

Abstract
Tumor surveillance requires the interaction of multiple molecules and cells that participate in innate and the adaptive immunity. Cathelicidin was initially identified as an antimicrobial peptide, although it is now clear that it fulfills a variety of immune functions beyond microbial killing. Recent data have suggested contrasting roles for cathelicidin in tumor development. Because its role in tumor surveillance is not well understood, we investigated the requirement of cathelicidin in controlling transplantable tumors in mice. Cathelicidin was observed to be abundant in tumor-infiltrating NK1.1+ cells in mice. The importance of this finding was demonstrated by the fact that cathelicidin knockout mice (Camp−/−) permitted faster tumor growth than wild type controls in two different xenograft tumor mouse models (B16.F10 and RMA-S). Functional in vitro analyses found that NK cells derived from Camp−/− versus wild type mice showed impaired cytotoxic activity toward tumor targets. These findings could not be solely attributed to an observed perforin deficiency in freshly isolated Camp−/− NK cells, because this deficiency could be partially restored by IL-2 treatment, whereas cytotoxic activity was still defective in IL-2-activated Camp−/− NK cells. Thus, we demonstrate a previously unrecognized role of cathelicidin in NK cell antitumor function.
The ability of the immune system to control tumor growth and thereby to function as an endogenous defense mechanism against cancer has received much attention (1–3). Multiple studies have highlighted both innate and adaptive immune cells as essential parts of the tumor surveillance system (4). As components of the innate immune system, NK cells have been shown to provide immune surveillance of certain tumors, including B16 melanoma and RMA-S lymphoma (5–9). Upon recognition of target cells, the contents of NK cell granules are released into the synapse formed between target and NK effector cells, and entry of granzymes and perforin into target cells is believed to ultimately mediate target cell death (10, 11). The importance of perforin is evident from studies showing that mice with a targeted deletion of the perforin gene are susceptible to microbial infections, fail to reject transplanted tumors, and spontaneously develop aggressive B cell lymphoma as they age, indicating a fatal lapse of tumor immune surveillance (11, 12).
Cathelicidins are a family of antimicrobial peptides that have been identified in several epithelial tissues and some myeloid cells and cell lines (13). Both the human (CAMP) and murine (Camp) cathelicidin genes are translated as propeptides that are further processed in a cell- and tissue-specific manner to a mature peptide, best known as LL-37 in humans (14) and murine cathelicidin peptide (mCRAMP) in mice (15). The relevance of cathelicidin to mammalian host defense has been demonstrated by targeted deletion of Camp in mice (Camp−/−), which results in increased susceptibility to infections in several organ systems (16–20).
Recent studies have suggested contrasting roles for human cathelicidin in human tumor development (21–23). Interestingly, cathelicidin is expressed in human NK cells (24), and like perforin, activated cathelicidin peptides function in part by disrupting membranes (25). However, the role of cathelicidins in NK cell function has not been studied. Therefore, we sought to determine the importance of cathelicidin to NK cell function and in vivo tumor defense in mice. We demonstrate for the first time that deficient expression of Camp is directly associated with the growth of specific tumor cell lines in mice and suggest a previously unsuspected role for cathelicidins in NK cell antitumor function.
Materials and Methods
Cells lines
RMA-S is a MHC class I (MHC-I)–negative variant of RMA, a mutagenized variant of Rauscher virus-induced T cell lymphoma of C57BL/6 origin (26). Yac-1 is a Moloney murine leukemia virus-induced lymphoma that lacks MHC-I expression and is sensitive to lysis by NK cells (27). B16.F10 is a murine melanoma cell line with high survival and growth potential.
Mice and tumor challenge experiments
Camp−/− mice in a C57BL/6 background were generated as described earlier, backcrossing was based on MaxBax analysis (Charles River Laboratories, Wilmington, MA) and congenicity to C57BL/6 was 97.73%. Mice were used at the age of 9–13 wk (18). All experiments involving animal work were in accordance with and with the approval of the Institutional Animal Care and Use Guidelines of the University of California San Diego (UCSD) (La Jolla, CA) and the VA San Diego Healthcare System (San Diego, CA). B16 or RMA-S cells were trypsinized, washed in PBS, and centrifuged at 1000 rpm for 15 min and were resuspended in sterile PBS. Cells were counted in a hemocytometer, and ~1 × 106 cells/mouse were injected s.c. into the hind flank. Wild type (wt) and Camp−/− mice were injected identically. For some experiments with RMA-S, some mice were depleted of NK cells by injections of anti-NK1.1 (clone PK136, purified from hybridoma) on days −2, 0, +2, and +7 relative to RMA-S challenge (day 0). For other experiments with RMA-S, polyinosinic:polycytidylic acid (poly[I:C]) (100 μg/injection; Sigma-Aldrich, St. Louis, MO) was injected i.p. 1 d before tumor challenge. Mice were defined as RMA-S tumor-bearing when tumors reached 16 mm2 in size. Tumors were measured as the two perpendicular diameters with a caliper, and the product of the diameters was taken as a measure for tumor size.
Cell isolation
For quantitative real-time RT-PCR, Western blot analysis, and surface-enhanced laser desorption ionization time-of-flight mass spectroscopy (SELDI-TOF-MS), fresh NK cells were purified from splenocytes by FACS sorting of NK1.1+CD3− cells (minimum of 97% purity; UCSD VA Flow Cytometry Core). For killing assays, NK cells were purified using DX-5 MicroBeads (Miltenyi Biotec, Auburn, CA), according to the manufacturer’s protocol. Purity was examined by flow cytometry and was 60–80% in all assays. For generation of lymphokine-activated killer (LAK) cells, purified NK cells (DX-5 MicroBeads) were kept in culture in the presence of 1000 U/ml IL-2 (eBioscience, San Diego, CA) for 7–10 days. FACS analysis revealed that LAK cells were ~100% NK1.1+ and CD3−. For other experiments, splenocytes were sorted for NK1.1+CD3+ (NKT cells), and NK1.1−CD3+ (T cells).
Human melanoma samples
Paraffin-embedded tissues were obtained from four patients diagnosed with malignant melanoma. Three of the four melanoma tumors were 1 mm or deeper, and one melanoma sample was 0.45 mm in depth. Samples were embedded in paraffin for histological analyses. All samples were collected by surgical excision from skin, following local institutional ethical committee guidelines. Procedures were in accordance with the Declaration of Helsinki principles and after approval by the Institutional Review Board. Informed consent was obtained from each patient.
Histology, immunohistochemistry, and immunofluorescence stainings
Paraffin sections were deparaffinized and microwaved treated to unmask epitopes. Purified NK cells were cultured in chamber slides (Thermo Fisher Scientific, Rochester, NY). Histology sections and chamber slides were stained with the following Abs, substrates, and substances: H&E (performed by the Histology Core Facility at UCSD); anti-CD31 (PECAM-1; BD Pharmingen, San Diego, CA); anti-NK1.1, anti-NKp46, anti-CD3, anti CD16/32, anti-perforin, and isotype IgG controls (eBioscience); unlabeled primary rabbit-anti-CRAMP and preimmune serum (28); primary rabbit anti-LL-37 or preimmune serum, FITC-conjugated or Alexa Fluor 568-conjugated secondary Ab (Sigma-Aldrich); biotinylated secondary Ab (Vector Laboratories, Burlingame, CA); and diaminobenzidine substrate (Sigma-Aldrich). Immunofluorescence stainings were mounted in Prolong Anti-Fade reagent containing DAPI (Molecular Probes, Eugene, OR). Sections were evaluated with an Olympus BX41 microscope or a laser confocal microscope (Olympus FV1000).
Quantitative real-time RT-PCR
Gene expression was normalized against mouse GAPDH. For SYBR Green detection of serine protease inhibitor 6 (SPI-6) primer pairs were used as described previously (29). TaqMan gene expression assays detecting perforin and granzyme B were purchased from Applied Biosystems (Foster City, CA). The expression of Camp was evaluated using FAM-CAGAGGA TTGTGACTTCA-MGB probe with primers 5′-CTTCACCAGCCC GTCCTTC-3′ and 5′-CCAGGACGACACAGCAGTCA-3′. For GAPDH expression, a VIC-CATCCATGACCACCCCTGGCCAAG-MGB probe with primers 5′-CTTAGCACCCCTGGCCAAG-3′ and 5′-TGGTCATG AGTCCTTCCACG-3′ were used.
Western blot and dot blot
For Western blot analysis, equal amounts of protein from resting or activated NK cells (NK1.1+CD3−) or the positive controls were loaded on a 16% Tris-Tricine Gel (Bio-Rad, Hercules, CA) for electrophoresis. Proteins were transferred to a polyvinylidene difluoride membrane; the membrane was blocked in 3% nonfat milk in 0.1% Tween TBS, then incubated with the primary Ab, anti-CRAMP (QCB, Hopkinton, MA), anti-perforin, or anti-granzyme B (eBioscience). For dot blot, supernatants from de-granulating NK cells or cell extracts were collected and loaded onto a nitrocellulose membrane, and anti-CRAMP was diluted in 5% nonfat milk and 3% BSA in PBS. After washing and incubation with the species-specific, HRP-conjugated secondary Ab (DakoCytomation, Carpinteria, CA), immunoreactive proteins were detected by Western Lightning system (PerkinElmer, Wellesley, MA).
SELDI-TOF-MS
Splenocytes from eight mice were FACS sorted for NK1.1+CD3− NK cells, NK1.1+CD3+ NKT cells, and NK1.1−CD3+ T cells. Proteins were extracted and SELDI-TOF was performed as previously described with slight modifications (30). Cathelicidin capture was done with a rabbit anti-CRAMP Ab described previously (31). Samples were analyzed on a SELDI mass analyzer PBS IIC with a linear TOF mass spectrometer (Ciphergen Biosystems, Fremont, CA) using time-lag focusing.
In vitro killing assays
For in vitro killing assays, murine melanoma cell lines B16.F10 and human A375 (American Type Culture Collection, Manassas, VA) were used. A nonradioactive MTT-like proliferation assay (Promega, Madison, WI) and a cytotoxicity detection kit based on measurement of lactate dehydrogenase (LDH) activity (Roche, Basel, Switzerland) were used according the manufacturers’ instructions. Apoptotic and necrotic cells were quantified by Annexin V and propidium iodide (PI) staining with FACS analysis (BD Pharmingen).
Flow cytometry
Single-cell suspensions from spleens were prepared. Briefly, RBCs were lysed with lysis buffer (Sigma-Aldrich). FcRs were blocked for 5 min with an unconjugated Ab against the low-affinity receptors FcγRII/III (CD16/32 Ab; eBioscience) and stained with lineage-specific conjugated mAbs to CD3, NK1.1, Mac-1, CD43, αV, CD49b, CD27, and NKG2D (eBioscience). Cell-associated fluorescence was acquired on a BD FACSCanto II (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Flow-based killing assay
A flow-based cytotoxicity assay that has been validated to be completely concordant with standard chromium release assays was performed (32–35). Yac-1 cells were stained with 1 μM CFSE (Molecular Probes), and 1 × 105 labeled target cells were seeded into a 96-well plate or FACS tube in complete RPMI 1640 medium with 10% FBS and 100 U/ml IL-2. Freshly purified NK or LAK effector cells were added at various E:T ratios and incubated for 5 h at 37°C with 5% CO2. As controls, effector and target cells were cultured alone or in the presence of ionomycin and/or 2.5 μM EGTA. Cells were stained with 7-aminoactinomycin, anti-CD3, and anti-NK1.1 and acquired on a FACSCanto II. Percent cell death was assessed by measuring the percentage of CFSE target cells that were 7-aminoactinomycin positive. Yac-1 cells treated with ionomycin as a positive control showed cell death of at least 50% (data not shown).
Degranulation assay
Fresh purified splenocytes (anti-DX5 MicroBeads) were incubated for 4 h in the presence of anti-CD107a Ab and GolgiStop (BD Biosciences) with or without Yac-1 targets (1:1 and 1:5 NK:target ratio), IL-2 (800 U/ml) plus IL-12 (100 ng/ml; eBioscience), or PMA (100 ng/ml; Calbiochem, San Diego, CA) plus ionomycin (1 μM; Calbiochem). Thereafter, cells were stained with anti-NK1.1 and anti-CD3. Cells were then fixed and permeabilized using Cytofix/Cytoperm solution (BD Biosciences) and stained with CD107a Ab in permeabilization buffer (BD Biosciences). CD107a+ cells were measured within the NK cell population (data not shown).
Statistical analysis
A paired t test was used to determine statistical significance between control and test groups. A value of p < 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism software 5.0.
Results
Cathelicidin is present in the melanoma microenvironment and expressed in mouse NK1.1+ and NKp46+ cells
An increase in the abundance of cathelicidin is a common observation in inflamed, injured, or infected tissues and is associated with the capacity to mount an effective defense against microbial invasion (18, 36, 37). To determine whether cathelicidin is also present during the host response to tumor invasion, we evaluated the expression of cathelicidin in skin surrounding the murine B16.F10 melanoma cell line after injection s.c. into the backs of C57BL/6 mice. Cathelicidin expression was abundantly detected by immunohistochemical staining of cells infiltrating the tumor (Fig. 1). These observations in mice were consistent with staining for cathelicidin in cells surrounding spontaneous primary melanomas in human skin (Fig. 2). Cells expressing cathelicidins surrounding the B16 melanoma were mostly NK1.1+ (Fig. 1A–C) and NKp46+ (Fig. 1D, 1E) and not CD3+ (Fig. 1F, 1G), suggesting that the presence of cathelicidins was a consequence of expression in NK cells. Prior studies showing that NK cells influence the growth of B16 melanoma (5–8, 38) led us to further test whether the survival and proliferation of B16.F10 and a human melanoma cell line (A375) could be directly influenced by the addition of synthetic mCRAMP or human cathelicidin peptide (LL-37). Both mCRAMP and LL-37 inhibited cell proliferation (Fig. 3A, 3B) and induced cytotoxicity (Fig. 3C, 3D) in these cells. Furthermore, mCRAMP increased PI uptake and induced an increase in the proportion of Annexin+ B16.F10 cells (Fig. 3E, 3F).
FIGURE 1.
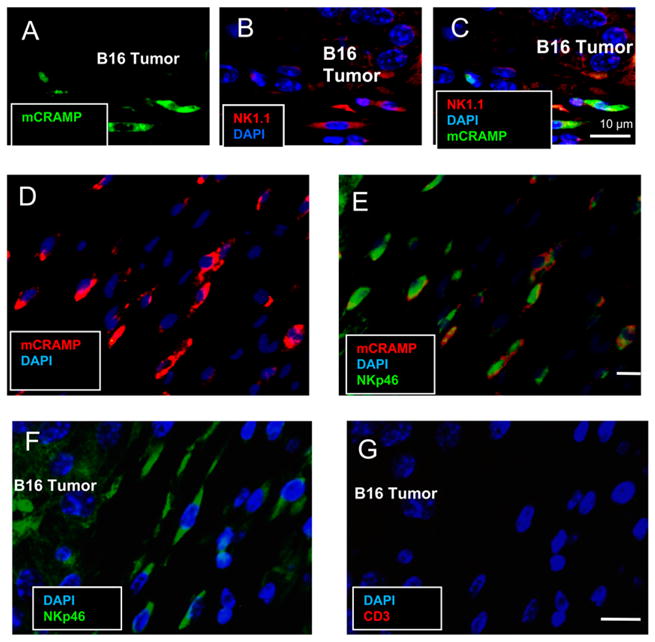
Cathelicidin is expressed in NK1.1+ and NKp46+ cells in the B16 tumor microenvironment. Immunofluorescence staining was performed on paraffin-enbedded sections from B16 tumor-bearing mice for mCRAMP (green), NK1.1 (red), and DAPI (blue). mCRAMP is present in the tumor microenvironment and colocalizes with NK1.1+ (AC). mCRAMP expression also colocalized with NKp46+ (D and E). The majority of cells that migrated to the B16 tumor were NKp46+ but CD3− (F and G). Scale bar: 10 μm.
FIGURE 2.
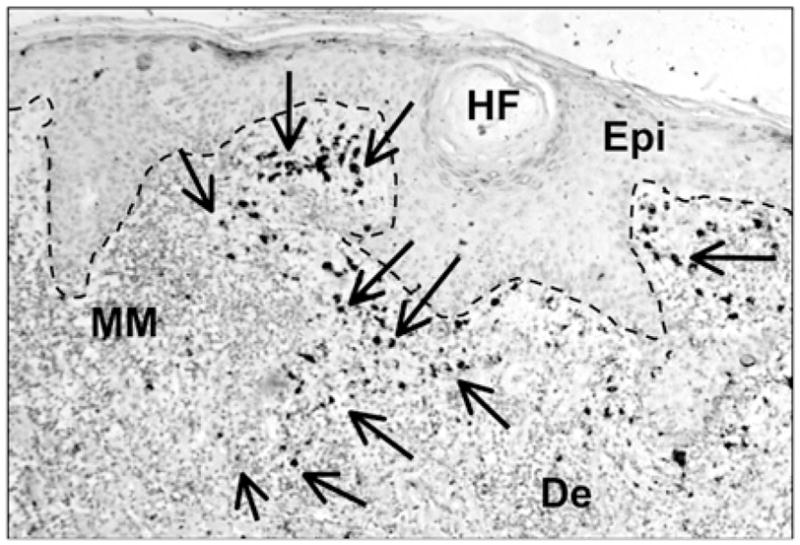
Cathelicidin is expressed in the microenvironment of human melanoma tissue. Expression of human cathelicidin in melanoma tumor-infiltrating immune cells. Immunohistochemistry was performed using rabbit-anti-LL-37 Ab and expression in melanoma tissue was visualized by diaminobenzidine substrate (indicated by arrows). Dotted line represents the basal membrane. Epidermal staining was weak as expected in noninflamed tissue. n = 4, one representative data from four patients is shown. Original magnification ×100. De, dermis; Epi, epidermis; HF, hair follicle; MM, melanoma.
FIGURE 3.

Cathelicidin peptides inhibit melanoma cell proliferation and induce LDH release and melanoma cell lysis in vitro. Murine B16 or human A375 melanoma cells were coincubated for 24 h with different concentrations of recombinant LL-37 or mCRAMP peptides, respectively. A and B, Cell proliferation was evaluated by a modified MTT assay and recorded as absorbance at 570 nm. C and D, Cell death was quantified by measuring LDH release from damaged cells and is plotted as percent cell death relative to Triton X-100 (Tx)–treated cells used as positive controls. Data are means + SD from triplicate measurements. One representative experiment out of three is shown. E and F, Annexin V and PI staining were performed on B16 cells that were incubated with recombinant mCRAMP peptides. E, Concentrations of mCRAMP that induced LDH release from B16 cells also showed double-positive Annexin V+ and PI+ staining. F, Double-positive Annexin V+ and PI+ cells were seen within 4 h of incubation with 128 μM CRAMP. Data shown are percentage of cells that were Annexin V+ and PI+ double positive.
Growth of B16 melanoma is increased in Camp−/− mice
The presence of cathelicidin in NK cells surrounding melanoma tumors, the capacity of this peptide to inhibit tumor cell growth in culture, and the prior observations that NK cells influence B16 tumor growth in mice all suggested that cathelicidin might be involved in suppressing tumor growth in this tumor model in vivo. To investigate the potential involvement of cathelicidin on B16 tumor growth in vivo, mice with a targeted deletion of cathelicidin (Camp−/−) and wt controls were given s.c. inoculations of B16. F10 cells into the hind flank. Tumors in Camp−/− mice were larger in size and were apparent 3–5 d earlier compared with tumors in wt controls (Fig. 4A–C). By day 8 after B16 tumor cell inoculation, nearly all Camp−/− mice developed tumors, whereas wt mice were tumor free (p = 0.0071; Fig. 4A, left panel). By day 12 after B16 inoculation (Fig. 4A, right panel), all mice showed established tumors, whereas the tumor sizes significantly differed between wt and Camp−/− mice (p = 0.0131). Growth of tumors in individual mice over time is shown in Fig. 4B. Photographic and histological evaluation of tumors including staining with H&E and CD31 is seen in Fig. 4C and revealed that the amount of the inflammatory infiltrate and blood vessel density around and within tumors were comparable between Camp−/− and wt mice.
FIGURE 4.

Accelerated B16 tumor growth in Camp−/− mice. Approximately 1 × 106 B16.F10 cells/mouse were inoculated s.c. in the hind flank of syngenic Camp−/− and wt mice. Mice were observed for tumor development, and tumors were measured with a caliber, and sizes in mm2 were calculated as described in Materials and Methods. A, Tumor formation occurred earlier and tumor sizes were larger in Camp−/− mice (n = 6) compared with wt controls (n = 6). Tumor sizes are plotted for day 8 and day 12 after tumor challenge, and each dot represents the size of one individual mouse. B, Tumor sizes and tumor development for individual mice are shown. *p < 0.05; **p < 0.01. Results are representative of three independent experiments. C, Images and histological evaluation of tumors in Camp−/− and wt mice. Immunohistochemistry reveals similar inflammation and degree of vascularization. Original magnification ×100 for H&E staining; ×200 for CD31 staining.
Cathelicidin does not colocalize with perforin
We next examined the subcellular localization, expression, and functional role of cathelicidin in populations of highly purified murine NK cells (NK1.1+CD3−). Cathelicidin gene expression in NK cells was detected by quantitative real-time PCR (Fig. 5A), and its abundance was higher compared with whole-cell populations of splenocytes or full-thickness skin samples (Fig. 5A). Immunofluorescence staining of lymphokine-activated NK cells showed that cathelicidin was expressed in a granular pattern distinct from perforin (Fig. 5B) and granzyme (data not shown). Western blot analyses further revealed that cathelicidin in freshly isolated NK cells was most abundantly present as the 18-kDa precursor protein (Fig. 5C), but more sensitive SELDI-TOF-MS also detected peptide forms of cathelicidins with a molecular mass of 4639.3 Da corresponding to a 42-aa peptide (Fig. 5D). In addition, SELDI-TOF-MS also detected cathelicidin in protein extracts of purified populations of NKT (NK1.1+CD3+) and T (NK1.1−CD3+) cells (Fig. 5E and data not shown).
FIGURE 5.

mCRAMP does not colocalize with perforin-containing granules and is processed to a mature peptide form. A, Transcript abundance of cathelicidin in various mouse tissues. Relative gene expression is shown for pooled samples from mouse skin (n = 2 mice), splenocytes (n = 6 mice), and FACS-sorted NK cells (CD3−NK1.1+, n = 8 mice). Relative gene expression of Camp was calculated and normalized to mouse skin. Data shown are means of triplicate measurements +SEM. B, LAK cells from B6 mice were stained for mCRAMP (green), perforin (red), and DAPI (blue). mCRAMP surrounds perforin-containing granules in a ring-shaped pattern. LAK cells were generated from purified NK cells kept in culture in the presence of 1000 U of IL-2 for 7–10 days and were ~99.9% pure. C, Western blot of total protein extracted from freshly isolated NK cells (NK1.1+CD3−) and femoral bone marrow (BM). For Western blot, equal amounts of total protein (10 μg) of NK cells or BM were analyzed and in both samples the 18-kDa proform of mCRAMP (18-kDa mCRAMP) was detected. The mature 5-kDa form of mCRAMP (5-kDa mCRAMP) was detected only in BM. Synthetic mCRAMP peptide was used as positive control. D, SELDI-TOF-MS detected mCRAMP peptide with a molecular mass of 4639.3 Da in murine NK.1.1+CD3− cells and in NK.1.1−CD3+ cells.
Camp−/− mice permit enhanced RMA-S tumor development
To further elucidate the role of cathelicidin in an NK cell-dependent tumor model, RMA-Slymphoma cells were used. RMA-S is deficient in MHC-I expression, and in vivo tumor development is controlled by NK cells (26), is not T cell dependent (7, 39), and has been used as a benchmark for in vivo NK cell activity (40). Wt and Camp−/− mice were injected s.c. with RMA-S in the hind flank, and tumor growth was monitored over time. All Camp−/− mice developed s.c. RMA-S tumors within 5 d, whereas only 25% of wt mice developed tumors up to day 24 (Fig. 6A). However, by day 40 after tumor challenge, all mice succumbed to tumor growth. To confirm the dependence on NK cells in this system, some wt mice were depleted for NK cells by anti-NK1.1 Ab before tumor challenge. All of these anti-NK1.1–treated mice developed early RMA-S tumors, thus confirming the role of NK cells in this model (data not shown). Furthermore, all anti-NK1.1–treated mice developed RMA-S tumors as early as Camp−/− mice (data not shown). In previous reports, such dramatic tumor kinetics were only observed in perforin deficient (11). Similar to observations made in the B16 tumor model, cathelicidin expression was abundant in perforin+- and NK1.1+-expressing cells in wt mice that migrated to the RMA-S tumor site, and mCRAMP was located in distinct granules within these cells (Fig. 7 and data not shown).
FIGURE 6.
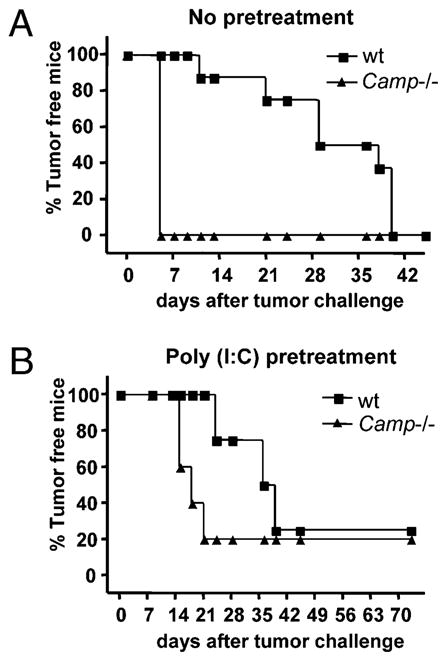
RMA-S tumor growth in Camp−/− mice. A, Wt (n = 8) and Camp−/− (n = 4) mice were injected s.c. into the hind flank with ~1 × 106 RMA-S cells. Data from one representative experiments out of two are shown with a total n = 14 wt mice and n = 10 Camp−/− mice. B, Wt (n = 4) and Camp−/− (n = 5) mice were injected with poly(I:C) (100 μg/mouse) 24 h before RMA-S tumor challenge to preactivate NK cells. Poly(I:C) treatment delayed onset of tumor growth, but the difference in tumor development between mouse groups remained.
FIGURE 7.
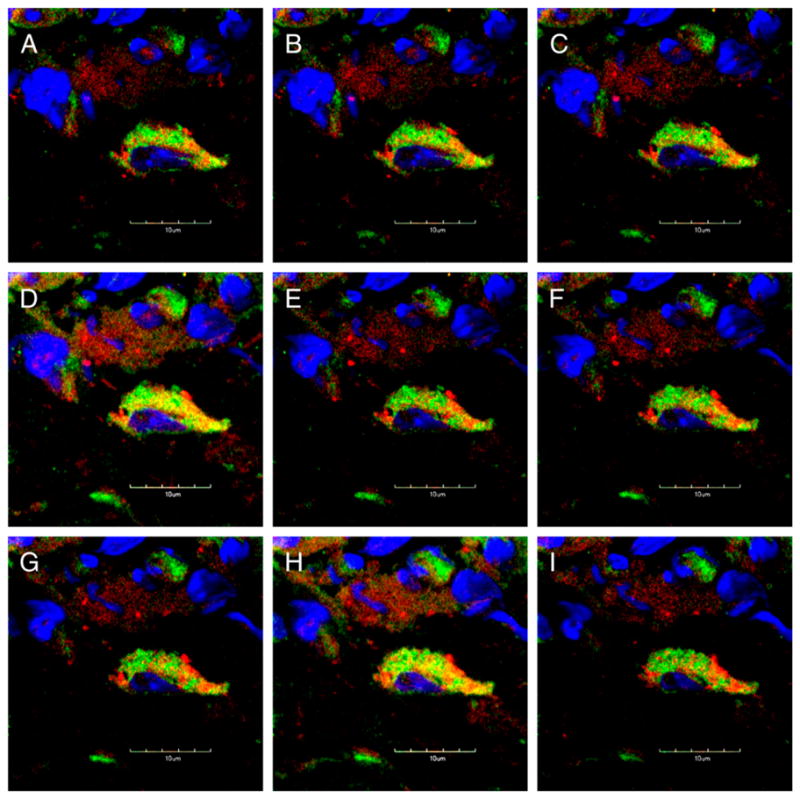
mCRAMP is expressed in perforin-expressing cells in RMA-S tumors. Immunofluorescence staining of established s.c. RMA-S tumors. Sections were stained for perforin (red), mCRAMP (green) and DAPI (blue). Projections of progressive confocal z-sections (0.2 μm) are shown revealing that mCRAMP is expressed in perforin+ cells, localized in distinct granules A–I, Original magnification ×300.
Killing efficiency exerted by activated NK cells on RMA-S cells has been demonstrated to be dependent on granule-induced cytotoxicity (41), and an effective way to stimulate this function in NK cells in vivo is the injection of poly(I:C). To determine whether the phenotype of RMA-S tumor development observed in Camp−/− mice could be overcome by preactivation of NK cells, poly(I:C) was injected once into the peritoneum 24 h before tumor challenge. Poly(I:C) delayed RMA-S tumor development similarly in wt and Camp−/− mice, but a difference between mouse groups remained (Fig. 6B).
NK cells from Camp−/− mice mature competently but show altered expression profiles of cytotoxicity-associated genes
Having shown that cathelicidin is expressed in the tumor microenvironment and is present in NK cells and that the lack of cathelicidin results in enhanced B16 melanoma and RMA-S lymphoma growth, we next sought to determine whether cathelicidin exerts its effects on NK cell cytotoxic function. The differentiation and maturation status impacts the NK cytotoxic potential (42), and hence, a difference in NK cell maturity may explain the results previously observed. Therefore, we examined whether the presence of Camp influences expression of Mac-1, CD49b, CD43, αV, CD27 NKG2D, and NKp46. No significant differences in maturation markers and surface receptor expression in NK cells derived from Camp−/− and wt mice were seen (Fig. 8A and data not shown). Thus, we concluded that wt and Camp−/− NK cells matured competently and thus acquired a similar surface receptor repertoire as wt NK cells. However, Camp−/− NK cells were observed to have altered expression of genes important for NK cell cytotoxic action. Camp−/− NK cells expressed less perforin mRNA compared with wt NK cells (Fig. 8B), a finding also seen in T cells (NK1.1−CD3+; data not shown), whereas the constitutive transcription of perforin in NKT cells (NK1.1+CD3+) was negligible (data not shown). In contrast, Camp−/− NK cells had moderately increased granzyme B mRNA levels and expression of the granzyme inhibitor, and SPI-6 was consistently downregulated in NK cells derived from Camp−/− mice compared with those from wt mice (Fig. 8C, 8D).
FIGURE 8.

Camp−/− NK cells express normal maturation markers but altered levels of perforin, granzyme B, and SPI-6. A, NK cell maturation is not defective in Camp−/− mice. Splenocytes from wt and Camp−/− mice were stained for NK1.1 and CD3, Mac-1, CD49b, CD43, αV, CD27, and NKG2D. Gated NK1.1+CD3− cell populations are shown. Percentages are given for dot blot quadrants. Representative dot blot quadrants are shown from five wt and five Camp−/− mice analyzed. Gene expression analyzes in isolated NK cells (NK1.1+CD3−) was determined by quantitative PCR or Western blot. NK cell from wt and Camp−/− mice were analyzed for their expression of perforin (B), granzyme B (C), and the physiological inhibitor of granzyme B, SPI-6 (D). Cells were pooled from three to eight mice. Histograms are representative of two to five independent experiments.
Cathelicidin is required for normal NK cell-mediated killing activity
To directly assess the role of cathelicidin in NK cell mediated cytotoxicity, flow-based killing assays using fresh and cytokine-activated NK cells from wt and Camp−/− mice against Yac-1 targets were performed. Yac-1 target cells are MHC-I deficient and very sensitive to NK cell-mediated lysis (43). Cytotoxic activity was dramatically diminished in NK cells derived from Camp−/− mice compared with wt cells in all E:T cell ratios tested. This effect was observed in freshly isolated NK cells (Fig. 9A). To determine whether a defect in perforin expression could explain the observed results, we tested whether the observed perforin deficiency could be overcome by IL-2 treatment, an effective method to increase perforin in cultured NK cells (44, 45). Perforin mRNA expression was measured by quantitative real-time PCR in NK cells isolated from wt and Camp−/− mice after treatment with 1000 U/ml IL-2 for 7–10 d. Treatment with IL-2 resulted in an increase of perforin mRNA levels in wt and Camp−/− cells such that a significant difference in expression no longer could be detected (Fig. 9B). These IL-2–activated NK cells (called LAK cells) continued to show deficient killing despite normalized perforin expression (Fig. 9C). As control, addition of EGTA to effector cells to inhibit granule release was seen to reduce cytotoxicity of wt and Camp−/− NK cells, confirming that cytotoxic activity against Yac-1 targets was granule-dependent (Fig. 9C).
FIGURE 9.
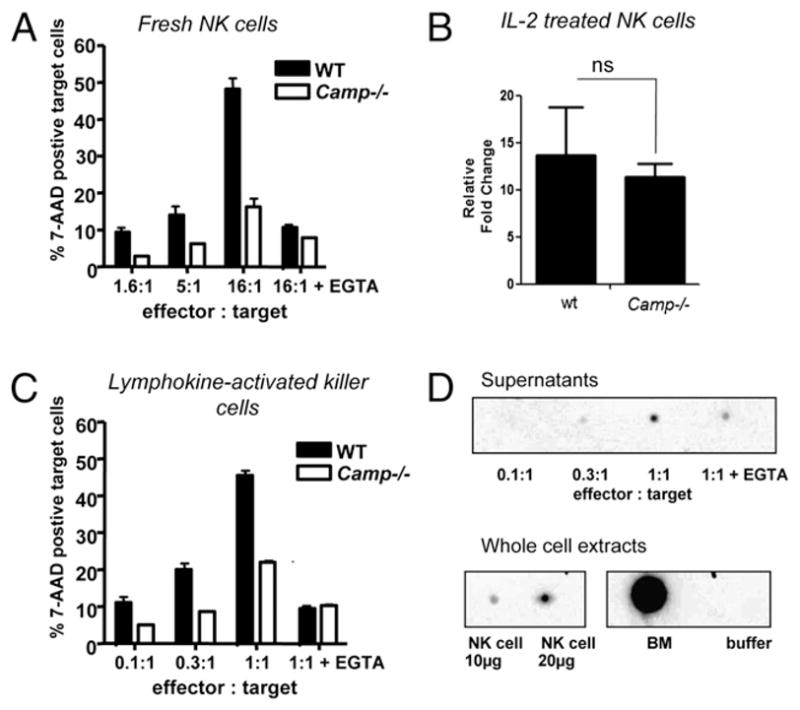
NK cells from Camp−/− mice are defective in killing MHC-I–negative Yac-1 target cells. A, Naive NK cells isolated from spleens with magnetic microbeads and were used in a flow-based 5-h killing assay against Yac-1 target cells at the E:T ratios indicated and in the presence or absence of EGTA. Data are presented as the mean + SEM of at two measurements and data shown representative of three independent experiments. B, NK cells were cultured in the presence of recombinant IL-2 (1000 U/ml) for 7–10 d, and perforin transcription was evaluated by quantitative real-time PCR. Wt and Camp−/− NK cells responded to IL-2 with increase in perforin mRNA. Fold change of perforin expression is expressed relative to each vehicle control. Results are representative of at least two independent experiments. Ns, nonsignificant by Student’s t test. C, LAK cells were used in a flow-based 5-h killing assay against Yac-1 target cells. Data are presented as the mean + SEM of two measurements, and data shown are representative of two independent experiments. D, Supernatants from degranulating LAK cells were analyzed by dot blot. As controls, NK cells and bone marrow (BM) and a negative control with buffer was included. mCRAMP was released by NK cells, and EGTA partially blocked this effect.
To further investigate whether wt NK cells release cathelicidin during the killing of Yac-1 targets, immunoblot analyses of cell supernatants were performed. In this study, mCRAMP was detected in an E:T cell ratio-dependent manner, suggesting that cathelicidin is involved in NK-mediated cytotoxcity (Fig. 9D). Release of mCRAMP was partially blocked by EGTA; thus, Ca2+-dependent “classic granule” release or other Ca2+-dependent mechanisms appear to be important for cathelicidin release from NK cells. To further test whether cathelicidin-dependent NK cell cytotoxicity correlates with a dysfunction in the release of classic granules, we evaluated CD107a expression as a marker of NK cell degranulation upon PMA/ionomycin stimulation. However, CD107a was not defective in Camp−/− NK cells compared with wt cells (data not shown).
Discussion
NK cells represent a specialized cell population that exerts strong cytotoxic activity against tumor and virally infected cells without prior exposure to an Ag. Their role in control of tumor growth has been clearly demonstrated in vitro and in vivo (46). In contrast, cathelicidins have been established as an essential element of host antimicrobial and inflammatory responses, but a function for these molecules in tumor biology is less clear (47–51). Contrasting observations on a role for the human cathelicidin LL-37 in ovarian cancer and other tumors have been published recently (21, 22, 52, 53). In the current study, the absence of cathelicidin is seen to result in enhanced growth of transplantable B16 melanoma and RMA-S lymphoma tumors in the skin, and the mechanism of action of cathelicidins in these models is associated with the capacity of cathelicidin to enable optimal NK cell function.
Multiple mechanisms could have contributed to the observation that Camp−/− mice have more rapid growth of B16 melanoma compared with wt controls. Cathelicidins are known to act as chemoattractive and angiogenic peptides and thus may influence tumor growth through these actions (54–57). These activities are beneficial during infection and wound healing because they promote inflammation and antimicrobial host defense, but their role in tumor control is unclear. The degree of inflammation and angiogenesis seen in the tumor environment of the B16 model was comparable between wt and Camp−/− mice, suggesting that the accelerated tumor growth seen in Camp−/− mice was not clearly because of its angiogenic and chemoattractive function.
Immunohistochemical studies identified cathelicidin in NK1.1+, a cell type known to influence the growth of these types of tumors. Cathelicidin staining was also abundant in NKp46+ cells, whereas CD3+ cells were almost absent in the B16 tumor micromilieu. The NK1.1+ marker could be present on NK, NKT, or other immune cells, which may contribute to B16 tumor surveillance. Indeed, cathelicidin was also found in murine NKT and T cells derived from the spleen from tumor nonbearing mice, but the function and relative contribution of cathelicidin in these lymphocytic cell populations compared with NK cells remain to be determined. To uncouple the role of NK from NKT cells, complex studies including Rag2−/− mice (which lack B, T, and NKT cells but have NK cells) and Rag2−/− Il2rg−/− mice (which lack all lymphocytes, including B, T, NKT, and NK cells) are needed. Also, an exhausting study of the tumor environment and the process involved in NK cell activation is needed to fully elucidate the role of cathelicidin in NK cell antitumor function. In this respect, it should be mentioned that cathelicidin fulfills different roles in and on various cells, including keratinocytes, neutrophils, and mast cells (24, 58–61), and presumably NK, NKT, and T cells.
In the current study, our data led us to focus on the function of cathelicidin in NK cells. Cathelicidin was abundantly detected in pure NK cell populations (NK1.1+CD3−) and was released during NK cytotoxic activity. The importance of these findings were related to findings of enhanced growth of B16 tumors in Camp−/− mice by the observation that growth of the MHC-I–deficient tumor RMA-S, a classical NK cell target cell line, is much favored in Camp−/− mice compared with wt mice. RMA-S tumor development could be delayed by poly(I:C) injections, but differences between wt and Camp−/− mice remained, thus further suggesting that cathelicidin is important to tumor control and is not dispensable by poly(I:C)-induced preactivation of NK cells. Activation of NK cells by poly(I:C) is presumably mediated by APCs (62, 63). However, recent studies indicated that poly(I:C) can also directly activate NK cells independent of APCs (64). In preliminary studies, we identified that pure populations of NK cells stimulated with poly(I:C) did not respond with increase of perforin levels (S. Morizane and R. L. Gallo, unpublished data), suggesting that it is more likely that NK cells become activated by an indirect, presumably APC-dependent effect. This indirect effect was not defective in Camp−/− mice, because the increased protective effect of poly(I:C) occurred in wt and Camp−/− mice. In this respect, functional in vitro studies further demonstrated that despite their cellular maturity, Camp−/− NK cells compared with wt NK cells had impaired cytotoxic activity against the tumor target cell line Yac-1, and expression analyses revealed that non-activated Camp−/− NK cells showed less perforin mRNA and protein levels compared with wt cells. This perforin deficiency could mostly be restored by IL-2 treatment, but this still was not sufficient to abrogate the defect in cytotoxic activity of Camp−/− NK cells. Thus, these data suggest that cathelicidin plays a relevant and nonredundant role in NK cell cytotoxic function that is independent of the unexplained decrease in perforin mRNA detected in freshly isolated Camp−/− NK cells.
In mice, mechanisms explaining transcriptional and translational regulation of perforin are incompletely understood. It has been suggested that epigenetic mechanisms and chromatin structure are important for perforin regulation (65) and cis-acting sequences that drive perforin transcription have been identified (66). Perforin is regulated during NK cell development, but our results show that no difference in maturation occurred in Camp−/− NK cells compared with wt control. Thus, the lack of perforin in non-activated Camp−/− NK cells is not likely due to aberrant NK cell development. The levels of granzyme B and its inhibitor SPI-6 were also altered in Camp−/− NK cells. Taken together, these effects may occur as a direct result of the lack of cathelicidin, or alternatively in an indirect manner. Nevertheless, the absence of endogenous cathelicidin precludes NK cells from optimal killing activity and reveals its complex role in NK cell biology.
Collectively, our results clearly show that Camp−/− mice permitted increased growth of two different murine xenograft tumor models that are relevant for human cancer and that these mice have deficient NK-mediated target cell killing. We envision at least two mechanisms by which cathelicidins may control tumor progression. One possibility is that cathelicidin functions as a cytotoxic membrane-permeating molecule acting with perforin and granzyme to limit cell growth. It should be noted that the concentration of cathelicidin required to inhibit tumor cell growth in vitro (128 μM; Fig. 2) is much higher than what is needed to kill bacteria. Thus, we speculate that cathelicidin must act in concert with other proteins released into the NK cell–target cell synapse to mediate its cytotoxic effect in vivo. The second mechanism by which cathelicidin could control NK-dependent tumor surveillance is via its regulation of gene expression, as suggested by altered levels of perforin, bcl-2, granzyme B, and SPI-6. Future studies will determine the exact nature of these novel functions of cathelicidin.
Our findings suggest that cathelicidin has a previously unrecognized role in NK cell biology and antitumor function. Because cathelicidin expression in humans is strongly influenced by vitamin D (67), and this nutritional requirement has been associated with a protective effect in several types of cancer (68), it is attractive to speculate that the proposed tumor-protective function of vitamin D may be due in part to the induction of cathelicidin as a tumor-protective host effector molecule. However, it is also reasonable to speculate that in some tumors (ovarian) that rely on the angiogenic properties of cathelicidins (22), or epithelial cancers, the increased cathelicidin could have detrimental effects. Thus, it will be important to further study the mechanisms responsible for enhanced tumor growth in Camp−/− mice and to investigate whether cathelicidin deficiency contributes to progression or regression of specific tumors in humans. Such findings could lead to novel therapeutic approaches for those tumors that may benefit from enhancement of NK cell activity.
Acknowledgments
This work was supported by a Veterans Affairs merit award and National Institutes of Health Grants AI052453 and AR45676 (to R.L.G.), by grants from the American Cancer Society, Cancer Research Coordinating Committee, V Foundation, Concern Foundation, and National Institutes of Health Grant CA128893 (to J.D.B.), by a grant (BMBF-LPD 9901/8-119) from the Deutsche Akademie der Naturforscher Leopoldina (to J.S.), and by a grant (BU 2212/1-1) from the Deutsche Forschungsgemein-schaft (to A.S.B.).
We thank the Flow Cytometry Research Core Facility of the VA San Diego Healthcare System for expert technical help in FACS sorting and Annexin V/PI staining. We thank the Histology Core Facility at UCSD for H&E. We also thank B. Brinkman from the Light Microscopy Facility of UCSD for help in confocal imaging. For help with SELDI-TOF-MS, we thank Dr. E. H. Koo and B. Cottrell from the Department of Neuroscience at UCSD and K. Yamasaki from our laboratory (Division of Dermatology, Department of Medicine, UCSD).
Abbreviations in this paper
- BM
bone marrow
- De
dermis
- Epi
epidermis
- HF
hair follicle
- LAK
lymphokine-activated killer
- LDH
lactatedehydrogenase
- mCRAMP
murine cathelicidin peptide
- MHC-I
MHC class I
- MM
melanoma
- PI
propidium iodide
- poly(I:C)
polyinosinic:polycytidylic acid
- SELDI-TOF-MS
surface-enhanced laser desorption ionization time-of-flight mass spectroscopy
- SPI-6
serine protease inhibitor 6
- Tx
Triton X-100
- UCSD
University of California San Diego
- wt
wild type
Footnotes
Disclosures
The authors declare no competing financial interests.
References
- 1.Bui JD, Schreiber RD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol. 2007;19:203–208. doi: 10.1016/j.coi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-γ-induced microRNA. J Immunol. 2009;182:39–43. doi: 10.4049/jimmunol.182.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smyth MJ, Swann J, Hayakawa Y. Innate tumor immune surveillance. Adv Exp Med Biol. 2007;590:103–111. doi: 10.1007/978-0-387-34814-8_7. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 5.Watzl C, Long EO. Exposing tumor cells to killer cell attack. Nat Med. 2000;6:867–868. doi: 10.1038/78624. [DOI] [PubMed] [Google Scholar]
- 6.Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- 7.Hayakawa Y, Kelly JM, Westwood JA, Darcy PK, Diefenbach A, Raulet D, Smyth MJ. Cutting edge: tumor rejection mediated by NKG2D receptor-ligand interaction is dependent upon perforin. J Immunol. 2002;169:5377–5381. doi: 10.4049/jimmunol.169.10.5377. [DOI] [PubMed] [Google Scholar]
- 8.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, et al. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 9.Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, Capanni M, Umansky V, Paschen A, Sucker A, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119:1251–1263. doi: 10.1172/JCI36022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 11.van den Broek MF, Kägi D, Zinkernagel RM, Hengartner H. Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol. 1995;25:3514–3516. doi: 10.1002/eji.1830251246. [DOI] [PubMed] [Google Scholar]
- 12.van den Broek ME, Kägi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J Exp Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- 14.Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem. 1996;238:325–332. doi: 10.1111/j.1432-1033.1996.0325z.x. [DOI] [PubMed] [Google Scholar]
- 15.Gallo RL, Kim KJ, Bernfield M, Kozak CA, Zanetti M, Merluzzi L, Gennaro R. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem. 1997;272:13088–13093. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 16.Chromek M, Slamová Z, Bergman P, Kovács L, Podracká L, Ehrén I, Hökfelt T, Gudmundsson GH, Gallo RL, Agerberth B, Brauner A. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12:636–641. doi: 10.1681/01.asn.0000926856.92699.53. [DOI] [PubMed] [Google Scholar]
- 17.Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol. 2006;117:836–841. doi: 10.1016/j.jaci.2005.12.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberger CM, Gallo RL, Finlay BB. Interplay between antibacterial effectors: a macrophage antimicrobial peptide impairs intracellular Salmonella replication. Proc Natl Acad Sci USA. 2004;101:2422–2427. doi: 10.1073/pnas.0304455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. 2005;174:4901–4907. doi: 10.4049/jimmunol.174.8.4901. [DOI] [PubMed] [Google Scholar]
- 21.Chuang CM, Monie A, Wu A, Mao CP, Hung CF. Treatment with LL-37 peptide enhances antitumor effects induced by CpG oligodeoxynucleotides against ovarian cancer. Hum Gene Ther. 2009;20:303–313. doi: 10.1089/hum.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, LaMarca HL, Tomchuck SL, Honer zu Bentrup K, Danka ES, Henkle SL, Scandurro AB. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci USA. 2009;106:3806–3811. doi: 10.1073/pnas.0900244106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coffelt SB, Waterman RS, Florez L, Höner zu Bentrup K, Zwezdaryk KJ, Tomchuck SL, LaMarca HL, Danka ES, Morris CA, Scandurro AB. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int J Cancer. 2008;122:1030–1039. doi: 10.1002/ijc.23186. [DOI] [PubMed] [Google Scholar]
- 24.Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jörnvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and α-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086–3093. [PubMed] [Google Scholar]
- 25.Gutsmann T, Hagge SO, Larrick JW, Seydel U, Wiese A. Interaction of CAP18-derived peptides with membranes made from endotoxins or phospholipids. Biophys J. 2001;80:2935–2945. doi: 10.1016/S0006-3495(01)76259-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 27.Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells: specificity and distribution according to genotype. Eur J Immunol. 1975;5:112–117. doi: 10.1002/eji.1830050208. [DOI] [PubMed] [Google Scholar]
- 28.Dorschner RA, V, Pestonjamasp K, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 2001;117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- 29.Cannarile MA, Lind NA, Rivera R, Sheridan AD, Camfield KA, Wu BB, Cheung KP, Ding Z, Goldrath AW. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol. 2006;7:1317–1325. doi: 10.1038/ni1403. [DOI] [PubMed] [Google Scholar]
- 30.Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL. Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology. 2006;118:509–519. doi: 10.1111/j.1365-2567.2006.02399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Nardo A, Yamasaki K, Dorschner RA, Lai Y, Gallo RL. Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. J Immunol. 2008;180:7565–7573. doi: 10.4049/jimmunol.180.11.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang L, Gusewitch GA, Chritton DB, Folz JC, Lebeck LK, Nehlsen-Cannarella SL. Rapid flow cytometric assay for the assessment of natural killer cell activity. J Immunol Methods. 1993;166:45–54. doi: 10.1016/0022-1759(93)90327-4. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg JE, Sherwood SW, Clayberger C. A novel method for measuring CTL and NK cell-mediated cytotoxicity using annexin V and two-color flow cytometry. J Immunol Methods. 1999;224:1–9. doi: 10.1016/s0022-1759(98)00038-6. [DOI] [PubMed] [Google Scholar]
- 34.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 35.Kane KL, Ashton FA, Schmitz JL, Folds JD. Determination of natural killer cell function by flow cytometry. Clin Diagn Lab Immunol. 1996;3:295–300. doi: 10.1128/cdli.3.3.295-300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Büchau AS, Gallo RL. Innate immunity and antimicrobial defense systems in psoriasis. Clin Dermatol. 2007;25:616–624. doi: 10.1016/j.clindermatol.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schauber J, Dorschner RA, Coda AB, Büchau AS, Liu PT, Kiken D, Helfrich YR, Kang S, Elalieh HZ, Steinmeyer A, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117:803–811. doi: 10.1172/JCI30142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dumitru CD, Antonysamy MA, Gorski KS, Johnson DD, Reddy LG, Lutterman JL, Piri MM, Proksch J, McGurran SM, Egging EA, et al. NK1.1+ cells mediate the antitumor effects of a dual Toll-like receptor 7/8 agonist in the disseminated B16-F10 melanoma model. Cancer Immunol Immunother. 2009;58:575–587. doi: 10.1007/s00262-008-0581-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci USA. 2000;97:2731–2736. doi: 10.1073/pnas.050588297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aguiló JI, Garaude J, Pardo J, Villalba M, Anel A. Protein kinase C-θ is required for NK cell activation and in vivo control of tumor progression. J Immunol. 2009;182:1972–1981. doi: 10.4049/jimmunol.0801820. [DOI] [PubMed] [Google Scholar]
- 42.Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, Yokoyama WM. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. 2002;3:523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- 43.Piontek GE, Taniguchi K, Ljunggren HG, Grönberg A, Kiessling R, Klein G, Kärre K. YAC-1 MHC class I variants reveal an association between decreased NK sensitivity and increased H-2 expression after interferon treatment or in vivo passage. J Immunol. 1985;135:4281–4288. [PubMed] [Google Scholar]
- 44.Chun M, Hoffmann MK. Modulation of interferon-induced NK cells by interleukin 2 and cAMP. Lymphokine Res. 1982;1:91–98. [PubMed] [Google Scholar]
- 45.Lotzová E, Savary CA. Interleukin-2 corrects defective NK activity of patients with leukemia. Comp Immunol Microbiol Infect Dis. 1986;9:169–175. doi: 10.1016/0147-9571(86)90009-3. [DOI] [PubMed] [Google Scholar]
- 46.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 47.Dennison SR, Harris F, Phoenix DA. The interactions of aurein 1.2 with cancer cell membranes. Biophys Chem. 2007;127:78–83. doi: 10.1016/j.bpc.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 48.Ohsaki Y, Gazdar AF, Chen HC, Johnson BE. Antitumor activity of magainin analogues against human lung cancer cell lines. Cancer Res. 1992;52:3534–3538. [PubMed] [Google Scholar]
- 49.Okumura K, Itoh A, Isogai E, Hirose K, Hosokawa Y, Abiko Y, Shibata T, Hirata M, Isogai H. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett. 2004;212:185–194. doi: 10.1016/j.canlet.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 50.Wong JH, Ng TB. Sesquin, a potent defensin-like antimicrobial peptide from ground beans with inhibitory activities toward tumor cells and HIV-1 reverse transcriptase. Peptides. 2005;26:1120–1126. doi: 10.1016/j.peptides.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 51.Papo N, Shai Y. New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry. 2003;42:9346–9354. doi: 10.1021/bi027212o. [DOI] [PubMed] [Google Scholar]
- 52.Coffelt SB, Scandurro AB. Tumors sound the alarmin(s) Cancer Res. 2008;68:6482–6485. doi: 10.1158/0008-5472.CAN-08-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber G, Chamorro CI, Granath F, Liljegren A, Zreika S, Saidak Z, Sandstedt B, Rotstein S, Mentaverri R, Sánchez F, et al. Human antimicrobial protein hCAP18/LL-37 promotes a metastatic phenotype in breast cancer. Breast Cancer Res. 2009;11:R6. doi: 10.1186/bcr2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Yang Q, Chen AP, Schmidt GM, Anderson JM, Wang J, Wooters JJ, Oppenheim, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elssner A, Duncan M, Gavrilin M, Wewers MD. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 β processing and release. J Immunol. 2004;172:4987–4994. doi: 10.4049/jimmunol.172.8.4987. [DOI] [PubMed] [Google Scholar]
- 56.Koczulla R, von Degenfeld G, Kupatt C, Krötz F, Zahler S, Gloe T, Issbrücker K, Unterberger P, Zaiou M, Lebherz C, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665–1672. doi: 10.1172/JCI17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–6265. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 58.Braff MH, Di Nardo A, Gallo RL. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J Invest Dermatol. 2005;124:394–400. doi: 10.1111/j.0022-202X.2004.23443.x. [DOI] [PubMed] [Google Scholar]
- 59.Di Nardo A, Braff MH, Taylor KR, Na C, Granstein RD, McInturff JE, Krutzik S, Modlin RL, Gallo RL. Cathelicidin antimicrobial peptides block dendritic cell TLR4 activation and allergic contact sensitization. J Immunol. 2007;178:1829–1834. doi: 10.4049/jimmunol.178.3.1829. [DOI] [PubMed] [Google Scholar]
- 60.Pütsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet. 2002;360:1144–1149. doi: 10.1016/S0140-6736(02)11201-3. [DOI] [PubMed] [Google Scholar]
- 61.Di Nardo A, Vitiello A, Gallo RL. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J Immunol. 2003;170:2274–2278. doi: 10.4049/jimmunol.170.5.2274. [DOI] [PubMed] [Google Scholar]
- 62.Djeu JY, Heinbaugh JA, Holden HT, Herberman RB. Role of macrophages in the augementation of mouse natural killer cell activity by poly I: C and interferon. J Immunol. 1979;122:182–188. [PubMed] [Google Scholar]
- 63.Djeu JY, Heinbaugh JA, Vieira WD, Holden HT, Herberman RB. The effect of immunopharmacological agents on mouse natural cell-mediated cytotoxicity and on its augmentation by poly I:C. Immunopharmacology. 1979;1:231–244. doi: 10.1016/0162-3109(79)90040-7. [DOI] [PubMed] [Google Scholar]
- 64.Schmidt KN, Leung B, Kwong M, Zarember KA, Satyal S, Navas TA, Wang F, Godowski PJ. APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J Immunol. 2004;172:138–143. doi: 10.4049/jimmunol.172.1.138. [DOI] [PubMed] [Google Scholar]
- 65.Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, Pipkin M, Lichtenheld M, Richardson B. DNA methylation and chromatin structure regulate T cell perforin gene expression. J Immunol. 2003;170:5124–5132. doi: 10.4049/jimmunol.170.10.5124. [DOI] [PubMed] [Google Scholar]
- 66.Pipkin ME, Ljutic B, Cruz-Guilloty F, Nouzova M, Rao A, Zúñiga-Pflücker JC, Lichtenheld MG. Chromosome transfer activates and delineates a locus control region for perforin. Immunity. 2007;26:29–41. doi: 10.1016/j.immuni.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 67.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. [Published erratum appears in 2004 J. Immunol. 173: 6489.] J Immunol. 2004;173:2909–2912. doi: 10.4049/jimmunol.173.5.2909. [DOI] [PubMed] [Google Scholar]
- 68.Garland CF, Gorham ED, Mohr SB, Garland FC. Vitamin D for cancer prevention: global perspective. Ann Epidemiol. 2009;19:468–483. doi: 10.1016/j.annepidem.2009.03.021. [DOI] [PubMed] [Google Scholar]