

Abstract
The grim prognosis of lung cancer, that has an overall 10–15% survival at 5 years, remains in the US the leading cause of cancer mortality, providing a compelling rationale for studying the molecular basis of this malignancy. Surmising the common, general association with smoking, lung cancers differ at the microscopic, anatomical, epidemiological and clinical level and harbor complex genetic and epigenetic alterations. Currently, lung cancer is divided into small cell lung carcinoma (SCLC) and non small cell lung carcinoma (NSCLC) for the purpose of clinical management. NSCLC constitutes 80–85% of lung cancers and is further divided into histological subtypes such as adenocarcinoma, squamous cell carcinoma, and large cell carcinoma, etc.
The ultimate goal for lung cancer research is to develop a strategy to block the tumor progression and improve the prognosis of lung cancer. This goal can realistically be achieved only when the biological complexity of this disease is taken into account. To this end, identification and understanding of molecular markers that are mechanistically involved in tumor progression are needed. Our recent studies suggest histological subtype-dependent distinct correlations between the expression and/or subcellular localization of tumor suppressive maspin with the progression and prognosis of NSCLC. Maspin is an epithelial specific member of the serine protease inhibitor (serpin) superfamily but recently identified as an endogenous inhibitor of histone deacetylase 1 (HDAC1). This novel biochemical activity coincides with a consensus emerged recently from the evidence that nuclear maspin confers better differentiated epithelial phenotypes, decreased tumor angiogenesis, increased tumor sensitivity to drug-induced apoptosis, and a more favorable prognosis. In the current review, we discuss the evidence that maspin may be a marker that stratifies the progression and prognosis of different subtypes of NSCLC.
Keywords: lung cancer, molecular marker, prognosis, histological subtypes, histone deacetylase 1, natural HDAC inhibitor, subcellular localization, differential expression, drug discovery
THE COMPLEXITY OF LUNG CANCER
Lung cancer is a leading cause of cancer morbidity and mortality worldwide, with an alarming increase in developing countries, largely as a result of tobacco smoking. In the US, death from lung cancer accounts for approximately 25% of all cancer deaths and exceeds the second, third, fourth and fifth most common causes of cancer death combined. Lung cancer can be generally divided into two types: small cell lung cancer (SCLC) and non small cell lung cancer (NSCLC). SCLC, of neuroendocrine origin, is highly aggressive and has a high potential for metastases. NSCLC constitutes 80–85% of lung cancers and can be further divided into several histological subtypes that differ clinically, epidemiologically and at the molecular level. Three major NSCLC subtypes are adenocarcinoma, squamous cell carcinoma and large cell carcinoma. Adenocarcinoma is the most frequently diagnosed NSCLC, representing 35–40% of all lung cancers and originates more commonly from epithelial cells at the terminal bronchioles/alveoli in the periphery of the lung. Adenocarcinoma is thought to progress through the morphological sequence atypical adenomatous hyperplasia→bronchiolo-alveolar carcinoma growth pattern→invasive carcinoma. Squamous cell carcinoma accounts for 25–30% of all lung cancers. The classic manifestation is a cavitary lesion in a proximal bronchus, and is characterized histologically by keratinization and/or intercellular bridges. Squamous cell carcinoma is thought to progress through the steps of normal bronchial mucosa→squamous metaplasia→low grade dysplasia→high grade dysplasia→invasive carcinoma. Large cell carcinoma, typically manifesting as a large peripheral mass on chest radiograph, accounts for 10–15% of lung cancers. Histologically, large cell carcinoma has sheets of highly atypical cells with focal necrosis, with no evidence of keratinization or gland formation.
The adoption of improved imaging techniques has made possible the detection of smaller and earlier stage lung cancers. However, since 1970, the 5-year survival rate has only increased from 7% to 15%. Currently, the strategy of lung cancer treatment is based on tumor stage as well as on the tumor type as either SCLC or NSCLC. SCLC tends to develop distant metastases early in the course of the disease, and are highly chemosensitive, therefore surgical resection is rarely considered in the management of the disease. First-line chemotherapy is the combination of platinum and etoposide. Although most patients are initially responsive, more than 90% of them develop resistance and eventually succumb to the disease. Treatment of NSCLC is based on its resectability. If NSCLC can be surgically removed, the cancer may be cured by surgery alone or by surgery followed by chemotherapy. If the tumor cannot be surgically removed, combination chemotherapy and radiation can be curative. For patients with metastatic NSCLC at presentation, chemotherapy remains the only therapeutic option and almost all the patients eventually succumb to their disease. It has been noted that the efficacy of pemetrexed (a folate antimetabolite) differs in patients with adenocarcinoma and squamous cell carcinoma [1]. In addition, high response rates have been observed with EGFR tyrosine kinase inhibitors (EGFR-TKIs) but this is limited to a histologically distinct subgroup of adenocarcinoma that have EGFR mutations [2]. Genetic studies have revealed multiple histological subtype-specific changes. For example, p16 inactivation is frequent and Rb mutations are infrequent in NSCLC, whereas the opposite is true in SCLC. In addition, both Rb and p53 are most commonly affected in SCLC. HER2 overexpression/mutation, EGFR mutations, and KRAS mutations occur almost only in adenocarcinomas, where they tend to be mutually exclusive [3, 4]. These data demonstrate the necessity and possibility of personalized therapeutic strategies, taking into account the complex etiology, histology and genetics/epigenetics of different subtypes of this disease.
To develop personalized therapeutic strategies of NSCLC, we need to better understand how lung cancer progression and prognosis can be stratified in addition to by histological subtypes. Specific molecular markers, in combination with other diagnostic tests (bronchoscopy, chest radiography, tissue biopsy, cytological tests of sputum, MRI, CT, and ultrasound), may be the key. Lung cancer cells may express carcinoembryonic antigen (CEA), NSE (neuron specific enolase), SCC (squamous cell carcinoma antigen-1 and -2), CYFRA21-1 (fragment of cytokeratin 19), TPA (tissue polypeptide antigen), or CA125 [5–9]. It is noted that many of these markers do not have a uniform marker value across different NSCLC subtypes. For example, CEA is significantly higher in adenocarcinoma than in large cell carcinoma, whereas NSE is low in adenocarcinoma but high in large cell carcinoma. Furthermore, these markers are not known to be functionally critical for lung cancer progression, and are not lung cancer-specific, thus have not been proven useful for either stratifying the progression or selecting treatment. Here we review the evidence that supports the clinical utility of maspin [10] as a marker for stratifying the prognosis of different subtypes of lung cancer and, possibly, predicting the effectiveness of lung cancer therapy.
THE EXPRESSION LEVEL AND/OR SUBCELLULAR LOCALIZATION OF MASPIN AS A MARKER FOR STRATIFYING THE PROGNOSIS OF DIFFERENT SUBTYPES OF LUNG CANCER
Although maspin expression has been detected in several types of cells, it is expressed predominantly by epithelial cells [11]. Since the discovery of the maspin gene [12], hundreds of published studies with human specimens indicate that maspin expression predicts a better prognosis for several types of carcinomas including breast, prostate, colon, and oral squamous cell carcinoma (for review, see [13]). The differential expression of maspin in tumor progression seems to be multifaceted and further dependent on the specific tumor types or subtypes.
Adenocarcinoma, in different tissues, seems to share a common differential maspin expression pattern in tumor progression [14, 15]. For example, in normal breast and prostate epithelial tissues, maspin protein was localized in the nucleus [12, 16]. In preneo-plastic or early stage of carcinogenesis, in the prostate, maspin expression is transiently up-regulated and the maspin protein is detected both in the nucleus and in the cytoplasm. As tumor further progresses to less differentiated states, maspin expression level is down-regulated or lost [16]. In lung cancer, nuclear localization, opposed to a combined nuclear and cytoplasmic localization of maspin, segregates with increased overall survival in early stage lung adenocarcinoma.
Since squamous cell carcinoma also originates from normal lung epithelial cells, i.e., bronchial ciliated epithelial cells, it remains a possibility that the switch from an exclusive nuclear maspin localization pattern to a combined nuclear and cytosolic pattern occurs prior to the step of carcinoma in situ. To this end, four other studies published on the differential expression of maspin in head and neck squamous cell carcinoma (oral squamous cell carcinoma and lyaryngeal squamous cell carcinoma), revealed a correlation of nuclear maspin with better differentiated phenotypes [17]. Interestingly, our earlier study of oral squamous cell carcinoma indicated a positive correlation of the maspin protein level with better cancer prognosis [18]. These data suggest that loss of maspin expression may still occur at a late stage of lung squamous cell carcinoma progression. To this end, Takanami et al recently showed that high levels of maspin expression predict favorable prognosis of lung squamous cell carcinoma [19]. It is important to understand the biological significance of the differential expression and/or subcellular localization of maspin as lung squamous cell carcinoma undergoes the full continuum of progression from normal epithelia to metastatic lesion.
In addition to the evidence for distinct patterns of maspin differential expression in histological subtypes of NSCLC, a critical consideration in favor of maspin as a marker for potential personalized lung cancer management is that maspin may directly regulate tumor progression. Earlier, we have shown that maspin curtails aggressive tumor phenotypes, inhibiting invasion and motility in vitro and inhibiting tumor growth and metastasis in experimental animal models [13, 20, 21]. Maspin was sufficient to induce prostate carcinoma cell redifferentiation in vivo [22]. The better differentiated phenotype of maspin-expressing tumor cells is also associated with increased sensitivity to apoptosis in vitro [23–25]. In preclinical biological studies, we showed that maspin re-expressed by both breast and prostate cancer cells sensitized cellular response to a broad spectrum of apoptosis stimuli [25] including TRAIL, TNF-α, endoplasmic reticulum stress inducer, and staurosporine (STS). It is important to note that the pro-apoptotic effect of maspin was tumor-specific. In addition, intracellular maspin, but not extracellular maspin, seems responsible for increased tumor cell sensitivity to drug-induced apoptosis [23]. A study with laryngeal carcinoma tissue specimens, nuclear maspin expression correlates with M30, a marker for epithelial cells at an early apoptotic stage [26].
Depending on the state of epithelial differentiation, maspin protein isolated from biological sources may be a 42 kDa monomer which is present as a secreted, a cytoplasmic, a nuclear, as well as a cell surface-associated protein [20]. While extracellular maspin seems to play a critical role in blocking the initiation step of the plasminogen activation cascade [22, 27, 28], intracellular maspin is specifically associated with a proapoptotic effect in tumor cells [ref]. Following the observation that nuclear maspin, but not the nuclear combined with cytoplasmic maspin, correlated with better survival of lung adenocarcinoma, we examined the correlation of maspin subcellular localization with tumor proliferation index [14]. Strictly nuclear maspin localization correlated with significantly lower proliferation index Ki67 in lung adenocarcinoma. Furthermore, maspin nuclear localization inversely correlated with VEGF-A. As compared to other factors implicated in tumor prognosis such as histological grade, p53, Ki67, VEGF-A, and tumor size, nuclear maspin was the most significant indicator for better survival. The inverse correlation of maspin with VEGF-A in lung adenocarcinoma is in line with our earlier evidence that maspin blocks the oxidative-stress induced VEGF-A production in tumor cells [29], and blocks tumor angiogenesis in xenograft tumor models [22, 30].
BEAS-2B is a non-transformed immortalized lung epithelial cells line that can be transformed by cigarette smoke condensate (CSC) [31–33]. BEAS-2B cells express maspin predominantly in the nucleus. To further clarify whether the translocation of maspin from the nucleus to the cytoplasm represents snap shots of tumor heterogeneity or a carcinogenesis-associated biological process, we examined the expression and subcellular distribution of maspin in BEAS-2B [15] and its CSC-transformed derivative. We have shown that CSC-transformed BEAS-2B clonal cell lines were significantly more invasive [15]. These CSC-transformed cells also expressed less maspin as compared to the parental BEAS-2B cells. Upon subcellular fractionation, nuclear maspin was found to be disproportionally further decreased in the CSC-transformed cells. This observation was consistent with the evidence from the resected lung adenocarcinomas studies by IHC.
A UNIQUE THERAPEUTIC OPPORTUNITY WITH MASPIN
Our recent data revealed that association of nuclear maspin with better differentiated epithelial phenotype is, at least in part, due to an inhibitory effect of maspin on histone deacetylase 1 (HDAC1) [24]. The identification of HDAC1 as a maspin-associated protein first came from a yeast two-hybrid screening [24]. This association is consistent with the nuclear localization of maspin. Indeed, the prostate tumor cell line PC3 that expresses maspin at a moderate level was used to show that maspin and HDAC1 interaction occurred in the nucleus [24]. Upon further structural analyses, the specific interaction between maspin and HDAC1 could be explained. HDAC1 catalyzes the removal of the ε-acetyl group from the side chain of a lysine in the protein substrate. The catalytic domain of HDAC1 contains a peptide recognition pocket and a catalytic triad (shown by the dashed line highlight in Fig. 1). The overall catalytic domain of HDAC1 is similar to that of a protease such as serine protease or metalloproteinase [34, 35]. The x-ray crystal structure of maspin [36, 37] suggests that maspin can use its reactive center loop (RCL) as pseudo substrate domain to interact with the catalytic domain of HDAC1. An arginine located at the center of maspin RCL could interact with catalytic triad of HDAC1, mimicking the substrate acetylated lysine and having both structural and electrostatic similarity. The molecular interaction of maspin with HDAC1 was confirmed in an array of normal and maspin-expressing tumorous prostate epithelial cell lines [24]. In paired human prostate tissues, maspin and HDAC1 levels were inversely correlated. Furthermore, more maspin-HDAC1 association was detected in the normal tissue of each pair.
Fig. 1. The Possible Mode of Molecular Interaction of Maspin and HDAC1.
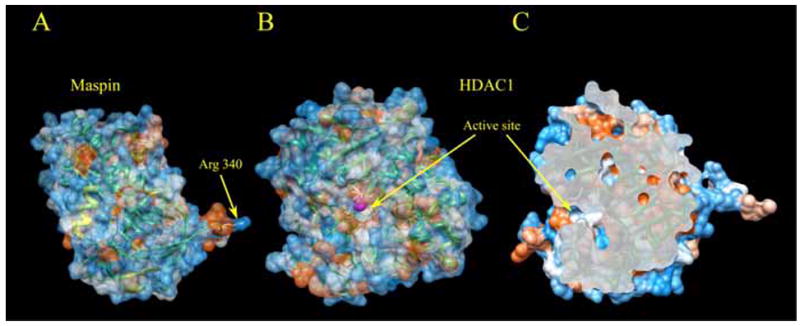
Surface representations of maspin and HDAC1, colored according to electrostatic potential. A. Crystal structure of maspin. Arg-340 is shown in stick representation. B. Model of HDAC1 structure produced basing on crystal structure of HDAC8. Catalytic triad residues are shown in stick representation and Zn2+ ion as magenta sphere. C. Slice of the molecular surface of HDAC1 active site shows the feasibility for maspin RSL to fit into the catalytic domain of HDAC1. View is rotated approximately 90° about the vertical axis of B.
We have tested a possibility that maspin may directly inhibit HDAC1. Purified recombinant protein not only binds HDAC1, but also inhibits its activity in vitro. Down-regulation of maspin led to increased activity of HDAC1 in PC3 cells [24]. Conversely, reintroduction of maspin to prostate cancer cell line DU145 that lost ability to express detectable endogenous maspin resulted in increased histone acetylation and increased expression of HDAC1 target genes Bax and p21. This data helped to explain our earlier finding that the pro-apoptotic effect of maspin was mediated, at least in part, by increased Bax expression and function [25]. Further, as predicted by the structural considerations, the interaction between maspin and HDAC1 depends on maspin RCL. Together, these findings allow one to postulate a discovery of the first endogenous inhibitor of HDAC1.
An unexpected finding that maspin inhibits the most abundant HDAC in mammalian cells, HDAC1, in fact represents a major breakthrough that helps to explain why maspin-expressing epithelial cells are less proliferative in vivo [38, 39], more sensitive to induced apoptosis [23, 25, 40] and maintain better differentiated phenotypes [20, 22, 41]. Briefly, the homeostasis of gene expression is, at least in part, controlled by the acetylation/deacetylation of chromatin. When the acetyl groups are removed by the action of housekeeping HDACs, chromatin will be packed into a closed structure, disallowing the access of transcriptional factors, thus repressing gene expression [42, 43]. It has been estimated that approximately 7% of total expressed genes in mammalian cells may be affected by HDAC1 [44]. Many of these targets (such as p21, p27, Bax, and cytokeratin 18) are involved in apoptotic response to adverse tissue microenvironment or growth inhibition. Several synthetic HDAC inhibitors show clinical activities with objective tumor regression in clinical trials [45–47]. Considering that malignant tumors acquire survival and drug resistance through multiple dysregulated networks of signaling events, the HDAC-targeted therapies have a conceivable advantage since chromosomal modulation by HDAC inhibition can lower the overall threshold of apoptotic sensitivity. In contrast, other therapeutic agents such as gefitinib (which inhibits the tyrosine kinase activity of EGFR [48, 49] are designed to target one specific molecule or pathway. Maspin is the only endogenous HDAC inhibitor identified thus far. Either maspin or HDAC1 knockout in mouse leads to embryonic lethality [38, 50], suggesting high biological significance of a fine balance between HDAC1 and maspin. It is of particular interest to note that maspin expression in tumor cell lines can be up-regulated by synthetic HDAC inhibitors [24]. It is likely that this positive feedback between the maspin effect on HDAC1 and maspin expression be a self-propelling mechanism to sustain effective HDAC1 inhibition.
Since maspin and synthetic HDAC inhibitors share similar biochemical and biological activities, it is intriguing to speculate that drug-resistant cancer cells that express low levels of maspin may become sensitized by synthetic HDAC inhibitors to the therapeutic agents. It is also possible that maspin combined with a synthetic HDAC1 inhibitor that has a broad spectrum of specificity may exert synergistic anti-tumor activity. In general, pharmacological HDAC inhibitors have a standard, modular construction with structural similarities to the HDAC acetyl-lysine substrate (Fig. 2) (also see reviews [51, 52]). The natural HDAC inhibitor, in the example of maspin, however, may have multiple contacts with HDAC at and near its catalytic site, as suggested by Fig. 1. It is known that under physiological and pathological conditions, HDAC-mediated gene transcription repression is dependent on the relative level of different HDAC isoforms and make-up of each HDAC protein complexes [53–55]. Furthermore, different HDAC isoforms in different classes may have distinct substrate specificities. While further structural-functional analysis is needed to address whether maspin only inhibits HDAC1 in specific molecular contexts, drug development to target the biochemcial and biological activities of specific HDAC isoforms may benefit from a better understanding of the mode of molecular interaction between HDAC1 and its natural inhibitor maspin.
Fig. 2.

The Substrate Acetylated Lysine as A Template for Synthetic HDAC inhibitors.
CONCLUDING REMARKS
Our data, albeit not all obtained with lung cancer specimens or lung cancer models, consistently support a role of maspin as a novel epithelial specific endogenous HDAC1 inhibitor that may both mark the better differentiated lung cancer phenotype and confer tumor cell apoptotic sensitivity. A unique and exciting direction for further research emerged from these studies is how maspin may be used to stratify different subtypes of NSCLC to predict the prognosis. On the other hand, tumor cells that no longer express maspin may become sensitive to drug-induced apoptosis if maspin expression can be restored. Since small molecular weight HDAC inhibitors and maspin shared the pro-apoptotic property, one may speculate further that pharmacological HDAC inhibitors may sensitize tumor cells that have lost maspin expression to drug-induced apoptosis.
Acknowledgments
The review was in part based on the work supported by NIH grants (CA127735 and CA084176, both to S. Sheng)
References
- 1.Scagliotti GV, De Marinis F, Rinaldi M, Crino L, Gridelli C, Ricci S, et al. The role of histology with common first-line regimens for advanced non-small cell lung cancer: a brief report of the retrospective analysis of a three-arm randomized trial. J Thorac Oncol. 2009;4:1568–71. doi: 10.1097/JTO.0b013e3181c06980. [DOI] [PubMed] [Google Scholar]
- 2.Costa DB, Kobayashi S, Tenen DG, Huberman MS. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR-mutant non-small cell lung cancers. Lung Cancer. 2007;58:95–103. doi: 10.1016/j.lungcan.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panani AD, Roussos C. Cytogenetic and molecular aspects of lung cancer. Cancer Lett. 2006;239:1–9. doi: 10.1016/j.canlet.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 4.Sy SM, Wong N, Lee TW, Tse G, Mok TS, Fan B, et al. Distinct patterns of genetic alterations in adenocarcinoma and squamous cell carcinoma of the lung. Eur J Cancer. 2004;40:1082–94. doi: 10.1016/j.ejca.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Blankenburg F, Hatz R, Nagel D, Ankerst D, Reinmiedl J, Gruber C, et al. Preoperative CYFRA 21–1 and CEA as prognostic factors in patients with stage I non-small cell lung cancer: external validation of a prognostic score. Tumour Biol. 2008;29:272–7. doi: 10.1159/000152945. [DOI] [PubMed] [Google Scholar]
- 6.Au NH, Cheang M, Huntsman DG, Yorida E, Coldman A, Elliott WM, et al. Evaluation of immunohistochemical markers in non-small cell lung cancer by unsupervised hierarchical clustering analysis: a tissue microarray study of 284 cases and 18 markers. J Pathol. 2004;204:101–9. doi: 10.1002/path.1612. [DOI] [PubMed] [Google Scholar]
- 7.Nisman B, Amir G, Lafair J, Heching N, Lyass O, Peretz T, et al. Prognostic value of CYFRA 21–1, TPS and CEA in different histologic types of non-small cell lung cancer. Anticancer Res. 1999;19:3549–52. [PubMed] [Google Scholar]
- 8.Ishida T, Kaneko S, Tateishi M, Oka T, Mitsudomi T, Sugimachi K, et al. Large cell carcinoma of the lung. Prognostic implications of histopathologic and immunohistochemical subtyping. Am J Clin Pathol. 1990;93:176–82. doi: 10.1093/ajcp/93.2.176. [DOI] [PubMed] [Google Scholar]
- 9.Brockmann M, Brockmann I, Herberg U, Muller KM. Adenocarcinoma of the lung. Immunohistochemical findings (keratin/CEA) J Cancer Res Clin Oncol. 1987;113:379–82. doi: 10.1007/BF00397723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Futscher BW, Oshiro MM, Wozniak RJ, Holtan N, Hanigan CL, Duan H, et al. Role for DNA methylation in the control of cell type specific maspin expression. Nat Genet. 2002;31:175–9. doi: 10.1038/ng886. [DOI] [PubMed] [Google Scholar]
- 11.Khalkhali-Ellis Z. Maspin: the new frontier. Clin Cancer Res. 2006;12:7279–83. doi: 10.1158/1078-0432.CCR-06-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–9. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 13.Sheng S. The promise and challenge toward the clinical application of maspin in cancer. Front Biosci. 2004;9:2733–45. doi: 10.2741/1432. [DOI] [PubMed] [Google Scholar]
- 14.Frey A, Soubani AO, Adam AK, Sheng S, Pass HI, Lonardo F. Nuclear, compared with combined nuclear and cytoplasmic expression of maspin, is linked in lung adenocarcinoma to reduced VEGF-A levels and in Stage I, improved survival. Histopathology. 2009;54:590–7. doi: 10.1111/j.1365-2559.2009.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lonardo F, Li X, Siddiq F, Singh R, Al-Abbadi M, Pass HI, et al. Maspin nuclear localization is linked to favorable morphological features in pulmonary adenocarcinoma. Lung Cancer. 2006;51:31–9. doi: 10.1016/j.lungcan.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Pierson CR, McGowen R, Grignon D, Sakr W, Dey J, Sheng S. Maspin is up-regulated in premalignant prostate epithelia. Prostate. 2002;53:255–62. doi: 10.1002/pros.10107. [DOI] [PubMed] [Google Scholar]
- 17.Marioni G, Staffieri C, Staffieri A, De Filippis C, Blandamura S. MASPIN tumour-suppressing activity in head and neck squamous cell carcinoma: emerging evidence and therapeutic perspectives. Acta Otolaryngol. 2009;129:476–80. doi: 10.1080/00016480802256079. [DOI] [PubMed] [Google Scholar]
- 18.Xia W, Lau YK, Hu MC, Li L, Johnston DA, Sheng S, et al. High tumoral maspin expression is associated with improved survival of patients with oral squamous cell carcinoma. Oncogene. 2000;19:2398–403. doi: 10.1038/sj.onc.1203535. [DOI] [PubMed] [Google Scholar]
- 19.Takanami I, Abiko T, Koizumi S. Expression of maspin in non-small-cell lung cancer: correlation with clinical features. Clin Lung Cancer. 2008;9:361–6. doi: 10.3816/CLC.2008.n.052. [DOI] [PubMed] [Google Scholar]
- 20.Sheng S. A role of novel serpin maspin in tumor progression: the divergence revealed through efforts to converge. J Cell Physiol. 2006;209:631–5. doi: 10.1002/jcp.20786. [DOI] [PubMed] [Google Scholar]
- 21.Lockett J, Yin S, Li X, Meng Y, Sheng S. Tumor suppressive maspin and epithelial homeostasis. J Cell Biochem. 2006;97:651–60. doi: 10.1002/jcb.20721. [DOI] [PubMed] [Google Scholar]
- 22.Cher ML, Biliran HR, Jr, Bhagat S, Meng Y, Che M, Lockett J, et al. Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in a model of prostate cancer bone metastasis. Proc Natl Acad Sci USA. 2003;100:7847–52. doi: 10.1073/pnas.1331360100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene. 2002;21:4089–98. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Yin S, Meng Y, Sakr W, Sheng S. Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Res. 2006;66:9323–9. doi: 10.1158/0008-5472.CAN-06-1578. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Yin S, Reddy N, Spencer C, Sheng S. Bax mediates the apoptosis-sensitizing effect of maspin. Cancer Res. 2004;64:1703–11. doi: 10.1158/0008-5472.can-03-2568. [DOI] [PubMed] [Google Scholar]
- 26.Marioni G, Giacomelli L, D’Alessandro E, Marchese-Ragona R, Staffieri C, Ferraro SM, et al. Nuclear localization of mammary serine protease inhibitor (MASPIN): is its impact on the prognosis in laryngeal carcinoma due to a proapoptotic effect? Am J Otolaryngol. 2008;29:156–62. doi: 10.1016/j.amjoto.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Biliran H, Jr, Sheng S. Pleiotrophic inhibition of pericellular urokinase-type plasminogen activator system by endogenous tumor suppressive maspin. Cancer Res. 2001;61:8676–82. [PubMed] [Google Scholar]
- 28.McGowen R, Biliran H, Jr, Sager R, Sheng S. The surface of prostate carcinoma DU145 cells mediates the inhibition of urokinase-type plasminogen activator by maspin. Cancer Res. 2000;60:4771–8. [PubMed] [Google Scholar]
- 29.Yin S, Lockett J, Meng Y, Biliran H, Jr, Blouse GE, Li X, et al. Maspin retards cell detachment via a novel interaction with the urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor system. Cancer Res. 2006;66:4173–81. doi: 10.1158/0008-5472.CAN-05-3514. [DOI] [PubMed] [Google Scholar]
- 30.Zhang M, Volpert O, Shi YH, Bouck N. Maspin is an angiogenesis inhibitor. Nat Med. 2000;6:196–9. doi: 10.1038/72303. [DOI] [PubMed] [Google Scholar]
- 31.Lonardo F, Dragnev KH, Freemantle SJ, Ma Y, Memoli N, Sekula D, et al. Evidence for the epidermal growth factor receptor as a target for lung cancer prevention. Clin Cancer Res. 2002;8:54–60. [PubMed] [Google Scholar]
- 32.Boyle JO, Langenfeld J, Lonardo F, Sekula D, Reczek P, Rusch V, et al. Cyclin D1 proteolysis: a retinoid chemoprevention signal in normal, immortalized, and transformed human bronchial epithelial cells. J Natl Cancer Inst. 1999;91:373–9. doi: 10.1093/jnci/91.4.373. [DOI] [PubMed] [Google Scholar]
- 33.Langenfeld J, Lonardo F, Kiyokawa H, Passalaris T, Ahn MJ, Rusch V, et al. Inhibited transformation of immortalized human bronchial epithelial cells by retinoic acid is linked to cyclin E down-regulation. Oncogene. 1996;13:1983–90. [PubMed] [Google Scholar]
- 34.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, et al. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–84. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–34. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Law RH, Irving JA, Buckle AM, Ruzyla K, Buzza M, Bashtannyk-Puhalovich TA, et al. The high resolution crystal structure of the human tumor suppressor maspin reveals a novel conformational switch in the G-helix. J Biol Chem. 2005;280:22356–64. doi: 10.1074/jbc.M412043200. [DOI] [PubMed] [Google Scholar]
- 37.Al-Ayyoubi M, Gettins PG, Volz K. Crystal structure of human maspin, a serpin with antitumor properties: reactive center loop of maspin is exposed but constrained. J Biol Chem. 2004;279:55540–4. doi: 10.1074/jbc.M409957200. [DOI] [PubMed] [Google Scholar]
- 38.Gao F, Shi HY, Daughty C, Cella N, Zhang M. Maspin plays an essential role in early embryonic development. Development. 2004;131:1479–89. doi: 10.1242/dev.01048. [DOI] [PubMed] [Google Scholar]
- 39.Zhang M, Shi Y, Magit D, Furth PA, Sager R. Reduced mammary tumor progression in WAP-TAg/WAP-maspin bitransgenic mice. Oncogene. 2000;19:6053–8. doi: 10.1038/sj.onc.1204006. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Chen D, Yin S, Meng Y, Yang H, Landis-Piwowar KR, et al. Maspin augments proteasome inhibitor-induced apoptosis in prostate cancer cells. J Cell Physiol. 2007;212:298–306. doi: 10.1002/jcp.21102. [DOI] [PubMed] [Google Scholar]
- 41.Seftor RE, Seftor EA, Sheng S, Pemberton PA, Sager R, Hendrix MJ. maspin suppresses the invasive phenotype of human breast carcinoma. Cancer Res. 1998;58:5681–5. [PubMed] [Google Scholar]
- 42.Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53:275–89. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- 43.Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet. 2001;10:693–8. doi: 10.1093/hmg/10.7.693. [DOI] [PubMed] [Google Scholar]
- 44.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006;26:7913–28. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vansteenkiste J, Canon JL, Riska H, Pirker R, Peterson P, John W, et al. Randomized phase II evaluation of aprinocarsen in combination with gemcitabine and cisplatin for patients with advanced/metastatic non-small cell lung cancer. Invest New Drugs. 2005;23:263–9. doi: 10.1007/s10637-005-6736-x. [DOI] [PubMed] [Google Scholar]
- 46.Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009;2:31. doi: 10.1186/1756-8722-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiedmann MW, Caca K. Molecularly targeted therapy for gastrointestinal cancer. Curr Cancer Drug Targets. 2005;5:171–93. doi: 10.2174/1568009053765771. [DOI] [PubMed] [Google Scholar]
- 48.Rubin BP, Duensing A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest. 2006;86:981–6. doi: 10.1038/labinvest.3700466. [DOI] [PubMed] [Google Scholar]
- 49.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–85. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marson CM. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med Chem. 2009;9:661–92. doi: 10.2174/187152009788679976. [DOI] [PubMed] [Google Scholar]
- 53.Mahlknecht U, Hoelzer D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med. 2000;6:623–44. [PMC free article] [PubMed] [Google Scholar]
- 54.Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3:300–9. doi: 10.4161/epi.3.6.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]