

Abstract
MLL (Mixed Lineage Leukemia) is the target of chromosomal translocations which cause leukemias with poor prognosis. All leukemogenic MLL fusion proteins retain the CXXC domain which binds to nonmethylated CpG DNA. We present the solution structure of the MLL CXXC domain in complex with DNA, showing for the first time how the CXXC domain distinguishes nonmethylated from methylated CpG DNA. Based on the structure, we designed point mutations which disrupt DNA binding. Introduction of these mutations into MLL-AF9 results in increased DNA methylation of specific CpG nucleotides in Hoxa9, increased H3K9 methylation, decreased expression of Hoxa9 locus transcripts, loss of immortalization potential, and inability to induce leukemia in mice. These results establish that DNA binding by the CXXC domain and protection against DNA methylation is essential for MLL fusion leukemia. They also provide support for this interaction as a potential target for therapeutic intervention.
The Mixed Lineage Leukemia (MLL) gene encodes a protein with homology to Drosophila trithorax (TRX) 1. MLL and TRX belong to an evolutionarily conserved family of proteins that positively regulate gene expression during development 2,3. These proteins counteract the effect of Polycomb family proteins which maintain the inactive state of target genes 4,5. ChIP-on-chip experiments indicate that MLL is associated with over 5000 human promoters suggesting that MLL may have a global role in transcription 6. The best studied downstream targets of MLL are homeobox (Hox) genes 3,7. MLL is required for the maintenance of spatial patterns of Hox gene expression during development and hematopoiesis 3,8,9.
MLL is a common target of chromosomal translocations found in human leukemias affecting both children 10 and adults 11. MLL leukemia accounts for up to 10% of AML and ALL in general. Translocations of MLL fuse an N-terminal fragment of MLL to one of more than 60 different fusion partners 9. Regardless of the fusion partner, the presence of MLL translocations is associated with early relapse and poor prognosis 12. In all MLL translocations, ~1400 amino acids from the N-terminus of MLL are fused in frame with the C-terminus of the fusion partner 13. Disruption of MLL by gene fusions upregulates expression of a subset of Hox genes leading to a block in hematopoietic differentiation 9,14. Despite the heterogeneity of fusion partners, the portion of MLL retained is very similar and includes two regions which have been shown to be indispensable for leukemogenic transformation: the N-terminal region which binds to menin and LEDGF (lens epithelium derived growth factor) 15,16 and the conserved CXXC domain which mediates binding to nonmethylated CpG DNA motifs and the co-repressor proteins HDAC1, Bmi-1 and CtBP 17–19.
MLL-related leukemias are associated with upregulation of Hox genes including Hoxa9 and Meis1 14. We have shown recently that MLL and MLL-AF4 bind to Hoxa9, protect a specific cluster of CpGs within a CpG island from methylation, and thereby maintain expression of Hoxa9 locus transcripts 20. The pattern of methylation of CpG islands differs between cell types and an abnormal methylation pattern is frequently associated with various diseases including multiple types of cancer 21. Methylated CpG dinucleotides are recognized by the well characterized methyl binding domain (MBD) proteins 22. To date, the only characterized domain capable of selective binding to unmethylated CpG dinucleotides is the CXXC domain. The CXXC domains from several proteins including MLL 17,18, MBD1 23 and CGBP 24 have been shown to bind DNA and recognize unmethylated CpG dinucleotides. Functionally, both CXXC and MBD domains in concert play a key role in decoding the methylation status of CpG islands and interpreting cytosine methylation, ultimately leading to gene transcription or silencing.
In order to gain further insight into the mechanism of methylation protection by MLL and to assess the importance of this function for MLL fusion leukemia, we solved the structure of the human CXXC domain in complex with DNA using solution NMR spectroscopy. Based on the structure, we identified point mutations in the CXXC domain which abrogate DNA binding to various extents without perturbing the structure. We introduced these mutations into MLL-AF9 and found that loss of DNA binding by the CXXC domain is correlated with increased Hoxa9 locus methylation. Importantly, introduction of these mutations into MLL-AF9 results in failure to immortalize primary bone marrow progenitor cells and failure to induce leukemia in mice. Our data provide new insights into the mechanism of transcriptional maintenance by MLL and MLL fusion proteins and provide support for the possible therapeutic utility of targeting the MLL CXXC-DNA interaction.
RESULTS
DNA binding specificity of the MLL CXXC domain
Recently, we have shown that the CXXC domain of MLL binds DNA oligonucleotides derived from a CpG island in the Hoxa9 locus 20. To determine whether the CXXC domain specifically recognizes DNA bases outside the core CpG motif, we tested binding to several designed oligonucleotides using isothermal titration calorimetry (ITC). This data (Supplementary Table 1) as well as our recently published results 20 suggest that the CXXC domain does not recognize DNA bases beyond the CpG dinucleotide and therefore is not solely responsible for specific localization to the Hoxa9 locus. This result is similar to that from a selected and amplified binding (SAAB) analysis of the binding specificity of the CGBP CXXC domain which was also selective for a CpG motif but showed some selectivity for the flanking bases 24. Interestingly, we observe 2–4 fold higher affinity for sites with multiple CpG motifs (Supplementary Table 1), possibly indicative of 1-dimensional diffusion along the DNA.
Structure of the CXXC domain in complex with CpG DNA
We determined the structure of the human MLL CXXC domain in complex with DNA using NMR spectroscopy. We designed a palindromic dodecameric oligonucleotide (DNApal) with a central CpG dinucleotide and prepared an equimolar complex with 13C,15N-labeled CXXC domain. To determine the structure of the complex, we measured an extensive set of experimental restraints including intra- and inter-molecular distances and residual dipolar couplings (RDCs) (see Table 1).
Table I.
NMR structural statistics for CXXC-DNA complex.
| CXXC-DNApal | |
|---|---|
| number of NOE distances | 912 |
| intraresidual | 429 |
| sequential (|i−j|=1) | 225 |
| medium-range (1 <|i−j|<5) | 115 |
| long range (|i−j|>4) | 143 |
| number of intermolecular NOE distances | 79 |
| number of hydrogen bond restraints a | 4 |
| number of dihedral angle restraints (Ψ+ φ)b | 10; 10 |
| number of 3JHNHα couplings | 40 |
| total number of RDCsc | 203 |
| 1DHN, 1DNC′, 2DHNC′, 1DC′Cα,1DCαHα | 44, 43, 38, 44, 34 |
| r.m.s. deviations from distance restraints (Å) | 0.01±0.001 |
| r.m.s. deviations for dihedral angles (deg) | 0.29±0.02 |
| r.m.s. deviations for J-couplings (Hz) | 0.77±0.13 |
| r.m.s. deviations for RDC (Hz) | |
| 1DHN | 1.05±0.05 |
| 1DNC′ | 0.27±0.01 |
| 2DHNC′ | 0.75±0.07 |
| 1DC′Cα | 0.59±0.04 |
| 1DHα′Cα | 4.8±0.16 |
| r.m.s. deviations for covalent geometry | |
| bond lengths (Å) | 0.0025±0.00006 |
| bond angles (deg) | 0.433±0.008 |
| impropers (deg) | 0.30±0.017 |
| Ramachandran plot statistics (%) d | |
| Residues in most favored regions | 90.5 e |
| Residues in additional allowed regions | 9.5 e |
| Residues in disallowed regions | 0 e |
| r.m.s.d. of the NMR ensemble (Å) | |
| backbone | 0.59±0.13 f |
| heavy atoms | 1.25±0.14 f |
two distance restraints for each hydrogen bond;
backbone dihedral angles from TALOS analysis 41;
RDCs measured in 50-S gel;
Ramachandran plot statistics according to PROCHECK analysis 42;
analysis for residues 1152-1198;
analysis for residues 1152-1198 of CXXC domain and 104-119,116-121 of DNA.
Examination of the DNA spectra indicated the presence of two sets of resonances for the central six base pair region of the palindromic oligonucleotide, consistent with the loss of symmetry of the DNA upon protein binding. We also observed exchange cross peaks between the two complementary DNA strands arising from the reversal of the orientation of the bound CXXC domain. A similar phenomenon has been observed for the binding of methylated CpG DNA to MBD1 22,25. Although a virtually complete assignment of DNA resonances was obtained, broadening caused by the exchange process made the quantitative analysis of NOESY spectra and de novo calculation of the DNA structure intractable. Importantly, the pattern of the NOEs observed for the DNA is very similar between free DNA and in the complex (such as sequential NOEs involving base H6/H8 protons and deoxyribose H1′ protons). We also observe relatively modest chemical shift changes for DNA upon protein binding (see Supplementary Table 2). The most significant chemical shift changes (more than 0.2 ppm) are observed only for protons that are in direct contact with the CXXC domain. These findings led us to conclude that the DNA conformation is not perturbed by the CXXC domain and we have used DNA in canonical B-form for structure calculations.
The structure of the MLL CXXC domain in the complex with DNA is very similar to that of the free protein (data not shown) 26. The CXXC domain binds DNA by inserting in a wedge-like manner into the major groove with the protein perpendicular to the DNA axis (Figure 1a). The CXXC domain contacts a short six base pair long sequence of the DNA centered on the CpG motif. Importantly, all of the base specific contacts are limited to the central CpG dinucleotide and are therefore consistent with the binding data described above. The surface area buried in the protein-DNA interface is relatively small (~1060 Å2) compared to the 1340 Å2 average contact area observed for protein-DNA complexes27, consistent with the moderate affinity of this interaction. The elongated shape of the CXXC domain results in residues distributed throughout the entire domain making contacts with DNA. These contacts are abundant in the major groove and reach the phosphate backbone in the minor groove (Figure 1). The majority of the intermolecular interactions are mediated by the 1182-1188 loop which forms the recognition element for the CpG motif. Several residues outside of the 1182-1188 loop participate in electrostatic and hydrophobic interactions with the sugar-phosphate backbone, contributing to binding.
Figure 1.

Structure of the CXXC domain – DNA complex and details of CpG recognition. a) representative conformer of the complex is shown in green (CpG binding loop 1182-1188 in magenta) and central 6 base pair region of DNA shown in blue and cyan; zinc atoms are shown as black spheres. b) Structure of the complex representing the view perpendicular to DNA axis; N- and C-terminal residues are labeled. Sidechains of Arg1150, Ser1152 and Leu1197 are shown in red. Cysteine resides coordinating zinc atoms are yellow. c) Schematic of protein-DNA contacts. Hydrogen bond, electrostatic, and van der Waals interactions of the protein backbone and sidechains with DNA are shown as red and blue arrows, respectively. The interaction of Arg1150 with minor groove is shown with green arrow. Ovals represent protein residues involved in base-specific (cyan) and electrostatic/van der Waals (green) contacts with DNA. d) Hydrogen bonds involving the carbonyls of Lys1185 and Lys1186 and N4-amine groups of Cyt106 and Cyt118. e) Hydrogen bonds formed between sidechains of Gln1187, Lys1186 and Gua107, Gua119, respectively. f) Close contacts between the protein backbone and cytosines in the CpG motif. The positions of H5 protons that are substituted by CH3 groups in methylated DNA are shown as gray spheres with the van der Waals radii of a methyl group. Backbone H and O atoms in intimate contact with the DNA are shown as magenta spheres with appropriate van der Waals radii.
The identity of the residues involved in contacts with the DNA major groove agrees well with a previous study using saturation transfer mapping to identify residues at the protein-DNA interface 26. However, we have additionally found interactions involving the protein (Arg1150, Ser1152, Leu1197) and the DNA minor groove (see Figure 1b) not identified in the previous study. This is corroborated by mutations of Arg1150 and Leu1197 to Ala which decrease binding 5-fold and 4-fold, respectively (Figure 2). We were unable to express the Ser1152Ala mutant in a soluble form, suggesting it may contribute to protein stability or folding. We also mutated Met1200 to Ala which gives rise to two weak NOEs to DNA. Interestingly, a moderate increase in DNA binding affinity was observed which may indicate relief of a steric clash of Met1200 with DNA.
Figure 2.

Design of mutations impairing interaction of the CXXC domain with DNA. a) Surface representation of the CXXC domain - DNA complex with indicated mutation sites. Residues involved in electrostatic interactions with DNA (Lys1185; Arg1154, Lys1193) are shown in green, Gln1187 forming hydrogen bond with guanine base in magenta, and Cys1188 located in close proximity to the DNA backbone in yellow. b) Relative dissociation constants (Kd) for binding of wildtype and mutant CXXC domains to DNApal determined using NMR titration. The colors of bars are the same as in panel a. Error bars indicate s.d. The Kd value for C1188D could not be determined due to very weak binding. The additional set of resides (shown in red) comprises mutation of N-terminal (Arg1150) and C-terminal (L1197 and M1200) residues to alanines; position of these residues on the structure of the complex (panel a) is omitted for clarity. c) Example of 15N-1H HSQC spectra showing titration of the Q1187A mutant (red) with increasing concentrations of DNApal (cyan - 1:1 protein-DNA ratio; blue - 1:4 ratio). d) Comparison of spectra of C1188D mutant without (red) and with DNApal in a 1:4 ratio (blue).
Structural basis for CpG recognition
The structure of the CXXC domain-DNA complex represents the first detailed insight into the molecular basis of recognition of CpG motifs by CXXC domains and explains the basis for discrimination between methylated and unmethylated DNA. Strikingly, recognition of the CpG motif involves a short 1182-1188 loop that mediates all base-specific contacts with DNA. Unlike the majority of protein-DNA complexes, these contacts are very limited and involve interactions with both side chains and the protein backbone. Specificity for CpG recognition is achieved by means of four intermolecular hydrogen bonds. The key element discriminating CpG versus other dinucleotides is the recognition of the two N4-amine groups (Cyt118 and Cyt106) by formation of hydrogen bonds with two protein backbone oxygens (Lys1185 and Lys1186, respectively) (Figure 1). In addition, the sidechain of Lys1186 is in position to form a hydrogen bond with Gua119 and the sidechain amide of Gln1187 forms hydrogen bonds with Gua107 (Figure 1d). Close contacts between DNA bases and the protein backbone play a major role in discrimination between methylated and unmethylated cytosines. The H5 protons of Cyt106 and Cyt118 are in very close proximity to the backbone Hα of Gln1187 (2.3 ± 0.1 Å) and carbonyl of Ile1184 (2.3 ± 0.3 Å), respectively (Figure 1). Methylation of either of these cytosines would result in a steric clash with the protein backbone, thus precluding binding of methylated DNA.
In addition to base-specific contacts, there are numerous interactions involving protein sidechains and the sugar-phosphate backbone of the DNA (Figure 1b). Electrostatic interactions with the DNA are mediated by the sidechains of Arg1150, Arg1154, Lys1176, Lys1178, Lys1185, Lys1190, Arg1192, Lys1193. In addition, Ile1184 contacts the hydrophobic part of the deoxyribose of Gua117. Interestingly, we also observe several weak NOEs involving the sidechains of Arg1150, Ser1152, Leu1197 and Met1200 with DNA H4′, H5′ and H5″ protons, indicative of an interaction of the CXXC domain with the minor groove of DNA as well.
Design of CXXC mutations to abrogate DNA binding
In order to probe the importance of protection of CpG motifs from methylation mediated by the CXXC domain, we developed mutations which specifically disrupt DNA binding without disruption of the protein fold. We selected three types of sites for mutagenesis: alanine mutations of residues involved in electrostatic interactions with the DNA (Arg1154, Lys1185, Lys1193), substitution of Gln1187 (Q1187A) involved in specific DNA contacts, and introduction of a repulsive interaction with the DNA backbone via a C1188D substitution (see Figure 2a). All of the expressed mutants were folded and had very similar HSQC spectra to the wild-type protein, indicating no structural changes. Their affinities toward the DNApal oligonucleotide were measured by NMR titration experiments (Figure 2c,d). The wild type protein yielded a Kd of 44 μM, in good agreement with the ITC results for other oligonucleotides with a single CpG element (Supplementary Table 1). The mutations interfering with electrostatic interactions decreased the binding affinity 9 to 15-fold (Figure 2b). A less pronounced effect (6 fold higher Kd) was observed for the Q1187A mutation abrogating interaction between the glutamine sidechain and the guanine base within the CpG motif. Our results are consistent with a previous qualitative assessment of DNA binding by CXXC mutants employing EMSA 26. To generate a mutant of the CXXC domain completely unable to bind DNA we selected Cys1188, a residue that is located in close proximity to the DNA backbone and thus could be used to introduce a repulsive interaction. While a C1188A mutation had no effect on DNA binding, the C1188D mutation led to the inability to detect any binding to DNA by NMR (Figure 2d).
MLL-AF9(C1188D) increases methylation of Hoxa9 CpG island
To investigate the role of the CXXC domain in methylation protection, we introduced the C1188D mutation into MLL-AF9. For comparison, we also included the inert C1188A mutation which does not affect DNA binding. After stable transfection of MLL-AF9(C1188A) or MLL-AF9(C1188D) constructs into Mll null MEF cells, we measured the methylation levels of CpG residues in the upstream region of the Hoxa9 locus. Genomic DNA samples underwent bisulfite treatment, then nested PCR was performed and PCR products were sequenced. Methylation levels were determined as we described previously 20. The Western blot in Supplementary Fig. 1 shows that all MLL-AF9 point mutation constructs are expressed.
We have analyzed the Hoxa9 locus CpG island methylation within cells expressing MLL-AF9(C1188A) and found a level of non-methylated CpG’s consistent with what we previously published regarding the effect of another MLL fusion, MLL-AF4, in a similar experiment 20. In contrast, the level of CpG methylation in cells expressing MLL-AF9(C1188D) was increased compared to MLL-AF9(C1188A) (Figure 3a). Therefore, a point mutation in the CXXC domain which abrogates DNA binding also leads to substantial loss of Hoxa9 methylation protection by MLL-AF9. Our findings provide direct evidence that the CXXC domain is required for maintenance of CpG DNA in the non-methylated state.
Figure 3.
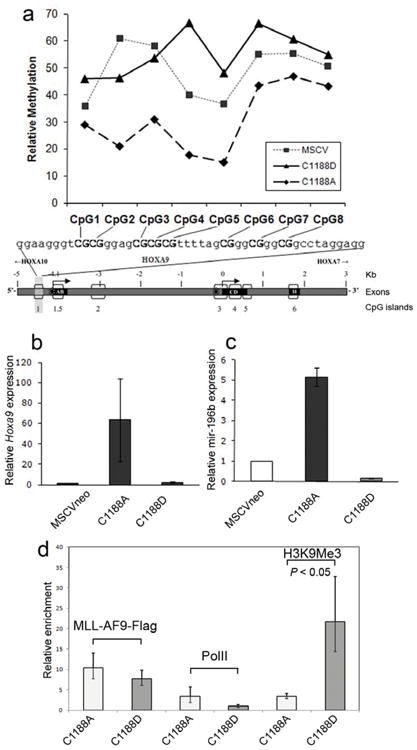
DNA binding activity of the MLL CXXC domain is required for MLL-AF9 to protect Hoxa9 from DNA methylation and induce Hoxa9 and mir-196b transcript expression, but not for binding to the locus. a) Relative methylation levels of CpGs in the upstream Hoxa9 locus in Mll null MEFs transfected with either MCSVneo, MSCVneo-MLL-AF9(C1188A), or MSCVneo-MLL-AF9(C1188D) and pSuper, after one week of puromycin selection. Bisulfite treatment, PCR and sequencing on genomic DNA samples were performed three times, and the results of one representative experiment are shown. Expression levels of Hoxa9 (b) and mir196b (c) in bone marrow progenitor cells transduced with MSCVneo vector, MLL-AF9(C1188A), or MLL-AF9(C1188D). Cells were harvested after one week culture in methylcellulose and expression levels of Hoxa9 and mir196b were quantified with real-time RT-PCR. Shown are average relative expression levels (s.d.). (d) ChIP assay performed on Phoenix cells transfected with FLAG tagged MSCV-MLL-AF9(C1188A) or MSCV-MLL-AF9(C1188D). Chromatin was immunoprecipitated with the indicated antibodies and real time PCR was performed with primers that localize near mir-196b in the upstream region of the HOXA9 locus. Samples were run in triplicate and were normalized to GAPDH and input chromatin, with error bars showing s.d.
MLL-AF9(C1188D) decreases levels of Hoxa9 and mir196b
The transforming potential of cells bearing MLL fusion proteins is correlated with high levels of Hoxa9 expression 9,14 as well as mir196b expression 28. which is within the Hoxa9 locus. We have demonstrated that cells expressing MLL-AF9(C1188D) have increased methylation of the Hoxa9 CpG island compared to MLL-AF9(C1188A). This should lead to decreased levels of expression for Hoxa9 and mir196b. In order to test this, we introduced MLL-AF9(C1188A) and MLL-AF9(C1188D) into bone marrow progenitors via retroviral transduction and measured the levels of Hoxa9 and mir196b by RT-PCR. Bone marrow cells expressing MLL-AF9(C1188A), with normal DNA binding activity, expressed significantly higher levels of Hoxa9 and mir196b compared to retroviral vector-expressing cells, as expected for MLL fusion proteins (Figure 3b,c). In striking contrast, bone marrow cells expressing MLL-AF9(C1188D) expressed Hoxa9 levels 30-fold lower and mir-196b levels 27-fold lower than MLL-AF9(C1188A) cells, levels similar to or below those expressed by cells expressing empty vector (Figure 3b,c).
MLL-AF9(C1188D) occupies Hoxa9, increases H3K9 methylation
We have examined the occupancy of MLL-AF9 mutants C1188A and C1188D at the Hoxa9 locus using chromatin immunoprecipitation (ChIP). Figure 3d shows that the occupancy of MLL-AF9 at this locus is not significantly affected by the introduction of the C1188D mutation which eliminates DNA binding by the CXXC domain. This is consistent with recent studies demonstrating a critical role for interaction with menin in localization of MLL to target genes 29. RNA Polymerase II (Pol II) occupancy at the Hoxa9 locus showed a trend toward decreased binding with the C->D mutation, although this did not reach statistical significance. In contrast, the level of trimethylated H3K9 increases significantly with introduction of the C1188D mutation. As this is a mark of repressive chromatin, this increase correlates very well with the decreased Hoxa9 expression seen by RT-PCR.
CXXC mutations abrogate transforming potential of MLL-AF9
Loss of protection of Hoxa9 CpG islands from methylation and subsequent downregulation of Hoxa9 and mir-196b expression should have a pronounced impact on the transforming properties of leukemogenic MLL-fusion proteins. To assess this, we introduced the mutations impairing DNA binding of the CXXC domain into MLL-AF9 and assessed their function in a colony formation assay 30. While normal murine bone marrow progenitor cells can form colonies when plated in semisolid medium in vitro, these cells differentiate and cannot be serially replated. In contrast, if MLL fusions are introduced into bone marrow progenitors the cells can be serially replated and become immortalized.
Bone marrow cells expressing MLL-AF9 exhibited high clonogenic activity as well as high proliferative capacity throughout four weeks of replating (Figure 4a). For example, after four platings almost 16% of the plated cells can each give rise to a colony, and the total cell numbers increased on average by 900 to 1000-fold (Figure 4a). A similar transforming potential was observed for MLL-AF9(C1188A) which does not alter the DNA binding affinity of the CXXC domain. Cells transduced with MLL-AF9(C1188A) showed similar number of colonies to MLL-AF9, and the colonies for both of these constructs were large, densely filled with cells, and had a uniform round shape, typical of progenitor or blast cells (Figure 4b). Cytospin and Wright-Giemsa staining of MLL-AF9 or MLL-AF9(C1188A) transduced cells after four platings revealed morphology expected for blasts, rather than differentiated cells (Figure 4b). In contrast, MLL-AF9(C1188D) lacks colony forming ability (the C1188D mutant cells proliferated on average only 1.7 fold) and behaved similarly to the MSCVneo negative control (Figure 4a). These colonies rarely survived into the fourth week of the colony assay and the surviving colonies were always very diffuse with small numbers of differentiated cells (Figure 4b).
Figure 4.

DNA binding activity of the MLL CXXC domain in MLL-AF9 is required for increased proliferative capacity and immortalization in a bone marrow progenitor serial replating assay. a) Average numbers of colonies for each of the four weeks after plating or re-plating in methylcellulose are shown for bone marrow progenitor cells expressing MLL-AF9 or MLL-AF9 with various CXXC domain point mutations, with error bars showing standard error. Also shown are the relative Kd values for binding of the wild type and mutated MLL CXXC domains to DNA. b) Digital photographs showing colony (above) and cell (below) morphologies of transduced bone marrow cells at the end of week 4 of the colony assay.
Deletion of the CXXC domain, mutation of cysteines that participate in zinc coordination, or mutation of the KFGG motif to AAAA all were shown previously to eliminate in vitro transformation by MLL-ENL 18. However, it is difficult to assess the specific role of these mutations since all of them disrupt the CXXC domain fold 26. It is known that the CXXC domain, in addition to DNA binding, also mediates protein-protein interactions with polycomb group proteins and histone deacetylases 19, therefore a misfolded CXXC domain could potentially interfere with multiple functions. Here, we have identified the C1188D mutation in the CXXC domain which disrupts DNA binding specifically while maintaining the protein structure. Therefore, we can specifically attribute the DNA binding capability of the CXXC domain as absolutely required for the transforming potential of MLL fusions.
We also assessed other mutations in the CXXC domain of MLL-AF9 which only partially decrease DNA binding affinity. Interestingly, introduction of the R1154A, K1185A, and Q1187A mutations into MLL-AF9 resulted in greatly reduced colony forming ability compared to MLL-AF9 (Figure 4). The number of colonies for these mutants in the final week of the assay varied and ranged from zero up to 300. However, all three mutants were vastly diminished in clonogenic activity compared to the average of 1500 colonies demonstrated by MLL-AF9 at the last replating time assessed. The reduced colony forming ability and proliferative capacity of the MLL-AF9 variants shows a good correlation with the DNA binding affinities of the CXXC mutants (Figure 4a).
Mice expressing MLL-AF9(C1188D) do not develop leukemia
Finally, to evaluate whether the disruption of the CXXC domain-DNA interaction will impact leukemogenic transformation in vivo, we have introduced MLL-AF9(C1188A) and MLL-AF9(C1188D) into mice. The c-Kit positive bone marrow progenitor cells retrovirally transduced with MSCV-MLL-AF9 containing point mutations C1188A or C1188D or with MSCVneo negative control were transplanted into lethally irradiated C57Bl/6 mice. All mice expressing the C1188A mutant developed acute myeloid leukemia within 46 days (n=8) as expected for MLL-AF9 infected mice (Figure 5a) 31,32. At the time of sacrifice due to leukemia-related morbidity, the MLL-AF9(C1188A) mice had elevated white blood cell counts (range: 12.28 K/ul to 4680 K/ul) (Figure 5b), pale femurs, and enlarged spleens and livers with infiltrating leukemia cells (see Supplementary Fig. 1). Peripheral blood cells expressed surface markers typical of MLL-AF9 myeloid leukemia. They were positive for c-Kit (CD117), CD11b and Gr-1, but negative for B220 and CD3. The mice infected with control MSCVneo (n=5) did not develop leukemia. Strikingly, no mice (n=9) transduced with MLL-AF9(C1188D) developed leukemia, and were sacrificed 200 days after transplant (Figure 5a). This shows that DNA binding by the CXXC domain in MLL-AF9 is required to induce leukemia in vivo.
Figure 5.
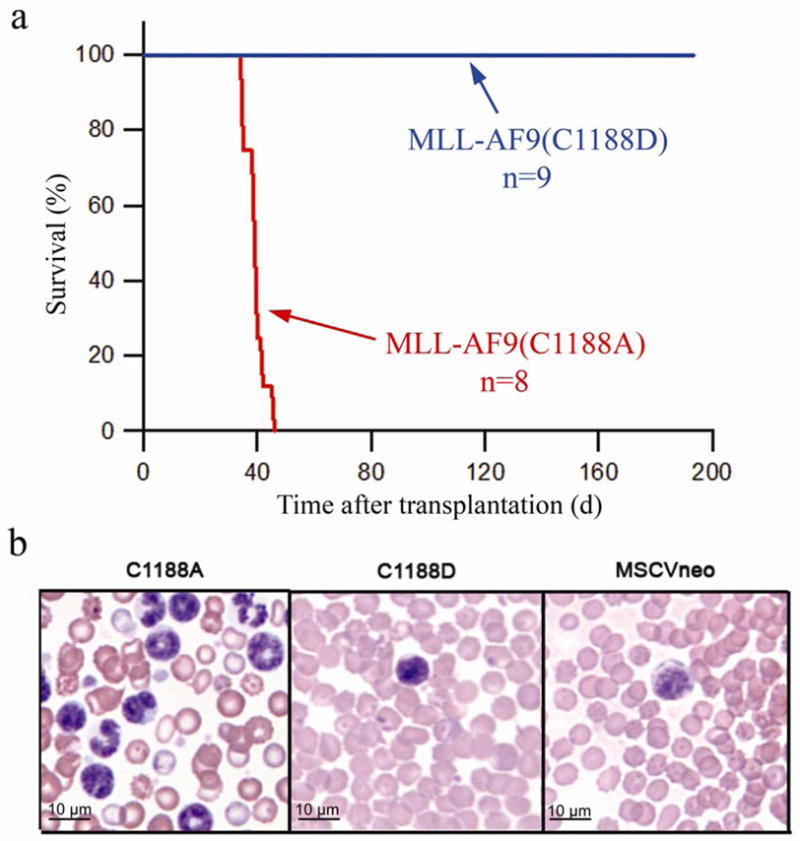
DNA binding activity of the MLL CXXC domain is required for MLL-AF9 to cause leukemia in vivo. a) Survival curve of mice transplanted with bone marrow progenitor cells infected with either MSCVneo-MLL-AF9(C1188A) or MSCVneo-MLL-AF9(C1188D). No mice transplanted with empty vector MSCVneo died (data not shown). b) Peripheral blood from mice at time of sacrifice (MLL-AF9(C1188A)) or at two months after bone marrow transplants (MSCVneo and MLL-AF9(C1188D)).
DISCUSSION
To shed light on the molecular mechanism of MLL fusions, we have solved the structure of the CXXC domain in complex with CpG DNA. This provided the first detailed structural account of how the CXXC domain discriminates between methylated and non-methylated DNA. Subsequently, we have used the structure of the CXXC-DNA complex to design point mutations in the CXXC domain which impair DNA binding without disrupting the fold of the domain. Introduction of these mutations into MLL-AF9 showed that the DNA binding function of the CXXC domain is indispensable for transformation of bone marrow progenitor cells and for leukemogenic transformation in mice. Consistent with this, cells expressing the non-oncogenic MLL-AF9(C1188D) variant with no CpG DNA binding have significantly increased CpG methylation of a specific region of the Hoxa9 locus, increased H3K9 methylation, and decreased expression of Hoxa9 locus transcripts, critical mediators of MLL fusion leukemia. To date, the role of the CXXC domain in leukemogenic transformation has only been assessed using in vitro colony forming ability18,33. In this work, for the first time, we have demonstrated that DNA binding by the CXXC domain is critically important for MLL-AF9 leukemogenesis in vivo.
Based on our studies, we are proposing a novel mechanistic role for the CXXC domain in maintaining expression of Hoxa9 by MLL-fusion proteins. It is well established that MLL-fusions are localized to the Hoxa9 locus 20,34. Recent studies indicate that interactions with menin and LEDGF are critical for this localization 29,16. Not surprisingly, based on the modest affinity of the CXXC domain for DNA, we have shown by ChIP that the CXXC domain has little to no role in localization to Hoxa9. Rather, our data clearly demonstrate that when the MLL fusion is localized to a specific site, the CXXC domain functions to maintain CpG motifs in the non-methylated state that is essential for maintenance of appropriate epigenetic marks and continued transcription. Thus, upon localization to the Hoxa9 locus, the presence of the CXXC domain is critical for maintenance of the high level of transcription of Hoxa9 and mir196b seen in MLL leukemias (Figure 6a). Indeed, we have shown that the C1188D mutation which abolishes DNA binding by the CXXC domain results in increased methylation of Hoxa9 and an increase in histone H3K9 methylation, leading to downregulation of Hoxa9 and mir196b transcription (Figure 6b). As a consequence, mutants of MLL-AF9 with impaired CXXC DNA binding fail to induce transformation or leukemia in mice.
Figure 6.

Model of the regulation of Hoxa9 locus transcription by the CXXC domain of MLL-AF9. a) The CXXC domain of MLL-AF9 protects specific CpG sequences within the Hoxa9 locus from methylation and maintains transcription within the locus; b) disruption of DNA binding function of CXXC domain by C1188D mutation results in increased methylation of the same CpGs, increased H3K9 trimethylation, and silencing of Hoxa9 and mir196b.
Patients with acute leukemias harboring MLL translocations have poor prognosis with current therapies and a very low survival rate 35. Therefore, there is a pressing need to develop novel approaches for treatment of MLL leukemias. Importantly, our results provide support for the idea that the interaction of the CXXC domain with DNA may constitute a useful drug target for MLL-related leukemias. Because the CXXC domain is preserved in all MLL translocations, inhibition of the CXXC domain-DNA interaction would be a general approach to reverse the oncogenic potential of MLL fusion proteins. Inhibition of the interaction of transcription factors with DNA is commonly considered a challenging task because of the typically very high DNA binding affinities of transcription factors (low nanomolar range). However, the CXXC domain binds DNA much more weakly (micromolar affinity) and our experiments demonstrate that even a 6-fold decrease in DNA binding affinity of the CXXC domain has a pronounced effect on the transforming potential of MLL fusions.
METHODS
Preparation of proteins and oligonucleotides
We cloned DNA encoding amino acids1147-1203 of MLL into the pGEX-4T-2 vector (Amersham Biosciences) and transformed into E. coli BL21 (DE3) cells (Stratagene) for expression. GST-fused CXXC domain was expressed and purified using glutathione affinity chromatography (Amersham-Pharmacia), cleavage of GST with thrombin, and ion exchange chromatography using SP-Sepharose (Amersham-Pharmacia). Point mutations in MLL were introduced using QuickChange PCR-based mutagenesis. All oligonucleotides were purchased from Integrated DNA Technologies. Samples were annealed and purified by ion-exchange chromatography purification using Q-Sepharose (Amersham-Pharmacia).
NMR spectroscopy
NMR spectra were measured on a Varian Inova 600 equipped with a cryogenically cooled probe. Unless otherwise indicated, all experiments were carried out at 25°C. All NMR spectra were processed using NMRPipe 36 and analyzed using Sparky (Goddard and Kneller, University of California, San Francisco) programs. All NMR spectra used for structure determination were collected using a 1 mM equimolar complex of 13C,15N CXXC domain (MLL 1147-1203) – palindromic CpG DNA (DNApal) in 25 mM deuterated bis-TRIS (pH 7.0) and 5 mM deuterated DTT. Further details are included in the Supplementary Material.
Measurement of RDCs for were done for 1 mM 13C,15N CXXC domain complexed with unlabeled DNApal DNA in 100 mM bis-TRIS (pH 7), 2 mM DTT and aligned in 50-S negatively charged gel 37. Following couplings have been measured 1DHN, 1DNCO, 2DHNCO, 1DCαCO, 1DHαCα at 35°C. The 1DCH couplings for DNA were measured for 15N CXXC domain-DNApal complex using 13C-coupled 1H-13C HSQC spectrum at natural abundance of 13C.
Determination of the dissociation constants by NMR for the CXXC domain (MLL 1147-1203) and mutants was performed by titration of 15N-labeled protein with concentrated solutions of DNApal oligonucleotide. Experiments were carried out in 50 mM phosphate buffer (pH 7.1), 300 mM NaCl, 2 mM DTT at 25 °C. Calculation of the dissociation constants was carried out using an established procedure 38.
Structure determination
For structure determination of the CXXC domain-DNA complex, we used a complex of 13C,15N CXXC domain (MLL 1147-1203) and unlabeled palindromic oligonucleotide (DNApal) with the sequence CCC TGC GCA GGG. The structure of the complex was determined using the simulated annealing protocol in CNS 39 employing 912 intramolecular and 79 intermolecular distance restraints, TALOS-based dihedral angle restraints, 3JHNHα couplings, and RDCs (see Table 1). All of the intermolecular distance restraints were employed as ambiguous distances to the two symmetry related protons in DNA. Due to significant overlap and broadening of DNA resonances in the complex, the complete quantitative analysis of NOESY spectra for DNA was intractable. Therefore, we employed DNA in a fixed B-DNA conformation. This assumption was based on the observed pattern and intensities of DNA NOEs, which is very similar to the spectrum of free DNA and consistent with a B-DNA conformation, and the lack of significant chemical shift changes for DNA upon binding to the CXXC domain. Details of the structure determination are provided in the Supplementary Material.
CpG methylation analysis by bisulphite treatment of DNA
Experiments were performed as we have previously described 20. Briefly, Mll −/− MEFs were co-transfected by electroporation with MSCV-MLL-AF9 C1188A or C1188D point mutation constructs and the pSuper plasmid to confer puromycin resistance at a 1:5 molar ratio. After one week of puromycin selection, genomic DNA was harvested (Puregene DNA kit, Gentra Systems). DNA was subjected to bisulfite treatment and clean-up with the EpiTect Bisulfite Kit (Qiagen). Nested PCR was performed on each sample, then PCR products were sequenced. Scion Image software was used to analyze each CpG peak in the sequence tracings. Area under the curve values were calculated for each CpG cytosine peak, which represents a methylated CpG residue, and its corresponding thymine peak, which represents an unmethylated CpG. Error due to background levels of conversion was determined by measuring non-CpG cytosine conversion. Relative methylation percentage at each CpG peak was then calculated.
Bone marrow colony replating assays
Bone marrow colony assays were performed as we have previously described with minor changes 30. Briefly, bone marrow cells were collected from 4–12 week old C57Bl6 mice followed by selection of c-kit positive progenitors. Progenitors were infected with retroviruses and plated in methylcellulose with cytokines and G418. Colonies were counted and replated each week for a total of four weeks. Experiments were performed in duplicate and were repeated at least six times. Cells collected from methylcellulose were processed by cytospin and stained with Hema 3 (Fisher Scientific). A FACSCanto flow cytometer (BD) was used to analyze colony assay cells or mouse peripheral blood and bone marrow cells stained with the following fluorochrome-conjugated antibodies: CD3e, CD11b, B220, Gr-1, CD117, Sca-1, and CD45.1 (e-Bioscience). Data was analyzed with FlowJo software. Full method description is included in Supplementary Material.
Quantitative RT-PCR
RNA was isolated from week 1 methylcellulose colony assay cells (see above) using TRI Reagent (Sigma) according to the manufacturer’s protocol. cDNA was analyzed using ABI Prism 7300. For microRNA quantification, reverse transcription was performed using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) using a total of 10 ng of RNA for each control and testing sample. Real-time PCR was performed at least in triplicate using the standard program. Relative mir-196b and Hoxa9 expression were normalized to RNU6B and hprt expression levels, respectively, and calculated using the 2−ΔΔCt method 40.
Chromatin Immunoprecipitation
The chromatin immunoprecipitation (ChIP) assay was performed using EZ-Magna ChIP G (Upstate/Millipore, Temecula, CA) according to the manufacture’s protocol. Phoenix cells (Orbigen, San Diego, CA) were transfected with MSCV-MLL-AF9-Flag constructs using the CalPhos Mammalian Transfection Kit (Clontech, Mountain View, CA). Cells were harvested and fixed with formaldehyde about 70 hours after transfection. Chromatin was immunoprecipitated using anti-Flag (Sigma, St. Louis, MO), anti- Histone H3 trimethyl K9 (Abcam, Cambridge, MA) and anti-RNA Polymerase II (Upstate/Millipore). Results were analyzed by qPCR in triplicate on an ABI 7300 real time PCR machine using iTaq SYBR Green Supermix with Rox (Bio-Rad, Hercules, CA). Enrichment was normalized to GAPDH and input chromatin, and calculated using the 2−ΔΔCt method 40. Primer sequences for the HOXA9 DNA are: 5′-GAAGCCACTAGTAAGCAAGCAGTC-3′ and 5′-GGCTGACTAGGAGATCTGATTAGG-3′. GAPDH primers were supplied in the ChIP kit (Upstate/Millipore).
In vivo mouse experiments
In vivo mouse experiments were carried out as previously described30. Bone marrow cells were collected from 5–6 week old B6.SJL (CD45.1+) mice, and C-Kit positive progenitor cells were isolated and infected with retrovirus as described above. The bone marrow was transplanted to lethally irradiated C57B1/6 recipient mice. To monitor for disease progression, peripheral blood samples were obtained periodically and complete blood counts were measured with a Hemavet 950 (Drew Scientific). The complete experiment description is in Supplementary Material.
Supplementary Material
Acknowledgments
This work was supported by NIH CA105049 and HL087188 and the Dr. Ralph and Marian Falk Medical Research Trust (NJZ-L), NIH Experimental Immunology training grant T32 AI007508-11A1 (LER), and by Leukemia and Lymphoma Society SCOR grant 7006-05 (JHB).
Footnotes
Accession codes. Protein Data Bank: Coordinates for the CXXC-DNA complex have been deposited with the accession code 2KKF.
AUTHOR CONTRIBUTIONS
T.C., L.E.R. and J.G. designed and performed experiments, analyzed data and wrote the manuscript; S.L., R.P., M.O., D.S.S. designed and performed experiments and analyzed data. N.J.Z.-L. and J.H.B. designed experiments, analyzed data and wrote the manuscript.
References
- 1.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 2.Sedkov Y, Tillib S, Mizrokhi L, Mazo A. The bithorax complex is regulated by trithorax earlier during Drosophila embryogenesis than is the Antennapedia complex, correlating with a bithorax-like expression pattern of distinct early trithorax transcripts. Development. 1994;120:1907–17. doi: 10.1242/dev.120.7.1907. [DOI] [PubMed] [Google Scholar]
- 3.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–8. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 4.Muller J, Gaunt S, Lawrence PA. Function of the Polycomb protein is conserved in mice and flies. Development. 1995;121:2847–52. doi: 10.1242/dev.121.9.2847. [DOI] [PubMed] [Google Scholar]
- 5.Orlando V, Jane EP, Chinwalla V, Harte PJ, Paro R. Binding of trithorax and Polycomb proteins to the bithorax complex: dynamic changes during early Drosophila embryogenesis. EMBO J. 1998;17:5141–50. doi: 10.1093/emboj/17.17.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guenther MG, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102:8603–8. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice KL, Licht JD. HOX deregulation in acute myeloid leukemia. J Clin Invest. 2007;117:865–8. doi: 10.1172/JCI31861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A. 1998;95:10632–6. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J Cell Biochem. 2005;95:234–42. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 10.Sorensen PH, et al. Molecular rearrangements of the MLL gene are present in most cases of infant acute myeloid leukemia and are strongly correlated with monocytic or myelomonocytic phenotypes. J Clin Invest. 1994;93:429–37. doi: 10.1172/JCI116978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox MC, et al. Chromosomal aberration of the 11q23 locus in acute leukemia and frequency of MLL gene translocation: results in 378 adult patients. Am J Clin Pathol. 2004;122:298–306. doi: 10.1309/RX27-R8GJ-QM33-0C22. [DOI] [PubMed] [Google Scholar]
- 12.Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–26. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 13.Meyer C, et al. The MLL recombinome of acute leukemias. Leukemia. 2006;20:777–84. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- 14.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birke M, et al. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002;30:958–65. doi: 10.1093/nar/30.4.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayton PM, Chen EH, Cleary ML. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol. 2004;24:10470–8. doi: 10.1128/MCB.24.23.10470-10478.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A. 2003;100:8342–7. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erfurth FE, et al. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc Natl Acad Sci U S A. 2008;105:7517–22. doi: 10.1073/pnas.0800090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baylin SB, et al. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–92. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 22.Ohki I, et al. Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell. 2001;105:487–97. doi: 10.1016/s0092-8674(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen HF, Ben-Porath I, Bird AP. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol Cell Biol. 2004;24:3387–95. doi: 10.1128/MCB.24.8.3387-3395.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Voo KS, Skalnik DG. Identification and characterization of the DNA binding domain of CpG-binding protein. J Biol Chem. 2001;276:44669–76. doi: 10.1074/jbc.M107179200. [DOI] [PubMed] [Google Scholar]
- 25.Inomata K, et al. Kinetic and thermodynamic evidence for flipping of a methyl-CpG binding domain on methylated DNA. Biochemistry. 2008;47:3266–71. doi: 10.1021/bi7019029. [DOI] [PubMed] [Google Scholar]
- 26.Allen MD, et al. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. Embo J. 2006;25:4503–12. doi: 10.1038/sj.emboj.7601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: A structural analysis. J Mol Biol. 1999;287:877–96. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 28.Popovic R, et al. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood. 2009;113:3314–22. doi: 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milne TA, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavau C, Du C, Thirman M, Zeleznik-Le N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000;19:4655–64. doi: 10.1093/emboj/19.17.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 32.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 33.Bach C, Mueller D, Buhl S, Garcia-Cuellar MP, Slany RK. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 2009;28:815–23. doi: 10.1038/onc.2008.443. [DOI] [PubMed] [Google Scholar]
- 34.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005;65:11367–74. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 35.Slany RK. When epigenetics kills: MLL fusion proteins in leukemia. Hematol Oncol. 2005;23:1–9. doi: 10.1002/hon.739. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–66. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 38.Tugarinov V, Kay LE. Quantitative NMR studies of high molecular weight proteins: application to domain orientation and ligand binding in the 723 residue enzyme malate synthase G. J Mol Biol. 2003;327:1121–33. doi: 10.1016/s0022-2836(03)00238-9. [DOI] [PubMed] [Google Scholar]
- 39.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.