

Abstract
Background
Mutations in the LMNA gene, encoding lamins A/C, represent a significant cause of dilated cardiomyopathy (DCM). We recently identified 18 protein-altering LMNA variants in a cohort of 324 unrelated patients with DCM. However, at least one family member with DCM in each of six pedigrees lacked the LMNA mutation (nonsegregation), while small sizes of five additional families precluded definitive determinations of segregation, raising questions regarding contributions by those variants to disease.
Methods and Results
We have, consequently, expressed, in COS7 cells, GFP-prelamin A (GFPLaA) fusion constructs incorporating the six variants in pedigrees with nonsegregation (R101P, A318T, R388H, R399C, S437Hfsx1, and R654X), the four variants in pedigrees with unknown segregation [R89L, R166P (in 2 families), I210S, R471H], and three additional missense variants (R190Q, E203K, L215P) that segregated with disease. Confocal immunofluorescence microscopy was used to characterize GFP-lamin A localization and nuclear morphology. Abnormal phenotypes were observed for 10/13 (77%) variants (R89L, R101P, R166P, R190Q, E203K, I210S, L215P, R388H, S437Hfsx1, R654X), including 4/6 demonstrating nonsegregation and 3/4 with uncertain segregation. All seven variants affecting coil 1B, and the lamin A-only mutation, R654X, exhibited membrane-bound GFP-lamin A aggregates and nuclear shape abnormalities. Unexpectedly, R388H largely restricted GFP-lamin A to the cytoplasm. Equally unexpected were unique streaked aggregates with S437Hfsx1, and giant aggregates with both S437Hfsx1 and R654X.
Conclusions
This work expands the recognized spectrum of lamin A localization abnormalities in DCM. It also provides evidence supporting pathogenicity of 10 of 13 tested LMNA variants, including some with uncertain or nonsegregation.
Keywords: dilated cardiomyopathy, genetics, lamin A/C
Introduction
The lamins are a family of intermediate filament proteins which localize to the inner nuclear envelope where they perform a number of crucial functions involving nuclear architecture, gene expression, mitosis, DNA replication, apoptosis, and signaling (reviewed in 1, 2). The major A-type lamins, lamins A and C, are produced through differential splicing of the 12 exon LMNA gene (1q21.2-q21.3). Since the alternative splice site is located in exon 10, lamin A and C transcripts encode proteins which differ along only a short C-terminal segment. The N-termini represent a series of alpha-helical coiled-coil domains necessary for lamin polymerization.3 The C-terminal tail domain, in contrast, adopts an immunoglobulin (Ig)-like structure and houses binding sites for DNA, chromatin, and other lamin-associated polypeptides (LAPs).4 Together, these domains permit the array of interactions necessary for maintaining integrity of the lamina.
Production of a mature lamin A polypeptide (664aa) necessitates a series of posttranslational modifications targeted to the prelamin A C-terminal CaaX motif, which is lacking in the truncated lamin C (572aa). Although the precise functions of prelamin A processing remain unknown, the sequential modifications may facilitate interactions with other lamina proteins or with the nuclear membrane.5
LMNA mutations have been implicated in at least eight distinct clinical phenotypes (laminopathies). Although recognized as unique entities, reports of patients6 or families7 exhibiting features of more than one disease and of individual mutations resulting in multiple laminopathies8, 9 suggest that the these diseases may be better considered as a phenotypic spectrum. Forms of muscular dystrophy with or without cardiac involvement [Autosomal Dominant Emery Dreifuss Muscular Dystrophy (AD-EDMD), Limb-Girdle Muscular Dystrophy Type 1B (LGMD1B)], diseases of adipose tissue and fat deposition [Familial Partial Lipodystrophy - Dunnigan type (FPLD), Mandibuloacral Dysplasia (MAD)], Restrictive Dermopathy (RD), Charcot Marie Tooth Disease Type 2 (CMT2), and premature aging syndromes such as Hutchinson-Gilford Progeria Syndrome (HGPS) and atypical Werner’s Syndrome (WS), are all part of the spectrum. LMNA mutations additionally represent the most frequent known genetic cause of dilated cardiomyopathy (DCM), occurring with a prevalence of ∼5–10% (familial) and 2–5% (sporadic).10 Missense mutations predominate; however, rare deletions, insertions, frameshifts, and nonsense mutations have all been reported.
Clinically, LMNA-related DCM is characterized by much inter- and intra-familial variability in onset and severity, but typically manifests as left ventricular enlargement (LVE) and reduced systolic function preceded by significant conduction system disease (CSD), particularly atrioventricular block and supraventricular arrhythmias. Sudden cardiac death (SCD) is also common and can represent the initial sign of disease (see 11,12 for review).
We recently identified 18 unique protein-altering LMNA variants in 19 probands from a cohort of 324 unrelated patients with idiopathic (non-ischemic DCM of unknown cause) or familial DCM.10 Identified variants showed usual patterns of age-dependent segregation with disease in many of the larger families (segregation pedigrees), supporting the pathogenicity of these mutations. However, the small sizes of other families precluded definitive assessments of segregation (unknown segregation pedigrees). We additionally observed that in six of the 19 families (32%), at least one family member with clinically evident DCM lacked the putatively causative LMNA variant (nonsegregation pedigrees), raising questions regarding the contribution of these variants to disease.10 A large number of studies have demonstrated abnormalities in nuclear morphology and lamin A/C localization in cells expressing LMNA variants.13–21 Therefore, in order to better determine their pathogenic potential, we generated GFP-prelamin A fusion constructs corresponding to 13 of the 18 identified LMNA variants (including variants lacking definitive segregation data) and constitutively expressed each in COS7 cells. These studies complement available molecular, family, and/or clinical data10, 22, 23 by providing evidence supporting pathogenicity for 10 of the 13 analyzed LMNA variants.
Materials and Methods
Plasmid Construction
Full-length human prelamin A (664 amino acids) was generated from HEK293 total RNA extract using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Applied Science) and was cloned into the pAcGFP1-C1 fluorescent protein expression vector (Clontech) using manufacturer’s protocols for the In-Fusion 2.0 Dry-down Kit (Clontech). The In-Fusion Primer Design Tool (http://bioinfo.clontech.com/infusion/convertPcrPrimersInit.do) was used to design sense (5’-GGACTCAGATCTCGACTGCCGGCCATGGAGAC-3’) and antisense (5’-GATCCCGGGCCCGCGGCCTGGCAGGTCCCAGAT-3’) primers. Vector linearization was accomplished using Kpn1 and Xho1. The cloning reaction was performed with a vector:insert molar ratio of 1:2 and the resulting wild type GFP-prelamin A fusion construct (GFPLaA-WT) was transformed into One Shot TOP10 E. coli (Invitrogen). Construct fidelity was confirmed first by restriction analysis with BglII and BamHI, and subsequently by dye-terminator sequencing using the ABI 3100 Automated Capillary DNA Sequencer (Applied Biosystems).
Mutagenesis
Thirteen mutant constructs were generated from GFPLaA-WT following manufacturer’s protocols for the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). Mutations and their flanking sequences were confirmed as above. Constructs generated included the six variants in pedigrees with nonsegregation (R101P, A318T, R388H, R399C, S437Hfsx1, and R654X) and an additional 7 variants (R89L, R166P, R190Q, G203K, I210S, L215P, R471H) representing all remaining missense variants in the cohort. To focus research efforts, analyses were restricted to these 13 variants, as the remaining five variants were considered likely to cause significant disruption of the LMNA gene [a splice site variant (357-1G>T) caused the loss of exon 210, two were nonsense mutations (R225X, Q234X), one was a frameshift mutation (E372RfsX107), and one was an insertion (D475insE) mutation], and clinical data were consistent with their pathogenicity10.
Cell Culture, Transfection, and Confocal Immunofluorescence Microscopy
COS7 (African green monkey kidney) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) + GlutaMAX (Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic (Gibco) at 37°C in a 5% CO2 water-jacketed incubator. The COS7 cell line was selected for its high transfectability and demonstrated utility in similar lamin A/C morphological studies.21 Because wildtype lamin A overexpression has been shown to form aggregates similar to those observed in some mutant samples,14, 17 transfection conditions were optimized to minimize aggregation of overexpressed GFP-lamin A in GFPLaA-WT samples prior to assessment of mutant constructs. Transfections were conducted using Lipofectamine 2000 reagent (Invitrogen) scaled for culture in 35mm diameter, 10mm microwell glass-bottom dishes (MatTek Corporation). Twenty-four hours prior to transfection, 400,000 cells were seeded in growth medium lacking antibiotic/microcytic and were cultured overnight to 80–90% confluency. Plasmid DNA (0.5ug) was complexed with lipofectamine in a ratio of 1:2.5. Transfection proceeded for 24 hours at 37°C.
The same investigator (JC) completed all experiments. A second investigator (DL) assigned unique identifiers prior to transfection, blinding the first investigator to all sample identities. The first investigator remained blinded until all images were acquired, qualified, and analyzed.
Prior to image acquisition, overnight culture media was aspirated and replaced with 1mL phenol-red free and antibiotic/antimycotic enriched DMEM and supplemented with 10uL Hoechst 33258 (0.2mM) nuclear stain. Images were acquired with a Zeiss LSM510/UV confocal microscope outfitted with a C-Apochromat 63x/1.2 W Corr water immersion objective lens, and separate UV (351nm) and GFP (488nm) emission filters. During acquisition fluorescent nuclei were qualitatively classified by observed nuclear morphology. Results of independent experiments were amalgamated for analysis. Post-acquisition image processing was accomplished with AxioVision 4.7 software (Zeiss Microimaging Inc.).
Statistical Analyses
Numbers of aggregate-containing and abnormally shaped nuclei for each variant LMNA construct were compared to corresponding wildtype counts using two-tailed Chi-Square (all cell counts >5) or Fisher’s Exact Tests (any cell count <=5). Graphpad InStat 3 statistical software (http://www.graphpad.com/) was used for all analyses.
Results
Clinical Data
Clinical and pedigree data from clinically affected individuals with confirmed or obligate LMNA mutations previously published by our group10, 22, 23 are summarized for each family (see Table 1, Figure 1).
Table 1.
Clinical characteristics of clinically affected LMNA variant carriers
| Individuals* | n | Family | Amino Acid |
Nucleotide | Disease Onset |
AV Block (n%) |
Aflut/A fib (n%) |
CS Other (n%) |
PCM /ICD (n%) |
SCD (n%) |
DCM (n%) |
HF (n%) |
HrtTx (n%) |
CSD† | DCM‡ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A.3 | 1 | A | R89L | 266G>T | 31 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | +++ | +++ |
| B.8* | 1 | B | R101P | 302G>C | 36 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | +++ | +++ |
| D.3 | 1 | D | R166P | 497G>C | 40 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | +++ | +++ |
| E.3 | 1 | E | R166P | 497G>C | 42 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | +++ | +++ |
| F.6, F.7, F.8 | 3 | F | R190Q | 569G>A | 26–45 | 1 (33) | 0 | 2 (67) | 0 | 0 | 1 (33) | 1 (33) | 1 (33) | ++ | +++ |
| G.3, G.5, G.8, G.10, G.14, G.15, G.16, G.18 G.19, G.21, G.23 |
11 | G | E203K | 607G>A | 30–46 | 8 (73) | 4 (36) | 5 (45) | 3 (27) | 0 | 3 (27) | 2 (18) | 1 (9) | +++ | +++ |
| H.3 | 1 | H | I210S | 629T>G | 45 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | +++ | +++ |
| I.2, I.4, I.5, I.10, I.13, I.16, I.18, I.19, I.20, I.21, I.22, I.26 |
12 | I | L215P | 644T>C | 25-<58 | 4 (33) | 5 (42) | 8 (67) | 7 (58) | 1 (8) | 7 (58) | 3 (25) | 0 | +++ | +++ |
| L.3* | 1 | L | A318T | 952G>A | 41 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | + | +++ |
| N.8*, N.9* N.15* |
3 | N | R388H | 1163G>A | 31–45 | 0 | 0 | 1 (33) | 0 | 1 (33) | 3 (100) | 2 (67) | 0 | +++ | +++ |
| O.3 | 1 | O | R399C | 1195C>T | 76 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | + | None |
| O.11* | 1 | 15 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | None | +++ | |||
| P.1, P.3, P.8, P.10, P.14, P.16, P.18, P.20 |
8 | P | S437Hf sx1 |
1307_1308i nsGCAC |
27–56 | 3 (34) | 3 (34) | 5 (62.5) | 4 (50) | 3 (34) | 3 (34) | 1 (13) | 1 (13) | +++ | +++ |
| P.23* | 1 | 22 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | ++ | +++ | |||
| Q.2 | 1 | Q | R471H | 1412G>A | 44 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | +++ | +++ |
| S.5, S.6 | 2 | S | R654X | 1960C>T | 62–65 | 1 (50) | 2 (100) | 1 (50) | 1 (50) | 1 (50) | 1 (50) | 1 (50) | 0 | +++ | +++ |
| S.9*, S.11* | 2 | 39–47 | 1 (50) | 1 (50) | 2 (100) | 2 (100) |
1 (50) | 2 (100) | 1 (50) | 0 | +++ | +++ |
N = # of clinically affected individuals. For the 6 families demonstrating LMNA variant nonsegregation, individuals marked with a * represent LMNA mutation carriers who are also at-risk for carrying a possible second causative genetic variant.
+++ = severe CSD characterized by ICD/PM placement or SCD, ++ = any degree AVB, AF, or Afib. + = any other conduction system abnormality.
+++ = severe IDC/FDC characterized by HF or transplantation, ++ = IDC/FDC without HF or transplantation, + = LVE or systolic dysfunction not meeting criteria for IDC/FDC24
Figure 1.


Partial pedigrees for families with LMNA variants. Numbering is consistent with tables and figures in this study and with past reports of these families (see 10, 22, 23 for clinical data). Probands are indicated with an arrow. Solid symbols represent IDC with or without heart failure. Shaded symbols represent any other cardiovascular abnormality. Open symbols indicate unaffected individuals. Mutation carrier status is shown by a + (presence), (+) (obligate) or − (absence). Absence of any symbol for mutation carrier status indicates lack of available DNA for analysis. Question marks (?) denote insufficient clinical data. Figure 1A: Pedigrees with nonsegregation. Figure 1B: Pedigrees with segregation or unknown segregation.
Morphological Data
Full-length wildtype and mutant prelamin A cDNAs were transiently expressed as GFP fusion constructs in COS7 cells. Each construct was analyzed by confocal immunofluorescence microscopy to determine nuclear morphology and GFP-lamin A localization. Results are summarized (Table 2, Table 3) and representative images are provided (Figure 2).
Table 2.
Statistical analyses for wildtype vs. variant LMNA constructs
|
LMNA Construct |
Total Cells (n)* |
# Nuclei with Aggregates (% n) |
p-value; Test† | # Nuclei with Abnormal Shape (% n) |
p-value; Test† |
|---|---|---|---|---|---|
| Wildtype (WT) | 415 | 22 (5) | N/A | 67 (16) ‡ | N/A |
| R89L | 108 | 106 (98) | <0.0001; FE | 32 (30) | 0.0023; CS |
| R101P | 84 | 83 (99) | <0.0001; FE | 55 (65) | <0.0001; CS |
| R166P | 95 | 92 (97) | <0.0001; FE | 52 (55) | <0.0001; CS |
| R190Q | 142 | 55 (39) | <0.0001; CS | 38 (27) | 0.0076; CS |
| E203K | 151 | 25 (17) | <0.0001; CS | 36 (24) | 0.0481; CS |
| I210S | 138 | 134 (97) | <0.0001; FE | 72 (52) | <0.0001; CS |
| L215P | 154 | 99 (64) | <0.0001; CS | 75 (49) | <0.0001; CS |
| A318T | 136 | 4 (3) | 0.3529; FE | 27 (20) | 0.3863; CS |
| R388H* | N/A | N/A | N/A | N/A | N/A |
| R399C | 143 | 8 (6) | 0.8934; CS | 28 (20) | 0.4158; CS |
| S437Hfsx1 | 105 | 105 (100) | <0.0001; FE | 11 (10) | 0.1935 ; CS |
| R471H | 115 | 9 (8) | 0.4258; CS | 25 (22) | 0.2068; CS |
| R654X | 114 | 108 (95) | <0.0001; CS | 15 (13) | 0.5259; CS |
Pooled data were derived from at least two independent experiments. The predominant cytoplasmic localization of GFP-R388H precluded statistical analysis.
CS = Chi-square test, FE = Fisher’s exact test.
Determinations of normal nuclear morphology were conservatively estimated, so some of this 16% likely represents normal variation.
Table 3.
Nuclear morphology and GFP-lamin A localization patterns for confocally imaged wildtype and variant LMNA constructs
|
LMNA Construct |
Family | Nuclear Morphology* | Lamin A Localization | Supports mutation pathogenicity? |
|---|---|---|---|---|
| Wildtype (WT) |
N/A | Smooth-edged, circular NE (∼85%). Mild nuclear shape abnormalities (∼15%) |
Homogenous along nuclear periphery (95%), Small NE associated agg. (5%) |
N/A |
| R89L | A | Mild to mod. abnormal nuclear shapes (∼30%) |
Small NE associated agg. (98%), homogenous along nuclear periphery (2%) |
Yes |
| R101P | B | Mod. to grossly abnormal nuclear shapes (∼65%) |
Mod. sized NE associated agg. (99%), Homogenous along nuclear periphery (1%) |
Yes |
| R166P | D/E | Mild to mod. abnormal nuclear shapes (∼55%) |
Small NE associated agg. (97%), homogenous distribution along nuclear periphery (3%) |
Yes |
| R190Q | F | Mild to mod. abnormal nuclear shapes (∼25%) |
Homogenous along nuclear periphery (61%), Small to mod. (rare) sized NE associated agg. (39%) |
Yes |
| E203K | G | Comparable to WT | Homogenous along nuclear periphery (83%). NE associated agg. (17%) |
Yes |
| I210S | H | Mod. to grossly abnormal nuclear shapes (∼50%) |
Mod. sized NE associated agg. (97%), Homogenous along nuclear periphery (3%) |
Yes |
| L215P | I | Mod. to grossly abnormal nuclear shapes (∼50%) |
Mod. sized NE associated agg. (64%), Homogenous along nuclear periphery (36%) |
Yes |
| A318T | L | Comparable to WT | Comparable to WT | No |
| R388H | N | Comparable to WT | 1) Diffuse, cytoplasmic 2) Large, nonspherical, signal-saturated cytoplasmic agg. 3) Intermediate to 1) and 2). 4) Diffuse, cytoplasmic with NE-associated agg. (rare) |
Yes |
| R399C | O | Comparable to WT. | Comparable to WT. | No |
| S437Hfsx1 | P | Comparable to WT | Variable agg. phenotypes (100%): giant agg. (∼50%), streaked agg. (∼43%), small agg. (∼35%) |
Yes |
| R471H | Q | Comparable to WT. | Comparable to WT | No |
| R654X | S | Comparable to WT | Small to giant agg. (95%, giant agg. in 5%), homogenous along nuclear periphery (5%) |
Yes |
NE = nuclear envelope.
Figure 2.
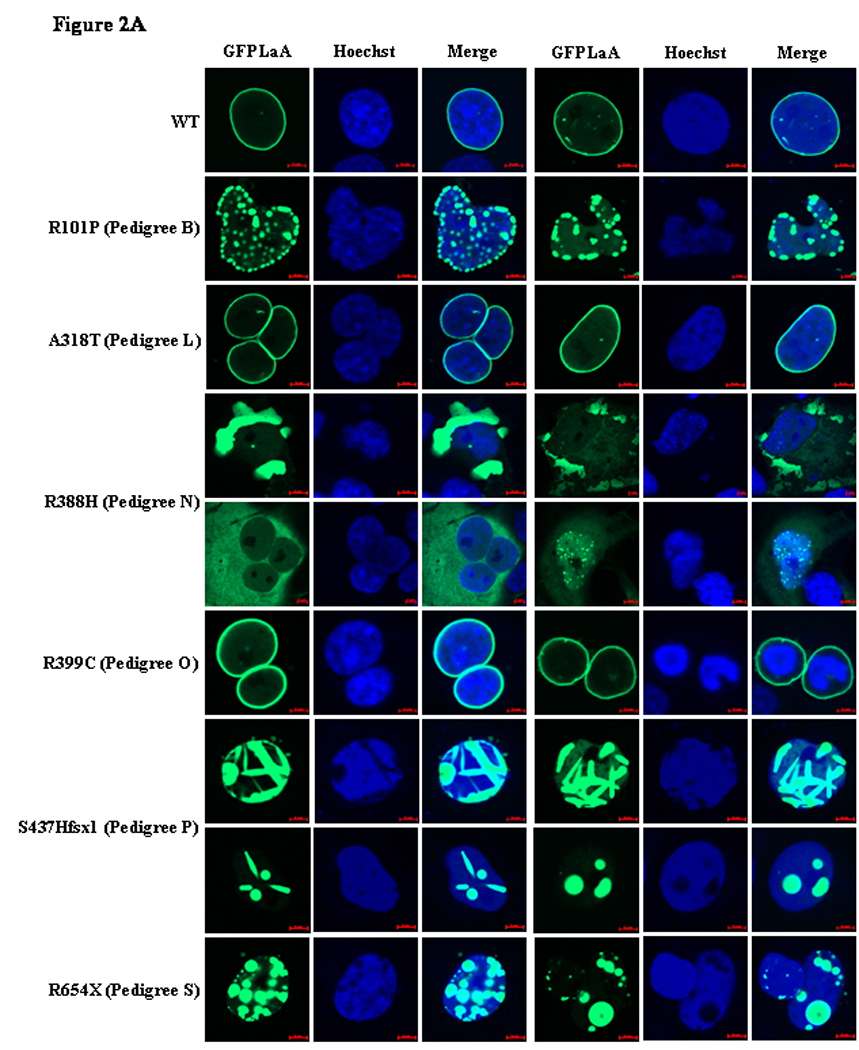
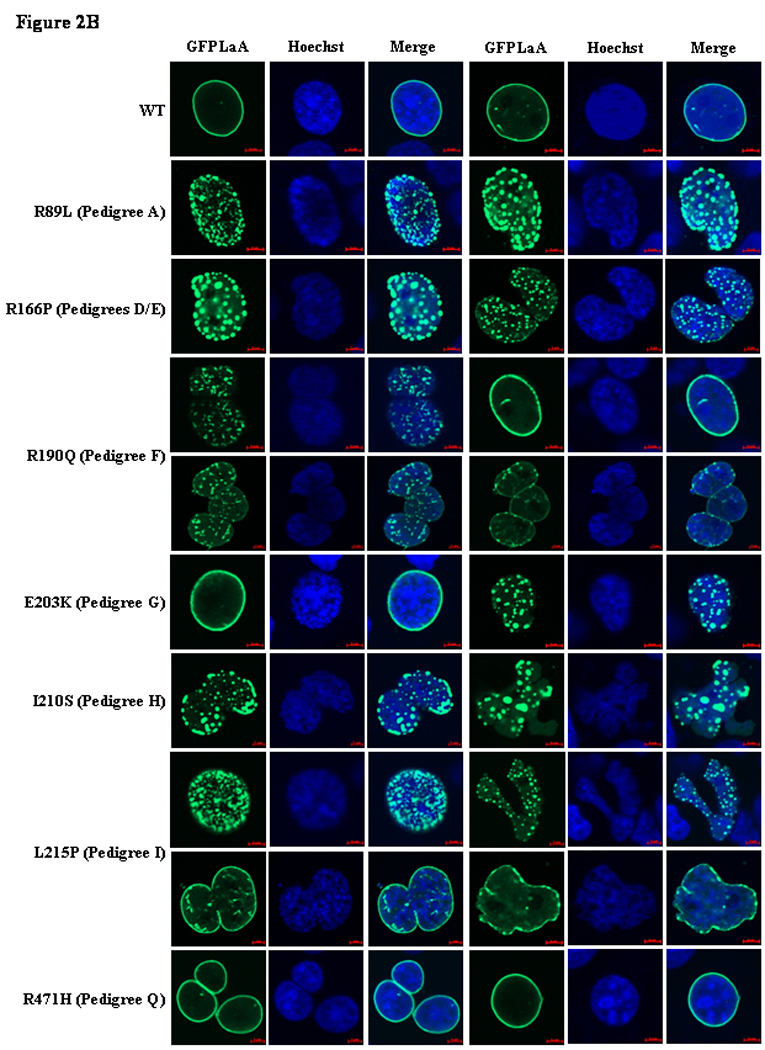
GFP-lamin A (GFPLaA) localization and nuclear morphology in COS7 cells transfected with wildtype/mutant fusion constructs and stained with Hoechst 33258. In all experiments, confocal immunofluorescence microscopy was performed 24 hours post-transfection. Scale bars are 5µm in length. Figure 2A. Representative images for the six LMNA variants in pedigrees with nonsegregation. Figure 2B. Representative images for the three variants with segregation and four variants with unknown segregation.
Nearly all nuclei in cells transfected with GFPLaA-WT exhibited homogenous GFP-lamin A localization throughout the nuclear periphery (95%). Rarely, nuclei contained small, nuclear envelope-associated aggregates (5%). Transfection with the 13 mutant constructs resulted in a number of GFP-lamin A localization phenotypes. The GFPLaA-A318T (Family L), -R399C (Family O), and -R471H (Family Q) constructs were morphologically comparable to GFPLaA-WT. A significantly greater proportion of nuclei with nuclear envelope-associated aggregates was observed with GFPLaA-E203K (Family G, 17%), and GFPLaA-R190Q (Family F, 39%) although the majority of nuclei remained comparable to GFPLaA-WT. Even more severe phenotypes, characterized by >50% aggregation levels and unique GFP-lamin A distribution patterns, were seen with the remaining constructs. Of these, rates of aggregation were lowest for GFPLaA-L215P (Family I, 64%), and significantly higher (>95%) for GFPLaA-R89L, -R101P, -R166P, and -I210S (Families A, B, D/E, and H, respectively). GFPLaA-R388H (Family N), GFPLaA-S437Hfsx1 (Family P), and GFPLaA-R654X (Family S) were most intriguing: GFPLaA-R388H was predominantly and dramatically restricted to the cytoplasm, either as a diffuse, low-fluorescence veil, or as highly saturated aggregates. Only very rarely was nuclear localization observed and, in these few instances, GFP-lamin A was as likely to form small aggregates as to remain homogenously distributed. Equally unexpected was the finding of unique streaked aggregates (43%) in the nuclei of S437Hfsx1 expressing cells, and the presence of giant aggregates in S437Hfsx1 (50%) and R654X (5%) expressing nuclei. Smaller aggregates were also prominent.
Consistent with previous reports,14–16, 19, 20, 25, 26 all coil 1B variants exhibited variable levels of aggregation. Compared to cells expressing GFPLaA-WT, significantly higher levels of mild to gross nuclear shape abnormalities, including nuclear envelope blebbing, were additionally observed. The extent and relative severity of these abnormalities are summarized in Table 3 and Figure 2.
GFPLaA-R399C (Family O) was uniquely notable for retraction of DNA from a clearly GFP-lamin A-demarcated nuclear lamina in rare transfected nuclei (see Figure 2). Though occurring in only a few cells, this phenotype was observed across multiple transfections and was considered to be an abnormal finding. Nevertheless, the vast majority of nuclei expressing GFPLaA-R399C, as well as all nuclei expressing GFPLaA-A318T (Family L) and GFPLaA-R471H (Family Q), were indistinguishable from wildtype.
Abnormal nuclear phenotypes were ultimately observed for 10/13 (77%) LMNA constructs (see Table 3), including 4/6 (67%) incorporating variants from pedigrees with nonsegregation and 3/4 (75%) incorporating variants from pedigrees with uncertain segregation, collectively supporting pathogenicity of these variants.
Morphological data for three LMNA variants (A318T, R399C, and R471H) were less revealing, with each demonstrating phenotypes comparable to wildtype, despite various associated manifestations of cardiovascular disease in carrier families. Families L (L.3) and Q (Q.2) both exhibited severe CSD and/or DCM (see Table 1). Two at-risk individuals in family Q (Q.4, Q.5) additionally demonstrated DCM/CSD of extremely early onset, with Q.5 notably suffering SCD at 18 years of age. Conversely, no symptoms were present in the mutation-positive mother of the proband in family O (O.9), while a maternal grandfather (O.3), who also carried the R399C mutation, exhibited only CSD. Paternally inherited DCM/CSD of unknown cause, however, was noteworthy, as was extremely aggressive, early onset DCM (requiring transplantation at 15 years) in the proband (O.11), who was also at risk of carrying a putative second, paternally inherited, causative genetic variant. These data suggest that the R399C variant may represent a low-risk allele acting in concert with an unknown paternal factor to cause the proband’s severe DCM. This mutation has been previously reported in a female patient with FPLD27; however, no evidence of DCM (or any other laminopathy) was present in that individual.
Discussion
To further delineate pathogenicity of LMNA variants previously identified in our DCM cohort10 we assessed nuclear morphology and GFP-lamin A localization in 13 variants. These included six variants in pedigrees, termed ‘nonsegregation’ pedigrees, where one or more family members with DCM did not carry the family variant, as well as four additional LMNA variants for which small family size (only one subject with DCM available for genetic analysis) precluded determination of segregation. These latter pedigrees were termed ‘pedigrees with unknown segregation’. Of the 13 variants, ten demonstrated abnormal GFP-lamin A localization and/or nuclear morphological abnormalities. Considered alongside clinical diagnoses of CSD and/or DCM in mutation carriers, these data provide evidence for pathogenicity of three of the four variants identified in families with unknown segregation and four of the six variants identified in families with nonsegregation. The absence of the variants observed in the nonsegregation pedigrees in 150 unrelated controls or in LMNA mutation databases (http://www.dmd.nl and http://www.umd.be:2000/IFAM.shtml), considered with the current morphological studies and prior molecular and pedigree data,10 argues for the existence of a second, unidentified, causative factor in clinically affected, but LMNA mutation-negative, family members. Whether these additional factors are genetically determined or environmentally imposed remains to be determined. However, the apparent heritable nature of DCM in some of the affected subjects who do not carry the LMNA variants (e.g. pedigrees N, S) suggests that a second genetic cause of DCM may be present in these families. Collectively, these data suggest a more complex basis for DCM in some multiplex pedigrees than has been previously appreciated (see also 10).
As previously described for this cohort,10 a number of mutation-positive individuals with no evidence of disease were present in several families (e.g. pedigrees G, I, and N), demonstrating age-dependent penetrance. Because LMNA-related DCM demonstrates age-dependent penetrance, (median 40.9 years in 10), this incomplete segregation is best explained by the relative youth of these individuals (23–41 years at time of assessment) rather than non-pathogenicity of the familial variant. We distinguish these individuals who show incomplete, age-dependent segregation (a LMNA mutation positive individual who has not yet manifest LMNA cardiomyopathy) from those subjects affected with DCM who do not carry the LMNA pedigree mutation, the latter of which are termed ‘nonsegregants’ in this and the prior10 work.
Since a number of previous studies have indicated that C-terminal mutations are significantly less likely to result in aggregation,19, 20, 26 the lack of an abnormal R399C nuclear phenotype does not preclude pathogenicity of this variant, nor of the similarly expressed A318T (Family L) and R471H (Family Q) variants. C-terminal missense mutations resulting in lamin A aggregation16, 17 have, nevertheless, been described. Caution is, consequently, necessary when considering results generated for these variants.
R388H, which borders the C-terminus of coil 2B and downstream non-helical regions, was unique among the missense variants studied. Unexpectedly, expressing cells predominantly demonstrated cytoplasmic GFP-lamin A localization and aggregation. The proximity of position 388 to the nuclear localization signal (NLS) at 416–42328 may offer some explanation for the dramatic localization defects; however, the fact that the R399C variant resulted in nuclear import indicates that at least a portion of the region proximal to the NLS has no influence on this process. An alternative explanation is loss of stable association with critical binding partners, such as LAPs, DNA, or chromatin. This hypothesis is consistent with the work of Strelkov et al.,29 which proposes that coil 2B is likely to interact with various lamin binding partners. Demonstration that the nearby R377H mutation resulted in cytoplasmic localization of the lamin B receptor (LBR)30 supports this proposal, as do fluorescence loss of intensity after photobleaching (FLIP) experiments demonstrating significantly increased mobility of R386K lamin A/C mutants.13
We additionally report two severe GFP-lamin A distribution defects resulting from non-missense genetic lesions. The first, a novel C-terminal S437Hfsx1 insertion and frameshift, unmasks a premature stop codon at position 438 leading to truncation of lamin A and lamin C by 227 and 135 amino acids, respectively. COS7 cells expressing this variant demonstrated a variety of defects, often within the same nucleus. Although ∼35% of nuclei exhibited small membrane-associated aggregates similar to those observed for the coil1B variants, unique streaked aggregates (∼43%) and giant aggregates (∼50%) were also prominent. The streaked aggregates, which varied in size, shape, and number between nuclei, are a novel finding. Giant aggregates have been observed in cells expressing exogenous mutant lamin C,13, 15 but have not, to our knowledge, been reported for lamin A.
The second lesion, a R654X nonsense mutation, truncates prelamin A by 11 amino acids, removing the conserved CaaX motif and elongating the mature protein by seven amino acids. This mutation was previously reported in a patient with HGPS and a homozygous null ZMPSTE24 mutation,31 and, notably, the mother and brother of that individual both displayed no signs of laminopathy, despite carrying the nonsense mutation. This variant appeared to be pathogenic in our cohort (Pedigree S), with carriers demonstrating significant CSD and DCM, including instances of SCD, PM/ICD placement, and HF. Ninety-five percent of expressing nuclei exhibited a variety of GFP-lamin A aggregates, including giant aggregates similar to those seen for S437Hfsx1(∼5%).
Although our study did not address pathogenic mechanisms, we can hypothesize that loss of critical binding domains for DNA, chromatin, and LAPs, may have resulted in the abnormal S437Hfsx1 aggregate phenotypes. Furthermore the inability of the truncated S437Hfsx1 and R654X transcripts to be post-translationally modified may also have contributed to disease, possibly through failure to translocate to the nuclear envelope5 and/or through accumulation of a toxic, incompletely processed precursor, as is seen in HGPS.32
Two laminopathy pathogenicity models have been proposed, each attempting to recognize both the phenotypic breadth of the laminopathies and the web of interactions existing between the lamins, lamin-associated proteins, DNA, and chromatin. The “structural” model proposes that LMNA mutations increase cellular susceptibility to mechanical strain through impairment of interactions critical for nuclear and cytoskeletal stability.33, 34 Because normal functioning places significant mechanical strain on individual muscle cells, the structural model has been particularly attractive for studying LMNA-related cardiomyopathy and muscular dystrophy. The alternative “gene-expression” model proposes that LMNA mutations impair critical signaling pathways through influence on gene expression at the nuclear periphery. This hypothesis is supported by abundant literature documenting heterochromatin loss or redistribution in patient fibroblasts for many of the laminopathies.35–37 These two hypotheses are not necessarily mutually exclusive: in which capacities and to what extent abnormalities in nuclear architecture and/or gene expression determine particular laminopathic phenotypes are important questions and the subject of current experimentation.
Limitations
While this in-vitro heterologous cell system has been shown to be a useful and sensitive tool for determining the potential pathogenicity of novel LMNA variants in this and other research studies,13–21 negative results for three variants carried by families with manifestations of cardiovascular disease suggest that more sophisticated approaches may reveal more subtle abnormalities. Additional nuclear morphologic studies, gene transfer experiments into small animals, or studies with human pluripotent cells harboring LMNA variants may help to further clarify the potential impact of LMNA variants of uncertain pathogenicity.
Conclusions
LMNA mutations, a significant cause of genetic DCM, were assessed with nuclear morphology and GFP-lamin A localization studies. Analyses of the LMNA variants in nonsegregation pedigrees identified in our DCM cohort support pathogenicity of 4/6 and argue for the existence of a second, unidentified causative factor in these families. In addition, demonstration of abnormal GFPLaA localization in 3/4 pedigrees for which segregation is uncertain indicate that nuclear morphological studies may also be of value in cases where LMNA-related DCM is suspected, but pedigree data is lacking.
Acknowledgements
We thank the many families and referring physicians for their participation in the Familial Dilated Cardiomyopathy Research Program, without whom these studies would not have been possible.
Funding Sources: This work was supported by NIH awards RO1-HL58626
Footnotes
Conflict of Interest Disclosure: None.
References
- 1.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7:940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- 2.Burke B, Stewart CL. The laminopathies: the functional architecture of the nucleus and its contribution to disease (*) Annu Rev Genomics Hum Genet. 2006;7:369–405. doi: 10.1146/annurev.genom.7.080505.115732. [DOI] [PubMed] [Google Scholar]
- 3.Stuurman N, Sasse B, Fisher PA. Intermediate filament protein polymerization: molecular analysis of Drosophila nuclear lamin head-to-tail binding. J Struct Biol. 1996;117:1–15. doi: 10.1006/jsbi.1996.0064. [DOI] [PubMed] [Google Scholar]
- 4.Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci. 2004;117:979–987. doi: 10.1242/jcs.01102. [DOI] [PubMed] [Google Scholar]
- 5.Holtz D, Tanaka RA, Hartwig J, McKeon F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell. 1989;59:969–977. doi: 10.1016/0092-8674(89)90753-8. [DOI] [PubMed] [Google Scholar]
- 6.Zirn B, Kress W, Grimm T, Berthold LD, Neubauer B, Kuchelmeister K, Muller U, Hahn A. Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am J Med Genet A. 2008;146A:1049–1054. doi: 10.1002/ajmg.a.32259. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473–476. doi: 10.1161/01.cir.101.5.473. [DOI] [PubMed] [Google Scholar]
- 8.Mercuri E, Brown SC, Nihoyannopoulos P, Poulton J, Kinali M, Richard P, Piercy RJ, Messina S, Sewry C, Burke MM, McKenna W, Bonne G, Muntoni F. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve. 2005;31:602–609. doi: 10.1002/mus.20293. [DOI] [PubMed] [Google Scholar]
- 9.Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, Hattersley A, Hudson J, Jardine P, Josifova D, Longman C, McWilliam R, Owen K, Walker M, Wehnert M, Ellard S. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A. 2008;146A:1530–1542. doi: 10.1002/ajmg.a.32331. [DOI] [PubMed] [Google Scholar]
- 10.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–780. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 12.Rankin J, Ellard S. The laminopathies: a clinical review. Clin Genet. 2006;70:261–274. doi: 10.1111/j.1399-0004.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- 13.Broers JL, Kuijpers HJ, Ostlund C, Worman HJ, Endert J, Ramaekers FC. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp Cell Res. 2005;304:582–592. doi: 10.1016/j.yexcr.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Hubner S, Eam JE, Wagstaff KM, Jans DA. Quantitative analysis of localization and nuclear aggregate formation induced by GFP-lamin A mutant proteins in living HeLa cells. J Cell Biochem. 2006;98:810–826. doi: 10.1002/jcb.20791. [DOI] [PubMed] [Google Scholar]
- 15.Sylvius N, Hathaway A, Boudreau E, Gupta P, Labib S, Bolongo PM, Rippstein P, McBride H, Bilinska ZT, Tesson F. Specific contribution of lamin A and lamin C in the development of laminopathies. Exp Cell Res. 2008;314:2362–2375. doi: 10.1016/j.yexcr.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motsch I, Kaluarachchi M, Emerson LJ, Brown CA, Brown SC, Dabauvalle MC, Ellis JA. Lamins A and C are differentially dysfunctional in autosomal dominant Emery-Dreifuss muscular dystrophy. Eur J Cell Biol. 2005;84:765–781. doi: 10.1016/j.ejcb.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Bechert K, Lagos-Quintana M, Harborth J, Weber K, Osborn M. Effects of expressing lamin A mutant protein causing Emery-Dreifuss muscular dystrophy and familial partial lipodystrophy in HeLa cells. Exp Cell Res. 2003;286:75–86. doi: 10.1016/s0014-4827(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 18.Gilchrist S, Gilbert N, Perry P, Ostlund C, Worman HJ, Bickmore WA. Altered protein dynamics of disease-associated lamin A mutants. BMC Cell Biol. 2004;5:46. doi: 10.1186/1471-2121-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holt I, Ostlund C, Stewart CL, Man N, Worman HJ, Morris GE. Effect of pathogenic mis-sense mutations in lamin A on its interaction with emerin in vivo. J Cell Sci. 2003;116:3027–3035. doi: 10.1242/jcs.00599. [DOI] [PubMed] [Google Scholar]
- 20.Raharjo WH, Enarson P, Sullivan T, Stewart CL, Burke B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J Cell Sci. 2001;114:4447–4457. doi: 10.1242/jcs.114.24.4447. [DOI] [PubMed] [Google Scholar]
- 21.Sylvius N, Bilinska ZT, Veinot JP, Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL, Tang AS, Dyack S, Grzybowski J, Ruzyllo W, McBride H, Tesson F. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J Med Genet. 2005;42:639–647. doi: 10.1136/jmg.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jakobs PM, Hanson EL, Crispell KA, Toy W, Keegan H, Schilling K, Icenogle TB, Litt M, Hershberger RE. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail. 2001;7:249–256. doi: 10.1054/jcaf.2001.26339. [DOI] [PubMed] [Google Scholar]
- 23.Hershberger RE, Hanson EL, Jakobs PM, Keegan H, Coates K, Bousman S, Litt M. A novel lamin A/C mutation in a family with dilated cardiomyopathy, prominent conduction system disease, and need for permanent pacemaker implantation. Am Heart J. 2002;144:1081–1086. doi: 10.1067/mhj.2002.126737. [DOI] [PubMed] [Google Scholar]
- 24.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 25.Brodsky GL, Bowersox JA, Fitzgerald-Miller L, Miller LA, Maclean KN. The prelamin A pre-peptide induces cardiac and skeletal myoblast differentiation. Biochem Biophys Res Commun. 2007;356:872–879. doi: 10.1016/j.bbrc.2007.03.062. [DOI] [PubMed] [Google Scholar]
- 26.Ostlund C, Bonne G, Schwartz K, Worman HJ. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci. 2001;114:4435–4445. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- 27.Lanktree M, Cao H, Rabkin SW, Hanna A, Hegele RA. Novel LMNA mutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660) Clin Genet. 2007;71:183–186. doi: 10.1111/j.1399-0004.2007.00740.x. [DOI] [PubMed] [Google Scholar]
- 28.Hegele R. LMNA mutation position predicts organ system involvement in laminopathies. Clin Genet. 2005;68:31–34. doi: 10.1111/j.1399-0004.2005.00447.x. [DOI] [PubMed] [Google Scholar]
- 29.Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H. Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins. J Mol Biol. 2004;343:1067–1080. doi: 10.1016/j.jmb.2004.08.093. [DOI] [PubMed] [Google Scholar]
- 30.Reichart B, Klafke R, Dreger C, Kruger E, Motsch I, Ewald A, Schafer J, Reichmann H, Muller CR, Dabauvalle MC. Expression and localization of nuclear proteins in autosomal-dominant Emery-Dreifuss muscular dystrophy with LMNA R377H mutation. BMC Cell Biol. 2004;5:12. doi: 10.1186/1471-2121-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denecke J, Brune T, Feldhaus T, Robenek H, Kranz C, Auchus RJ, Agarwal AK, Marquardt T. A homozygous ZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNA gene causes Hutchinson-Gilford progeria syndrome (HGPS): insights into the pathophysiology of HGPS. Hum Mutat. 2006;27:524–531. doi: 10.1002/humu.20315. [DOI] [PubMed] [Google Scholar]
- 32.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broers JL, Peeters EA, Kuijpers HJ, Endert J, Bouten CV, Oomens CW, Baaijens FP, Ramaekers FC. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–2580. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- 35.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabatelli P, Lattanzi G, Ognibene A, Columbaro M, Capanni C, Merlini L, Maraldi NM, Squarzoni S. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2001;24:826–829. doi: 10.1002/mus.1076. [DOI] [PubMed] [Google Scholar]
- 37.Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res. 2003;291:352–362. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]