. Molecular basis of fascioscapulohumeral muscular dystrophy. Cell. Mol. Life. Sci. 2004;61:557–566. doi: 10.1007/s00018-003-3285-3. doi:10.1007/s00018-003-3285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabellini D., Green M.R., Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. doi:10.1016/S0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 19.Kriaucionis S., Paterson A., Curtis J., Guy J., Macleod N., Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. doi:10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heilstedt H.A., Shahbazian M.D., Lee B. Infantile hypotonia as a presentation of Rett syndrome. Am. J. Med. Genet. 2002;111:238–242. doi: 10.1002/ajmg.10633. doi:10.1002/ajmg.10633. [DOI] [PubMed] [Google Scholar]
- 21.Belichenko P.V., Wright E.E., Belichenko N.P., Masliah E., Li H.H., Mobley W.C., Francke U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 2009;514:240–258. doi: 10.1002/cne.22009. doi:10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- 22.Levy S.E., Chen Y.S., Graham B.H., Wallace D.C. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene. 2000;254:57–66. doi: 10.1016/s0378-1119(00)00252-3. doi:10.1016/S0378-1119(00)00252-3. [DOI] [PubMed] [Google Scholar]
- 23.Brower J.V., Lim C.H., Han C., Hankowski K.E., Hamazaki T., Terada N. Differential CpG island methylation of murine adenine nucleotide translocase genes. Biochim. Biophys. Acta. 2009;1789:198–203. doi: 10.1016/j.bbagrm.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guy J., Gan J., Selfridge J., Cobb S., Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. doi:10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rylski M., Amborska R., Zybura K., Konopacki F.A., Wilczynski G.M., Kaczmarek L. Yin Yang 1 expression in the adult rodent brain. Neurochem. Res. 2008;33:2556–2564. doi: 10.1007/s11064-008-9757-y. [DOI] [PubMed] [Google Scholar]
- 26.He Y., Dupree J., Wang J., Sandoval J., Li J., Liu H., Shi Y., Nave K.A., Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. doi:10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rijkers T., Deidda G., van Koningsbruggen S., van Geel M., Lemmers R.J., van Deutekom J.C., Figlewicz D., Hewitt J.E., Padberg G.W., Frants R.R., van der Maarel S.M. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J. Med. Genet. 2004;41:826–836. doi: 10.1136/jmg.2004.019364. doi:10.1136/jmg.2004.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yusufzai T.M., Wolffe A.P. Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res. 2000;28:4172–4179. doi: 10.1093/nar/28.21.4172. doi:10.1093/nar/28.21.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams V.H., McBryant S.J., Wade P.A., Woodcock C.L., Hansen J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 2007;282:15057–15064. doi: 10.1074/jbc.M700855200. doi:10.1074/jbc.M700855200. [DOI] [PubMed] [Google Scholar]
- 30.Nikitina T., Ghosh R.P., Horowitz-Scherer R.A., Hansen J.C., Grigoryev S.A., Woodcock C.L. MeCP2–chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 2007;282:28237–28345. doi: 10.1074/jbc.M704304200. doi:10.1074/jbc.M704304200. [DOI] [PubMed] [Google Scholar]
- 31.Sharer J.D. The adenine nucleotide translocase type 1 (ANT1): a new factor in mitochondrial disease. IUBMB Life. 2005;57:607–614. doi: 10.1080/15216540500217735. doi:10.1080/15216540500217735. [DOI] [PubMed] [Google Scholar]
- 32.Kaukonen J., Zeviani M., Comi G.P., Piscaglia M.G., Peltonen L., Suomalainen A. A third locus predisposing to multiple deletions of mtDNA in autosomal dominant progressive external ophthalmoplegia. Am. J. Hum. Genet. 1999;65:226–234. doi: 10.1086/302445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaukonen J., Juselius J.K., Tiranti V., Kyttala A., Zeviani M., Comi G.P., Keranen S., Peltonen L., Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. doi:10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 34.Graham B.H., Waymire K.G., Cottrell B., Trounce I.A., MacGregor G.R., Wallace D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. doi:10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 35.Bauer M.K.A., Schubert A., Rocks O., Grimm S. Adenine nucleotide translocase-1, a component of the permeability pore, can dominantly induce apoptosis. J. Cell. Biol. 1999;147:1493–1501. doi: 10.1083/jcb.147.7.1493. doi:10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tawil R., Van Der Maarel S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34:1–15. doi: 10.1002/mus.20522. doi:10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 37.Lyle R., Wright T.J., Clark I.N., Hewitt J.E. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arm of the acrocentric chromosomes. Genomics. 1995;28:389–397. doi: 10.1006/geno.1995.1166. doi:10.1006/geno.1995.1166. [DOI] [PubMed] [Google Scholar]
- 38.Guy J., Hendrich B., Holmes M., Martin J.E., Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001;27:322–326. doi: 10.1038/85899. doi:10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 39.Mari F., Azimonti S., Bertani I., Bolognese F., Colombo E., Caselli R., Scala E., Longo I., Grosso S., Pescucci C., et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. doi:10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- 40.Bertani I., Rusconi L., Bolognese F., Forlani G., Conca B., De Monte L., Badaracco G., Landsberger N., Kilstrup-Nielsen C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006;281:32048–32056. doi: 10.1074/jbc.M606325200. doi:10.1074/jbc.M606325200. [DOI] [PubMed] [Google Scholar]
- 41.Li L.C., Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. doi:10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.