

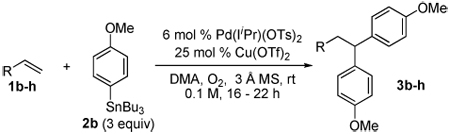
Abstract

Evaluation of the scope of a PdII-catalyzed oxidative 1,1-diarylation reaction of terminal olefins using aryl stannanes is reported. The reaction is shown to be tolerant of functionality commonly encountered in organic synthesis, however the reaction outcome was found to be dependent on the nature of the aryl stannane used. A mechanistic rationale for the observation of this influence is provided.
The rapid introduction of molecular complexity through the formation of multiple carbon-carbon bonds in a single synthetic step can be a powerful tool for organic chemists. In order to accomplish this using palladium catalysis, the interception of reactive PdII-alkyl intermediates is typically required prior to β-hydride elimination.1,2,3,4,5,6 Recently, we reported a catalytic system that installs two aryl groups on an olefin substrate (Scheme 1a).7 The proposed mechanism of this reaction is similiar to that of the oxidative Heck reaction8,9 in that after initial transmetalation, an alkene inserts into the Pd-aryl bond. Critical to the success of the diarylation reaction is the exploitation of the stability of the proposed Pd-π-benyzl intermediate A.10 This interaction slows β-hydride elimination, allowing for a second transmetalation, ultimately delivering the 1,2-diarylation product.
Scheme 1.

Diarylation of styrenes
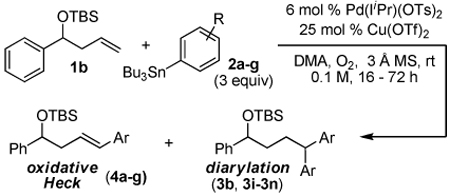
During this study, an interesting byproduct was observed arising from A slipping to an η1 Pd-alkyl (Scheme 1c), followed by β-hydride elimination and olefin reinsertion to give a new Pd-π-benzyl D. This is proposed to undergo a second transmetalation followed by reductive elimination to give the 1,1-diarylation product. The observation of a linear free energy relationship between the electronic nature of the styrene substrates and the ratio of these two products revealed the reaction’s high level of sensitivity to the stability of π-benzyl intermediates. This led to the hypothesis that alkyl-substituted olefins would give exclusively 1,1-diarylated products, since the substrate cannot provide π-benzyl stabilization, but this interaction would be accessible with the arene originating from the aryl stannane via palladium migration. To evaluate this hypothesis, nonene was subjected to the conditions optimized for 1,2-diarylation of styrenes and this indeed resulted in the formation of the expected 1,1-diaryl product 3a (Scheme 2).7a The proposed mechanism for this reaction is initiated by a Heck insertion, followed by β-hydride elimination to give F. Hydride insertion at the position β to the arene gives G which can be stabilized as a π-benyzl intermediate. A second transmetalation is proposed to occur followed by reductive elimination to provide the 1,1-diarylation product. Since the diarylmethine core structure is a common motif in biologically active compounds,11 we believed it would be valuable to pursue this initial result and to determine the scope and limitations of this new methodology.
Scheme 2.

1,1-Diarylation of nonene
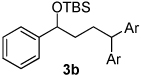
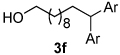
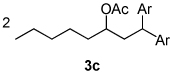
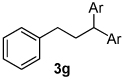
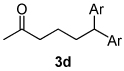
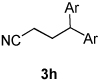
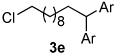
Using the optimal conditions previously reported, this reaction was found to be quite tolerant of functional groups commonly encountered in organic synthesis (Table 1). A substrate bearing a protected homoallylic alcohol leads to a good yield of the desired product (3b). Similiarly, a protected allylic alcohol (3c) undergoes the diarylation reaction cleanly with no products derived from Pd-π-allyl chemistry observed. Substrates containing a ketone (3d) and an ester (3c) are well tolerated, as are primary chlorides (3e). A substrate with a distal free alcohol resulted in moderate yields of the 1,1-diarylation product (3f) and allyl benzene gives a high yield of diarylation products as a mixture of regioisomers (3g). Allyl cyanide is a poor substrate for this reaction, providing a 27% yield (3h). Importantly, an enantioenriched sample of 1c suffered no erosion in enantiomeric excess when converted to 3c using these conditions (eq 1).
Table 1.
Scope of 1,1-diarylation
Ar=p-MeOC6H4.
Yield is average of two experiments performed on 0.5 mmol scale. The remainder of the mass balance in largely an oxidative Heck product, which was not quantified for these reactions.
Isolated as a 7.2:1 mixture with 1,3-diaryl product.
Next, the scope of the aryl stannane component of the reaction was evaluated (Table 2). The reaction proceeds well using p-alkyl phenyl stannanes (entry 2) and PhSnBu3 (entry 3). p-Fluoro phenyl stannane gave a slightly diminished yield of 3k, while use of more electron poor p-chloro phenyl stannane 2e resulted in a further decrease in yield. Using the highly electron deficient aryl stannane bearing a trifluromethyl group gave very low yield with a greater amount of the Heck product, and poor tolerance of steric hindrance in the aryl stannane was observed (2f). The major byproduct for these reactions is oxidative Heck products 4, presumably arising from Pd-dissociation from intermediate F (Scheme 2), the yields of which are also reported in Table 2.
Table 2.
Scope of 1,1-diarylation
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 2 | % 3a | % 4a | entry | 2 | % 3a | % 4a |
| 1 | 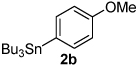 |
82 3b |
13 4a |
4 | 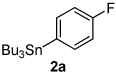 |
63 3k |
18 4d |
| 2 | 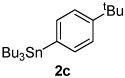 |
80 3i |
13 4b |
5b | 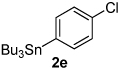 |
46 3l |
36 4e |
| 3 | 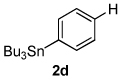 |
75 3j |
9 4c |
6c | 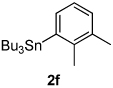 |
26 3m |
26 4f |
Yields are average of two experiments performed on 0.5 mmol scale except where noted. The remainder of the mass balance is isomerized starting material.
One experiment performed on 0.42 mmol scale.
Recovered 47% starting material.
 |
(1) |
Evaluation of the scope of the aryl stannane revealed a clear trend between the electronic nature of the aryl stannane and the yields of both the 1,1-diarylation and oxidative Heck products. Specifically, as the aryl stannane became more electron deficient, the ratio of diarylation product (3) to the oxidative Heck product (4) decreased. Intrigued, we questioned whether there was a direct relationship between these parameters. The ratios of 1,1-diarylation to Heck products was determined by 1H NMR integration performed on a mixture where the tin byproducts had been chromatographically removed. Plotting log[1,1-diarylation]/[Heck] as a function of the Hammett parameters of 2 (σ) indeed revealed a linear free energy relationship with a ρ = −0.91 (Figure 1). This relationship can be explained by noting that the cationic metal center is stabilized by the π-electrons of the aryl group in π-benzyl structure G, and that electron rich arenes provide more stability and impart a longer lifetime to this intermediate than electron poor arenes. This slows β-hydride elimination, allowing for a second transmetalation, which results in a higher ratio of 1,1-diarylation to oxidative Heck products when electron rich aryl stannanes are used. It is interesting to note that despite the different products delivered by the 1,2- and 1,1-diarylation reactions, the slope of the two Hammett plots are nearly identical (ρ = −0.88 for work described in reference 7a), suggesting that they are affected to a similar degree by the stability of Pd-π-benzyl intermediates. This catalyst control can presumably be manupulated by the nature of the dative ligand and counterions on the metal for future applications.
Figure 1.

Hammett plot
In conclusion, we have found that the PdII-catalyzed oxidative diarylation reaction of terminal olefins is tolerant of diverse functionality, and does not erode enantiomeric excess in a substrate with a proximal stereocenter. The reaction performs well using electron rich aryl stannanes, but yield and selectivity suffer when the aryl stannane is electron deficient, or sterically hindered. We have reported a linear free energy relationship quantifying this observation and demonstrating the reaction’s high sensitivity to Pd-π-benzyl stability. Future work will be directed toward the development of a diarylation reaction capable of installing two distinct aryl groups on a terminal olefin.
Supplementary Material
Acknowledgment
This research was supported by the National Institutes of Health (NIGMS RO1 GM3540).
Footnotes
Supporting Information Available: Detailed experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For an overview of palladium-catalyzed difunctionalization reactions, see: Jensen KH, Sigman MS. Org. Biomol. Chem. 2008;6:4083–4088. doi: 10.1039/b813246a. For an example of palladium-catalyzed arylhalogenation, see: Kalyani D, Sanford MS. J. Am. Chem. Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798.
- 2.For examples of palladium-catalyzed dialkoxylation see Chevrin C, Le Bras J, Henin F, Muzart J. Sythesis. 2005:2615–2628. Schultz MJ, Sigman MS. J Am. Chem. Soc. 2006;128:1460–1461. doi: 10.1021/ja0579053. Thiery E, Chevrin C, Le Bras J, Harakat D, Muzart J. J. Org. Chem. 2007;72:1859–1862. doi: 10.1021/jo062491x. Zhang Y, Sigman MS. J. Am. Chem. Soc. 2007;129:3076–3077. doi: 10.1021/ja070263u. Jensen KH, Pathak TP, Zhang Y, Sigman MS. J. Am. Chem. Soc. 2009;131:17074–17075. doi: 10.1021/ja909030c.
- 3.For examples of hydrofunctionalization using a similar strategy, see: Gligorich KM, Schultz MJ, Sigman MS. J. Am. Chem. Soc. 2006;128:2794–2795. doi: 10.1021/ja0585533. Gligorich KM, Cummings SA, Sigman MS. J. Am. Chem. Soc. 2007;129:14193–14195. doi: 10.1021/ja076746f. Podhajsky SM, Sigman MS. Organometallics. 2007;26:5680–5686. doi: 10.1021/om700675z. Iwai Y, Gligorich KM, Sigman MS. Angew. Chem., Int. Ed. 2008;47:3219–3222. doi: 10.1002/anie.200705317. Urkalan KB, Sigman MS. J. Am. Chem. Soc. 2009;131:18042–18043. doi: 10.1021/ja908545b.
- 4.For examples of palladium-catalyzed diamination reactions see: Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J. Am. Chem. Soc. 2005;127:7308–7309. doi: 10.1021/ja051181d. Du H, Zhao B, Shi Y. J. Am. Chem. Soc. 2007;129:762–763. doi: 10.1021/ja0680562. Du H, Yuan W, Zhao B, Shi Y. J. Am Chem. Soc. 2007;129:11688–11689. doi: 10.1021/ja074698t. Xu L, Shi Y. J. Org. Chem. 2008;73:749–751. doi: 10.1021/jo702167u. Du H, Yuan W, Zhao B, Shi Y. J. Am. Chem. Soc. 2007;129:7496–7497. doi: 10.1021/ja072080d. Du H, Zhao B, Shi Y. J. Am. Chem. Soc. 2008;130:8590–8591. doi: 10.1021/ja8027394.
- 5.For examples of nucleopalladation-alkene insertion stratagies see: Arai MA, Kuraishi M, Arai T, Sasai H. J. Am. Chem. Soc. 2001;123:2907–2908. doi: 10.1021/ja005920w. Minami K, Kawamura Y, Koga K, Hosokawa T. Org. Lett. 2005;7:5689–5692. doi: 10.1021/ol052377l. Kawamura Y, Imai T, Hosokawa T. Synlett. 2006:3110–3114. Yip K-T, Yang M, Law K-L, Zhu N-Y, Yang D. J. Am. Chem. Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. Scarborough CC, Stahl SS. Org. Lett. 2006;8:3251–3254. doi: 10.1021/ol061057e.
- 6.For difunctionalization reactions using a PdII-PdIV catalytic cycle see: Streuff J, Hovelmann CH, Nieger M, Muniz K. J. Am. Chem. Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. Muniz K. J. Am. Chem. Soc. 2007;129:14542–14543. doi: 10.1021/ja075655f. Muniz K, Hovelmann CH, Streuff J. J. Am. Chem. Soc. 2008;130:763–773. doi: 10.1021/ja075041a. Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. Desai LV, Sanford MS. Angew. Chem., Int. Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. Tong X, Beller M, Tse MK. J. Am. Chem. Soc. 2007;129:4906–4907. doi: 10.1021/ja070919j. Welbes LL, Lyons TW, Cychosz KA, Sanford MS. J. Am. Chem. Soc. 2007;129:5836–5837. doi: 10.1021/ja071204j. Li Y, Song D, Dong VM. J. Am. Chem. Soc. 2008;130:2962–2964. doi: 10.1021/ja711029u.
- 7.Urkalan KB, Sigman MS. Angew. Chem., Int. Ed. 2009;48:3146–3149. doi: 10.1002/anie.200900218. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example of palladium-catalyzed diarylation of a chelating vinyl ether see: Trejos A, Fardost A, Yahiaoui S, Larhed M. Chem. Commun. 2009:7587–7589. doi: 10.1039/b918358b.
- 8.For reviews of the Heck reaction, see: Heck RF. Org. React. 1982;27:345–390. Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. Whitcombe NJ, Hii KK, Gibson SE. Tetrahedron. 2001;57:7449–7476. Kondolff I, Doucet H, Santelli M. Tetrahedron Lett. 2003;44:8487–8491.
- 9.For examples of oxidative Heck reactions, see: Du X, Suguro M, Hirabayashi K, Mori A, Nishikata T, Hagiwara N, Kawata T, Okeda T, Wang HF, Fugami K, Kosugi M. Org. Lett. 2001;3:3313–3316. doi: 10.1021/ol016529y. Parrish JP, Jung YC, Shin SI, Jung KW. J. Org. Chem. 2002;67:7127–7130. doi: 10.1021/jo020159p. Jung YC, Mishra RK, Yoon CH, Jung KW. Org. Lett. 2003;5:2231–2234. doi: 10.1021/ol034458s. Andappan MMS, Nilsson P, von Schenck H, Larhed M. J. Org. Chem. 2004;69:5212–5218. doi: 10.1021/jo049434t. Yoo KS, Yoon CH, Jung KW. J. Am. Chem. Soc. 2006;128:16384–16393. doi: 10.1021/ja063710z. Delcamp JH, Brucks AP, White CM. J. Am. Chem. Soc. 2008;130:11270–11271. doi: 10.1021/ja804120r.
- 10.(a) Becker Y, Stille JK. J. Am. Chem. Soc. 1978;100:845–850. [Google Scholar]; (b) Ling YS, Yamamoto A. Organometallics. 1998;17:3466–3478. [Google Scholar]; (c) Ling YS, Yamamoto A. Bull. Chem. Soc. Jpn. 1998;71:723–734. [Google Scholar]; (d) Johns AM, Utsunomiya M, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2006;128:1828–1839. doi: 10.1021/ja056003z. [DOI] [PubMed] [Google Scholar]; (e) Johns AM, Tye JW, Hartwig JF. J. Am. Chem. Soc. 2006;128:16010–16011. doi: 10.1021/ja067084h. [DOI] [PubMed] [Google Scholar]
- 11.(a) Moriconi A, Cesta MC, Cervellera MN, Aramini A, Coniglio S, Colagioia S, Beccari AR, Bizzarri C, Cavicchia MR, Locati M, Galliera E, Di Benedetto P, Bigilante P, Bertini R, Allegretti M. J. Med. Chem. 2007;50:3984–4002. doi: 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]; (b) Chen J-J, Chen P-H, Liao C-H, Huang I-S. J. Nat. Prod. 2007;70:1444–1448. doi: 10.1021/np070186g. [DOI] [PubMed] [Google Scholar]; (c) Liang H, Wu X, Yalowich JC, Hasinoff BB. Mol. Pharmacol. 2008;73:686–696. doi: 10.1124/mol.107.041624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.