

Abstract
We identify and investigate several critical parameters in the fabrication of single-stranded DNA conjugated poly(ethylene glycol) (PEG) microparticles based on replica molding (RM) for highly uniform and robust nucleic acid hybridization assays. The effects of PEG-diacrylate, probe DNA, and photoinitiator concentrations on the overall fluorescence and target DNA penetration depth upon hybridization are examined. Fluorescence and confocal microscopy results illustrate high conjugation capacity of probe and target DNA, femtomole sensitivity, and sequence specificity. Combined these findings demonstrate a significant step toward simple, robust, and scalable procedures to manufacture highly uniform and high capacity hybridization assay particles in a well-controlled manner by exploiting many advantages that the batch processing-based RM technique offers. We envision that the results presented here may be readily applied to rapid and high throughput hybridization assays for a wide variety of applications in bioprocess monitoring, food safety, and biological threat detection.
Introduction
Nucleic acid hybridization-based assays represent a highly selective and reliable route for biosensing applications in food- or water- borne pathogen monitoring1, 2, biological threat detection3, and disease diagnostics4. These assays are traditionally carried out in two array formats: planar or suspension. While routinely providing high throughput capacity, planar arrays (e.g. DNA microarrays with patterned probe DNAs on flat surfaces) can pose critical limitations for rapid and cost-efficient assays, such as long incubation times, low probe titers, and extensive equipment and software needs5. Particle-based suspension arrays offer attractive platforms for high throughput biosensing with low sample volume and rapid detection via scanning6-8. Combined with the ability to conjugate acrylate-modified probe DNA via UV-induced co-photopolymerization6, 7, polymeric hydrogel particles provide porous 3D networks that offer more favorable solution kinetics than 2D surfaces, leading to rapid hybridization and improved selectivity8.
High-throughput fabrication of hydrogel particles with well-defined shapes down to the submicrometer level may be achieved using photolithography9; for example, Meiring et al. reported on fabrication of shape-encoded hydrogel particle biosensor array platforms10. While offering straightforward and well-established procedures from the microfabrication technology, equipment cost and cumbersome recovery for such photolithographic fabrication presents hurdles for simple and robust routes to produce particles. Alternatively, continuous and stop-flow lithography11, 12 provide rapid and continuous production of monodisperse hydrogel particles with potent encoding schemes13 and multiplexing capabilities14 by taking advantage of the laminar nature of the microfluidic flow. Yet certain limitations exist with these microfluidic methods, such as the requirement of rapid polymerization without deformation, the liquid properties (e.g. viscosity) bound by the need for consistent flow, or adherence to channel walls11, 12, 15. Meanwhile, soft-lithographic fabrication of polymeric structures via replica molding (RM) has recently gained substantial attention due to many inherent advantages rising from the batch-processing based nature of the fabrication procedure15-17. These advantages include simple, robust, and inexpensive procedures16 as well as scalability9 and reliable duplication of complex structures with nanometer resolution16, 17.
As shown in the schematic diagram of Figure 1a, fabrication of ssDNA-conjugated poly(ethylene glycol) (PEG)-based microparticles via RM consists of first placing preparticle solution into PDMS micromolds (Step 1). Step 2 involves covering the filled micromolds with a PDMS-coated glass cover. Next, the micromolds are exposed to UV light for photopolymerization as shown in Step 3 of Figure 1a. Specifically, UV light reacts with initiator molecules to provide primary radical species through a photocleavage process that initiates radical chain polymerization18. The radicals add to monomer molecules and form new radicals or react with unconverted double bonds (i.e. Acrydite™) present on oligomer molecules. Polymer growth is terminated by destruction of the reactive center by an appropriate reaction, such as that of two radical species forming a longer chain19. Finally, Step 4 depicts resulting microparticles of different shapes for hybridization with target nucleic acids in either a 2D in-mold or 3D suspension array format. As shown in the fluorescence micrograph and SEM image of Figures 1b and c, practically unlimited dimensions and shapes of uniform particles can be fabricated in a single batch. A majority of recent studies using a similar soft lithographic RM technique, known as particle replication in nonwetting templates (PRINT), have focused mainly on nanoscale fabrication of polymeric hydrogel particles in an effort to harness the simplicity and robustness of the RM procedure for drug delivery applications9, 20-22. However, fabrication of uniform probe DNA-conjugated hydrogel particles with dimensions relevant to high throughput biosensing (i.e. 10-100 μm) via RM has not been demonstrated.
Figure 1.

DNA–conjugated Microparticle Fabrication via Replica Molding (RM) for Hybridization Assays. (a) Schematic diagram illustrating the batch-processing based RM procedure for production of DNA-conjugated microparticles. (b) Fluorescence micrograph of various shaped microparticles upon hybridization with fluorescently labeled target DNA. The yellow scale bar represents 100 μm. (c) Scanning Electron Microscopy (SEM) image showing consistent shape and dimension of disc-shaped microparticles fabricated via RM. The white scale bar represents 50 μm.
In this report, we identify and examine several critical parameters for fabrication of ssDNA-conjugated PEG-based microparticles. Specifically, we evaluate the effects of humidity as well as PEG-diacrylate (DA), probe DNA, and photoinitiator (PI) concentrations on the overall fluorescence and penetration depth of target DNA via fluorescence and confocal microscopy. First, minimizing rapid evaporation is an important factor for uniformity. Second, PEG-DA concentration substantially impacts the penetration depth, leading to significant differences in accessible probe DNA capacity. Third, the optimized conditions yield consistently uniform particles and femtomole quantity detection under standard conditions enlisted in this study. Combined these findings represent a significant step toward simple and robust procedures to manufacture highly uniform and high capacity nucleic acid hybridization assay particles in a controlled manner. We envision that the results and the fabrication methods presented in this study can be readily adapted to a variety of other systems for facile fabrication of potent biosensing or functional particles.
Experimental Section
Materials and Experimental Conditions
Poly(ethylene glycol) diacrylate (PEG-DA, Mn=700), PEG (Mw=200), and 2-hydroxy-2-methylpropiophenone (Darocur 1173, photoinitiator (PI)) were purchased from Sigma-Aldrich (St. Louis, MO). Probe DNA sequences (Table 1) with a 5’ end Acrydite™ modification were purchased from Integrated DNA Technologies (Coralville, IA) and target DNA sequences (Table 1) with a 5’end fluorescein isothiocyanate (FITC) modification were purchased from Gene Probe Technologies (Gaithersburg, MD). All of these materials were used without further purification.
Table 1.
List of probe and target DNA sequences for each particle shape used in this work.
| Shape | Probe DNA Sequences* |
|---|---|
| Circle | 5’-ATG ATG ATG ATG ATG ATG-3’ |
| Square | 5’-CAC TAC CGA TAC GTA CTC AG-3’ |
| Triangle | 5’-TTT TTC GGC AGG TCG GTA AC -3’ |
| Shape | Target DNA Sequences† |
|---|---|
| Circle | 5’-CAT CAT CAT CAT CAT CAT-3’ |
| Square | 5’-CTG AGT ACG TAT CGG TAG TG-3’ |
| Triangle | 5’-GTT ACC GAC CTG CCG AAA AA-3’ |
Probe DNA sequences are all Acrydite™ modified at the 5’end.
Target DNA sequences are all modified with fluorescein isothiocyanate (FITC) at the 5’end.
The composition of the preparticle solution was as follows: 20-60% (v/v) PEG-DA, 10% PEG 200, 0.5-3% PI, 10% Probe DNA (1-100 μM final concentration), and 18-58% TE Buffer (10 mM Tris, pH 8.0, Sigma-Aldrich and 1 mM EDTA, Sigma-Aldrich) containing 0.02% (v/v) of 10% (w/w) sodium dodecyl sulfate (SDS, Sigma-Aldrich). The humidity controlled environment consisted of a plastic glove box (Thermo Fisher Scientific) containing a 0.5 gallon ultrasonic humidifier. The humidity was manually controlled based on readings from a standard laboratory Traceable™ hygrometer/thermometer (VWR). An environment of 90% relative humidity was maintained for microparticle fabrication.
Fabrication of microparticles
The four major steps of soft lithography, as described by others17, were followed to prepare the microparticles. Briefly, AutoCAD was used to design the shaped patterns. High-resolution printing was used to generate photomasks on transparency films for fabrication of silicon masters with photolithography. PDMS elastomeric micromolds were formed with Sylgard 184 (Dow Corning) following a 48 hr cure at 65 °C. Finally, microparticles were fabricated from the PDMS elastomeric micromolds via replica molding (RM).
The details of the RM procedure used for this work are as follows: Preparticle solution (~75 μL) was first placed on the surface of the PDMS micromolds (1600 wells per 1 cm × 1 cm mold). Disposable plastic pipette tips were then used to rub the molds to remove air bubbles in the microwells. The excess preparticle solution was simply removed by a pipette tip and reserved for reuse in subsequent batches of particles requiring the same preparticle formulation. The filled PDMS micromolds were then sealed with a glass slide coated with a thin layer of PDMS obtained via spin-coating for 30 sec at 2000 rpm. A square section (same size as the microwell square region within the micromold) of PDMS was removed from the glass slide to provide a small gap between the glass surface and the top portion of the microwells. The sealed micromolds were then placed on an aluminum mirror (Thorlabs, Newton, NJ) and exposed to 365 nm UV light with an 8 W handheld UV lamp (Spectronics Corp., Westbury, NY) for 15 min. The polymerized particles were released from the microwells by first physically bending the mold, then placing water containing 0.5% (v/v) Tween 20 on the mold surface. The microparticles were collected by pipetting up and down a few times before transferring into a storage vial. Particle removal with water was repeated 3 times for complete collection of particles.
Hybridization Procedure
Before hybridization, microparticles were rinsed with 5× saline sodium citrate (SSC) buffer (75 mM sodium citrate, 750 mM sodium chloride, pH 7.0) containing 0.05% (v/v) Tween 20. The rinsing process involved pipetting the particles in solution to mix, then allowing particles to settle to the tube bottom. The supernatant was removed and the wash procedure was repeated 4 times. For hybridization, the microparticles (maximum number of particles per batch ~1000) were placed in solution with 200 nM target DNA (Table 1) in a total volume of 100 μL. Following hybridization for 30 min on a rotator at room temperature, unbound target DNA was rinsed 5 times using the rinsing procedure described above with gravity settling. This 30 min hybridization provided sufficient time for consistent signals14, 23, while hybridization of short DNA strands in hydrogel formats is rapid and reaches saturation within 10 min (data not shown).
Imaging Analysis
The hybridized microparticles were visualized with an Olympus BX51 microscope using a standard green filter set U-N31001 (Chroma Technology Corp., Rockingham, VT), and the fluorescence micrographs were captured with a DP70 microscope digital camera. Fluorescence intensity was evaluated with ImageJ software24. Confocal images were acquired on a Leica DMIRE2 microscope with a TCS SP2 scanner (Wetzlar, Germany). The particles were analyzed with a 63× (NA 1.2) water immersion objective at 488 nm excitation and 500-530 nm emission. The depth scan increment was 3 μm for the z-scan images shown in Figure 3. Analysis was performed with the Leica Confocal software (Wetzlar, Germany).
Figure 3.

Effects of PEG-DA Concentration on Target DNA Hybridization. Fluorescence micrographs (a-d) and confocal z-scan images (e-h) of microparticles following hybridization with target DNA. Scale bars represent 100 μm (a-d) and 50 μm (e-h). (i) Plots of average fluorescence intensity (a-d) and penetration depth (e-h) vs. PEG-DA concentration. Red circles (a-c) represent particle center areas used for fluorescence intensity quantification. Error bars represent standard deviation from 5 particles.
Equilibrium Binding Model
To generate the dotted red curve of Figure 7g, the equilibrium binding relationship between two complementary oligomers was considered as described by Pregibon et al.14:
where T represents target, P represents probe, and TP represents the double stranded complex. From the equilibrium binding relationship and assuming [T]o>>[P]o, the following equation is obtained with the equilibrium constant Kd:
where the initial target DNA concentration in the bulk solution is represented as [T]o while the initial probe DNA concentration and the target-probe duplex in the hydrogel matrix of the particles are represented as [P]o and [TP], respectively. A double reciprocal plot (also known as a Lineweaver-Burk plot) was generated to determine the constants Kd and [P]o from the linearized equation:
Kd and [P]o were found to be 228 nM and 139 nM, respectively. Note that [P]o represents the initial probe concentration within the particles estimated from fluorescence intensity values, and not bulk concentration or a direct measure of the actual probe concentration. These constants were then used with the equation above to generate the red curve shown in Figure 7g. Since the response was linear for low target concentrations, the red line in the inset of Figure 7g was generated from a linear fit of the low target concentration data.
Figure 7.

Target DNA Sensitivity. Fluorescence micrographs (a-f) of microparticles upon target DNA hybridization. Scale bars represent 200 μm. (g) Plots of average fluorescence intensity vs. target DNA concentration. The dotted red lines indicate the equilibrium binding model and linear fit for the main plot and inset, respectively. Error bars represent standard deviation from 5 particles.
Results and Discussion
Minimizing Negative Effects from Evaporation
One of the most important practical considerations toward monodisperse microparticle fabrication by replica molding (RM) is preventing rapid evaporation of small volume fluids. As shown in Figure 2a, probe DNA-conjugated microparticles prepared in ambient laboratory conditions (15% RH, 21 °C) appear similar in brightfield micrographs (and SEM image of Figure 1c). Yet, the fluorescence micrograph in Figure 2b reveals significant non-uniformity upon hybridization with fluorescently labeled target DNA. Fluorescence micrographs of particles in molds following in-mold hybridization with target DNA shows that this non-uniformity arises largely from rapid evaporation of water in the preparticle solution, particularly at the outer region of the molds (Figure 2c). Specifically, a substantial portion of water in the 400 pL PEG-DA/probe DNA mixture can evaporate from each microwell within seconds at ambient conditions25. For our system, evaporation mainly occurs during removal of excess preparticle solution and before placement of a PDMS-coated glass slide to seal the microwells (Figure 1a, between Step 1 and 2). As a result, the time spent between Steps 1 and 2 at ambient conditions has substantial impact on the uniformity and fluorescence; for example, samples exposed in ambient conditions for 5 sec had dramatically higher uniformity than those exposed for 30 sec (data not shown). Evaporation of a small fraction of water may lead to substantial loss of overall fluorescence by consequentially increasing the PEG-DA concentration in the preparticle solution (shown in Figure 3). This issue is easily addressed by completing Steps 1 and 2 of Figure 1a in a high humidity chamber (90% relative humidity in our case)26. Figures 2d to f show that this simple modification yields highly uniform fluorescence upon hybridization, particularly for particles on the outer edge of the mold. This modification is straightforward to implement and may be tailored for large scale fabrication without arduous or costly equipment. Importantly, the results shown in Figure 2 suggest that in-mold hybridization (Figures 2c and f) with target DNA as small marker molecules may provide a simple route to examine detailed microparticle formation and characteristics based on their locations within the mold, that are otherwise challenging via standard optical microscopy and SEM27. While RM offers an inherently simpler and more robust particle fabrication procedure than flow lithography techniques, exposure to low humidity environments during fabrication results in dramatically poor uniformity as well as probe DNA conjugation capacity, two important aspects for facile biosensing. Combined, these results illustrate a simple means to minimize negative effects of rapid evaporation for improved uniformity, and suggest the potential utility of in-mold hybridization for evaluating microparticles. microparticle formation and characteristics based on their locations within molds. Scale bars represent 500 μm.
Figure 2.
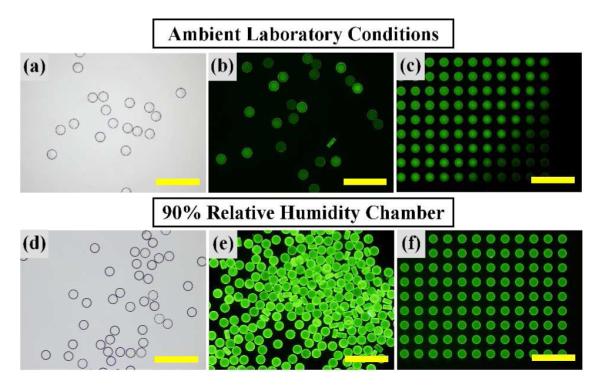
Minimizing Negative Effects from Evaporation. Brightfield micrographs (a,d) show similar appearances from particles prepared under different environments. Fluorescence micrographs following hybridization with fluorescently labeled target DNA (b,e) illustrate how evaporation of water from microwells within the micromolds significantly affects the overall fluorescence and uniformity. Fluorescence micrographs following in mold-hybridization with fluorescently labeled target DNA (c,f) identifies major sources of nonuniformity, and illustrates a simple method for examining detailed
Effects of PEG-DA Concentration on Target DNA Hybridization
We next examined the effect of PEG-DA concentration on the total fluorescence and penetration depth upon hybridization with target DNA as shown in Figure 3. For this, probe DNA-conjugated particles were fabricated with a range of PEG-DA concentrations (20-60% (v/v)) with all the other components in the preparticle solution identical. Upon hybridization with 200 nM complementary target DNA, total fluorescence and 2D profiles of the particles were examined via fluorescence and confocal microscopy.
The fluorescence micrographs in Figures 3a to d show that lower PEG-DA concentration yields brighter fluorescence, with 30% PEG-DA providing the highest and 60% PEG-DA providing negligible fluorescence. Formation of particles containing 20% PEG-DA was incomplete (data not shown). Importantly, fluorescence intensity was relatively uniform among the particles under all the PEG-DA conditions examined, confirming the benefit of simple humidity control (Figure 2). In-depth examination of particle fluorescence via confocal microscopy revealed significant differences in the target DNA penetration depth as shown in the 3 μm z-scan images obtained at the center of each particle (Figures 3e to h). Specifically, target DNA appears to penetrate 6 μm into the microparticles containing 30% PEG-DA while fluorescence is confined only to the outer surface of 60% PEG-DA particles. In addition, the fluorescence intensity of these z-scan images differs for each PEG-DA concentration tested, with 30% PEG-DA particles showing the brightest fluorescence. Overall, the results in Figure 3 clearly show that PEG-DA concentration in the microparticle formulation can significantly impact the quantity and depth of hybridization.
To further assess the effects of PEG-DA concentration on the fluorescence intensity and penetration depth quantitatively, we next plotted average results as shown in Figure 3i. Cropped center regions of the microparticles (~98 μm in diameter) were used to quantify the fluorescence intensity in order to avoid artifacts from the outer edges where more surface area from the sidewalls (50 μm height) provides stronger fluorescence (red circles of Figures 3a to c). Both data sets exhibit similar linear behavior, with 30% PEG-DA showing the brightest fluorescence and deepest penetration. Similarly, high PEG-DA concentration provides the lowest fluorescence intensity and minimal penetration confined to the outer particle surface. It is expected that high PEG-DA concentrations in the preparticle solution provides highly dense crosslinking leading to smaller pore sizes19. In this study, fluorescently labeled target DNA served as a small model molecule (6 kDa and linear) to semi-quantitatively examine the hydrogel pore size. For all the cases evaluated, the fluorescence is strictly confined to the outer regions of the microparticles. This result contrasts that for microparticles fabricated via flow lithography, where the presence of oxygen at particle surfaces is critical for continuous operation of microfluidic devices6-9. The requirement of inhibited photopolymerization at the particle surfaces by oxygen diffusion through PDMS for flow lithography processes leads to limited probe DNA density on the particle surfaces. This suggests an advantage of the batch processing-based RM technique that allows for high capacity probe DNA conjugation on the very outer edges while not being limited by the constraints posed by the need of continuous flow in microfluidic channels. In conclusion, the results in Figure 3 illustrate how PEG-DA concentration affects hybridization density of target DNA as well as its penetration into microparticles.
Effect of Probe DNA Concentration
As shown in Figure 4, we next examined the effects of probe DNA concentration on target DNA hybridization. For this, microparticles were prepared with six different concentrations of probe DNA from 1 to 100 μM, hybridized with 200 nM target DNA for 30 min at ambient conditions, and evaluated via fluorescence microscopy. First, fluorescence micrographs of Figures 4a to d illustrate a gradual increase in fluorescence intensity as probe DNA concentration increased from 1 to 50 μM. The particles were monodisperse and their fluorescence was relatively uniform in this probe range. Next, Figures 4e and f show that the fluorescence intensity reached a plateau at probe concentrations above 50 μM. To quantitatively compare the fluorescence intensity, average fluorescence intensity from five randomly selected particles was plotted as shown in Figure 4g. The plotted data display a linear trend up to a probe DNA concentration of 50 μM with small error bars, further confirming the uniformity of hybridization signals among particles. Within this linear region, the fluorescence intensity, representing the amount of target DNA hybridized to the microparticle, is proportional to the probe DNA concentration. The fluorescence intensity reaches saturation above 50 μM, where there is also less uniformity (i.e. larger error bars). Overall, this result provides a correlation between probe DNA concentration and fluorescence intensity; based on this, we used 50 μM probe concentration throughout this study.
Figure 4.

Effect of Probe DNA Concentration. Fluorescence micrographs (a-f) of microparticles upon target DNA hybridization. Scale bars represent 200 μm. (g) Plot of average fluorescence intensity vs. probe DNA concentration. Error bars represent standard deviation from 5 particles.
Effects of Photoinitiator (PI) Concentration
To evaluate the effects of photoinitiator (PI) concentration, we fabricated square shaped microparticles with varying PI concentrations between 0.5 and 3% (v/v) then examined particle formation and hybridization behavior as shown in Figure 5. First, the brightfield (Figure 5a) and fluorescence micrograph (Figure 5d) of particles with 0.5% PI show incomplete particle formation as well as minimal fluorescence upon hybridization. This result suggests that the quantity of free radicals generated at this PI concentration is not adequate to fully polymerize the preparticle solution or to incorporate sufficient probe DNA under the polymerization condition employed. At 1% PI, Figures 5b and e show complete and uniform particle formation with significant fluorescence. Among all the PI concentrations examined, 2% PI provides the strongest fluorescence intensity, as shown in Figure 5c. Finally, 3% PI yielded relatively dim fluorescence upon hybridization compared to lower PI concentrations, as shown in Figure 5f. As expected, particle formation with sufficient PI (2 and 3%) remained consistent with the 1% condition (brightfield images not shown). To further assess the trend of fluorescence intensity quantitatively, the average fluorescence intensities from the particle centers were plotted as shown in Figure 5g. This plot confirms that 2% PI concentration provides the highest fluorescence intensity for the conditions examined.
Figure 5.
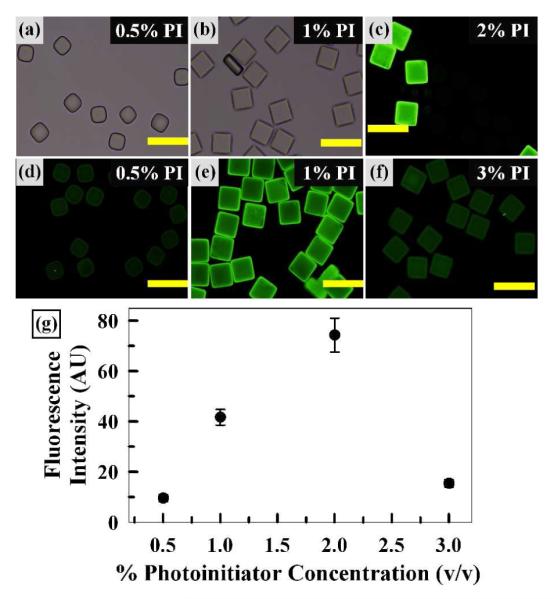
Effects of Photoinitiator (PI) Concentration. Brightfield (a,b) and fluorescence (c-f) micrographs of microparticles following hybridization with target DNA (200 nM). Scale bars represent 200 μm. (g) Plot of average fluorescence intensity vs. PI concentration. Error bars represent standard deviation from 5 particles.
Of all the PI concentrations examined, the 3% condition should provide the most rapid polymerization kinetics due to the greater availability of free radicals at this concentration. In addition, Lee et al. described how the penetration depth of photons decreases as PI concentration is raised, resulting in localization of the free radical initiation closer to the exposed surface28. The combination of increased quantity of free radicals and their localization provides a tightly crosslinked hydrogel network at the sample surface yielding ~1 nm pore size29-31, substantially smaller than the size of probe DNA. Therefore, results shown in Figure 5 suggest that the lower fluorescence at 3% PI largely occurred from the inability of target DNA to thoroughly penetrate the tightly crosslinked hydrogel network. Among the PI concentrations examined here, 2% PI provided the brightest fluorescence and was used to carry out the remaining studies in this work.
DNA Sequence Specificity
To further confirm the sequence-specific hybridization along with consistency and equivalent capacity among particles, we examined three types of particles differing by probe DNA sequence and shape (Table 1), as shown in Figure 6. All three particle types were mixed and divided into three batches, then hybridized with one of three fluorescently labeled complementary target DNAs. The brightfield micrographs of Figures 6a to c show the presence of three uniform microparticle types despite their difference in shape. The fluorescence micrographs of Figures 6d to f show that target DNA hybridization occurs only with particles containing complementary probe DNA. Importantly, minimal fluorescence from particles with non-complementary sequences hybridized under ambient conditions and minimal rinsing (i.e. 30 min, room temperature, TE buffer) clearly illustrates a highly selective hybridization assay afforded by the suspension array format with 3D hydrogel scaffolds unlike planar arrays32, 33. In addition, the fluorescence images show highly uniform and consistent fluorescence within each batch of hybridized particles as well as for different shapes. Average fluorescence intensities acquired from the particle centers for each shape (thus sequence) were also compared in a bar graph format, as shown in Figure 6g, indicating equivalent hybridization for each shape and sequence evaluated. This result is consistent with previous studies10, 14, 33, 34 on encoded particle-based suspension assays, and further attests to the robust nature of batch processing-based RM fabrication for rapid and selective assays via 3D hydrogel arrays.
Figure 6.
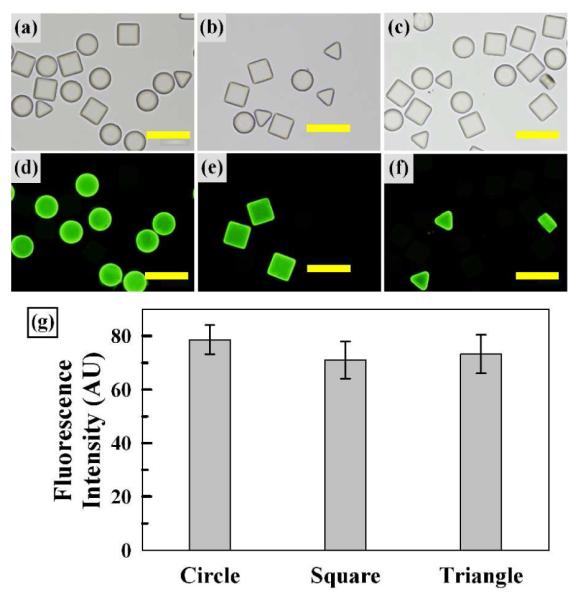
Sequence Specificity. Brightfield micrographs (a-c) of three microparticle types, all differing by the conjugated probe DNA sequence. Fluorescence micrographs (d-f) following hybridization with fluorescently labeled target DNA (200 nM) complementary to each particle type. Scale bars represent 200 μm. (g) Average fluorescence intensity graph for each particle shape. Error bars represent standard deviation from 3 particles.
Target DNA Sensitivity
Finally, we examined the responsiveness and sensitivity of the DNA-PEG microparticles produced under optimized fabrication conditions for target DNA, as shown in Figure 7. Probe DNA-conjugated microparticles were fabricated with 30% (v/v) PEG-DA, 50 μM probe DNA, 2% PI, 10% PEG 200, and hybridized with target DNA concentrations ranging between 0.01 nM and 2 μM. As shown in the fluorescence micrographs of Figures 7a to e, fluorescence intensity upon hybridization increased with target DNA concentration. In addition, Figures 7e and f show that fluorescence intensity reached saturation above 1 μM. Importantly, all of the micrographs clearly show uniformity among the microparticles and throughout their cross-sections. Overall, these images clearly show that the particles are responsive to target DNA concentrations and that low concentration target DNA is readily detectable.
To further quantitatively assess the sensitivity, we plotted the average fluorescence intensities as above and fit the data to an equilibrium binding model (Experimental Section), as shown in Figure 7g. First, every data point shows a small error bar throughout the target DNA range examined, confirming the uniformity of the particles and the robust nature of replica molding (RM)-based particle fabrication under optimized conditions. Second, the results illustrate typical equilibrium binding behavior where the response is linear at low target concentrations and reaches saturation at high target concentrations, similar to monolayer surface adsorption behavior represented by the Langmuir isotherm. The dotted red curve in Figure 7g represents the equilibrium binding model constructed from the fluorescence intensity measurements via a double reciprocal plot, and indicates a reasonably good fit of the data to the model. The trend of the data suggests that the hydrogel network provides favorable solution kinetics8 which may enable the probe DNA sites to behave like equal monolayer binding sites for hybridization. Third, the red linear regression line in the inset of Figure 7g further indicates the linear response behavior at low target concentrations. The exposure time used to obtain the images of Figures 7a to f was relatively low (0.5 sec) in order to ensure that the fluorescence was not at a saturating level for the higher target concentrations. Thus the strong fluorescence and saturation obtained at high target concentrations represents the extent of hybridization and not saturation of the microscope camera’s CCD detector (Olympus DP70). Furthermore, distinct uniformity and linear responses down to single nanomolar concentrations (i.e. high femtomole quantities) are readily detectable under the standard conditions employed in this study, involving one fluorescein molecule per target DNA and a 0.5 sec exposure time (1sec exposure for Figure 7g inset) with a standard epifluorescence microscope. In conclusion, the results shown in Figure 7 indicate robust, uniform, and sensitive features of our particles fabricated via RM for potential biosensing applications based on nucleic acid hybridization.
Conclusions
Particle-based suspension arrays offer attractive platforms for high throughput biosensing with low sample volume and rapid detection. Soft-lithographic fabrication of polymeric structures via replica molding (RM) possesses many inherent advantages including simple, robust, and inexpensive procedures as well as scalability and reliable duplication of complex structures with nanometer resolution. The simplicity of the microparticle fabrication strategy via RM presented in this report is shown by the use of a handheld UV lamp for photopolymerization rather than requiring delicate alignment and control of a high intensity UV source and other equipment necessary for photolithographic or microfluidic fabrication procedures10, 12, 35-37. In addition, the RM procedure presented in this report allows for simple collection of particles without the presence of unreacted monomer solution or other immiscible fluids. Extensive rinsing of particles fabricated via photolithography or microfluidics is required due to the presence of unreacted monomer solution or a continuous phase material, such as oil, with the fully polymerized particles9, 10, 33, 38. The robust nature of RM arises from the ability to examine of a wide range of parameters while not being limited by the viscosity, phase separation, or flow rate, which must be accounted for with microfluidic methods. Further, the ability to reuse excess preparticle solution along with all other components (e.g. silicon master molds, PDMS micromolds, etc.) attests to the cost efficiency of this procedure when considering the expense of synthesized probe DNAs for biosensing applications. For the RM procedure described here, 100 μL of preparticle solution will provide over 100 batches (1600 particles per batch) of particles where photolithographic and microfluidic applications would require significantly more preparticle solution to produce the same number of particles.
In this report, we examined several important parameters toward fabrication of uniform DNA-conjugated microparticles. Specifically, a simple humidity controlled environment minimized negative effects of rapid evaporation and showed the potential utility of in-mold hybridization for evaluating microparticles. Total fluorescence and 2D profiles of the hybridized particles showed how PEG-DA concentration affects hybridization and target DNA penetration depth. Probe DNA and photoinitiator (PI) concentrations were also examined to evaluate their effects on target DNA hybridization along with particle formation. Clear uniformity, sequence-specificity, and responsiveness down to single nanomolar concentrations (i.e. high femtomole quantities) under the standard conditions employed in this study support the simple, robust, and controllable nature of the replica molding process for fabrication of nucleic acid hybridization assay platforms. In-depth polymerization kinetics and multifunctional particle fabrication studies are currently underway. We envision that the results presented in this work may be readily scaled-up and/or applied to other systems involving other prepolymers (e.g. acrylamide, methacrylates, etc.) and low micrometer dimensions for rapid and high throughput assays.
Acknowledgement
This study was supported in part by the Converging Research Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2009-0082087), a grant from the Korea Health 21 R&D Project, Ministry of Health and Welfare (Project No: A062254), and the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean Government (MEST) (MEST No. R01-2008-000-11260-0). This study was also supported in part by award number K12GM074869 (TEACRS) from the National Institute of General Medical Sciences (Y.L). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- (1).Feng P. Mol. Biotechnol. 1997;7:267–278. doi: 10.1007/BF02740817. [DOI] [PubMed] [Google Scholar]
- (2).Gilbride KA, Lee DY, Beaudette LA. J. Microbiol. Methods. 2006;66:1–20. doi: 10.1016/j.mimet.2006.02.016. [DOI] [PubMed] [Google Scholar]
- (3).Ivnitski D, O’Neil DJ, Gattuso A, Schlicht R, Calidonna M, Fisher R. Biotechniques. 2003;35:862–869. doi: 10.2144/03354ss03. [DOI] [PubMed] [Google Scholar]
- (4).Hadd AG, Brown JT, Fandruss BF, Ye F, WalkerPeach CR. Expert Rev. Mol. Diagn. 2005;5:409–420. doi: 10.1586/14737159.5.3.409. [DOI] [PubMed] [Google Scholar]
- (5).Call DR. Crit. Rev. Microbiol. 2005;31:91–99. doi: 10.1080/10408410590921736. [DOI] [PubMed] [Google Scholar]
- (6).Rehman FN, Audeh M, Abrams ES, Hammond PW, Kenney M, Boles TC. Nucleic Acids Res. 1999;27:649–655. doi: 10.1093/nar/27.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kenney M, Ray S, Boles TC. Biotechniques. 1998;25:516–521. doi: 10.2144/98253pf01. [DOI] [PubMed] [Google Scholar]
- (8).Fotin A, Drobyshev A, Proudnikov D, Perov A, Mirzabekov A. Nucleic Acids Res. 1998;26:1515–1521. doi: 10.1093/nar/26.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Merkel TJ, Herlihy KP, Nunes J, Orgel RM, Rolland JP, DeSimone JM. Langmuir. 2009 doi: 10.1021/la903890h. In Press, 10.1021/la903890h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Meiring JE, Schmid MJ, Grayson SM, Rathsack BM, Johnson DM, Kirby R, Kannappan R, Manthiram K, Hsia B, Hogan ZL, Ellington AD, Pishko MV, Willson CG. Chem. Mater. 2004;16:5574–5580. [Google Scholar]
- (11).Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Nature Mater. 2006;5:365–369. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- (12).Dendukuri D, Gu SS, Pregibon DC, Hatton TA, Doyle PS. Lab Chip. 2007;7:818–828. doi: 10.1039/b703457a. [DOI] [PubMed] [Google Scholar]
- (13).Chapin SC, Pregibon DC, Doyle PS. Lab Chip. 2009;9:3100–3109. doi: 10.1039/b909959j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pregibon D, Toner M, Doyle PS. Science. 2007;315:1393–1396. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- (15).Zhao XM, Xia YN, Whitesides GM. J. Mater. Chem. 1997;7:1069–1074. [Google Scholar]
- (16).Xia YN, Whitesides GM. Ann. Rev. Mater. Sci. 1998;28:153–184. [Google Scholar]
- (17).Qin D, Xia YN, Whitesides GM. Nat. Protoc. 2010;5:491–502. doi: 10.1038/nprot.2009.234. [DOI] [PubMed] [Google Scholar]
- (18).Dendukuri D, Panda P, Haghgooie R, Kim JM, Hatton TA, Doyle PS. Macromolecules. 2008;41:8547–8556. [Google Scholar]
- (19).Odian G. Principles of Polymerization. 4th ed John Wiley & Sons Inc.; Hoboken: 2004. [Google Scholar]
- (20).Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. J. Am. Chem. Soc. 2005;127:10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- (21).Zhang H, et al. New J. Phys. 2009;11:075018. [Google Scholar]
- (22).Gratton SEA, Williams SS, Napier ME, Pohlhaus PD, Zhou Z, Wiles KB, Maynor BW, Shen C, Olafsen T, Samulski ET, DeSimone JM. Acc. Chem. Res. 2008;41:1685–1695. doi: 10.1021/ar8000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yi H, Wu L-Q, Ghodssi R, Rubloff GW, Payne GF, Bentley WE. Anal. Chem. 2003;76:365–372. doi: 10.1021/ac034662t. [DOI] [PubMed] [Google Scholar]
- (24).Rasband WS. U.S. National Institutes of Health: Bethesda; Maryland, USA: 1997-2009. http://rsb.info.nih.gov/ij/ [Google Scholar]
- (25).Jackman RJ, Duffy DC, Ostuni E, Willmore ND, Whitesides GM. Anal. Chem. 1998;70:2280–2287. doi: 10.1021/ac971295a. [DOI] [PubMed] [Google Scholar]
- (26).Berthier E, Warrick J, Yu H, Beebe DJ. Lab Chip. 2008;8:852–859. doi: 10.1039/b717422e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Badaire S. p., Cottin-Bizonne C. c., Woody JW, Yang A, Stroock AD. J. Am. Chem. Soc. 2006;129:40–41. doi: 10.1021/ja067527h. [DOI] [PubMed] [Google Scholar]
- (28).Lee JH, Prud’homme RK, Aksay IA. J. Mater. Res. 2001;16:3536–3544. [Google Scholar]
- (29).Kurdikar DL, Peppas NA. Polymer. 1995;36:2249–2255. [Google Scholar]
- (30).Mellott MB, Searcy K, Pishko MV. Biomaterials. 2001;22:929–941. doi: 10.1016/s0142-9612(00)00258-1. [DOI] [PubMed] [Google Scholar]
- (31).Russell RJ, Axel AC, Shields KL, Pishko MV. Polymer. 2001;42:4893–4901. [Google Scholar]
- (32).Yershov G, Barsky V, Belgovskiy A, Kirillov E, Kreindlin E, Ivanov I, Parinov S, Guschin D, Drobishev A, Dubiley S, Mirzabekov A. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4913–4918. doi: 10.1073/pnas.93.10.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pregibon DC, Doyle PS. Anal. Chem. 2009;81:4873–4881. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tan WS, Lewis CL, Horelik NE, Pregibon DC, Doyle PS, Yi H. Langmuir. 2008;24:12483–12488. doi: 10.1021/la802089q. [DOI] [PubMed] [Google Scholar]
- (35).Revzin A, Russell RJ, Yadavalli VK, Koh W-G, Deister C, Hile DD, Mellott MB, Pishko MV. Langmuir. 2001;17:5440–5447. doi: 10.1021/la010075w. [DOI] [PubMed] [Google Scholar]
- (36).Dendukuri D, Tsoi K, Hatton TA, Doyle PS. Langmuir. 2005;21:2113–2116. doi: 10.1021/la047368k. [DOI] [PubMed] [Google Scholar]
- (37).Xu S, Nie Z, Seo M, Lewis P, Kumacheva E, Stone HA, Garstecki P, Weibel DB, Gitlin I, Whitesides GM. Angew. Chem. Int. Ed. 2005;44:724–728. doi: 10.1002/anie.200462226. [DOI] [PubMed] [Google Scholar]
- (38).Yuet KP, Hwang DK, Haghgooie R, Doyle PS. Langmuir. 2009;26:4281–4287. doi: 10.1021/la903348s. [DOI] [PubMed] [Google Scholar]