

Abstract
This review encourages the reader to consider cerebral vascular disease beyond the traditional clinical end points of major motor and speech strokes and to consider the possible impact of embolic cerebral vascular disease on vascular cognitive decline. The paper examines the issue of “silent” strokes in the relationship between the structural stability of atherosclerotic carotid plaque and the development of nonmotor symptomatology, including cognitive decline. It addresses the question of the role of carotid emboli in “silent” stroke and their cognitive sequelae. In a study of endarterectomy patients, we relate plaque elasticity and its development of mechanical strain features and thinning of stabilizing fibrous cap at the point of these mechanical strain features. The possibility that microemboli from such mechanically unstable carotid plaques could contribute to “silent strokes” lead to a study of cognitive function in such patients. A linear relationship between the process of mechanically unstable areas of carotid plaques and cognitive decline suggests a contributory role for such a process in “silent strokes”.
Keywords: aging, atherosclerosis, carotid endarterectomy, cognitive decline, dementia, stroke
This review is meant to encourage those involved in the treatment of cerebrovascular disease to look beyond traditional clinical endpoints of motor and speech stroke. Our study examines the relationship between the structural stability of carotid atherosclerotic plaque forming at the bifurcation of the common internal/external carotids and the symptomatology of such lesions. The theory behind this body of work is the hypothesis that carotid atherosclerosis stroke presents not only as a classical episodic clinical condition, but may also involve elements of a continuous process involving large and small vessel circulations, microcirculatory changes, cellular metabolic resistance to ischemia and micro embolic events. Recent studies suggest for every recognized clinical stroke, 5 silent strokes take place1. The patient implications are enormous as modern imaging suggests 11 million “silent strokes” occur yearly in the US with poor understanding of the pathophysiology or cognitive consequences for our patients2. Within this framework, we choose to study the hypothesis that carotid artery atherosclerosis is likely to cause microemboli, as well as, classic macroemboli, which may result in more subtle disturbances than those ordinarily detected by more obvious clinical events such as stroke and transient ischemia attacks. Understanding the structural plaque abnormalities, which render a carotid plaque unstable and at risk of embolization would help to predict and treat individuals who are likely to suffer not only classic episodic major strokes, but also cognitive impairment from the contribution of microemboli to this overall disease process. We have previously described a non-invasive ultrasound based measure of plaque structure to investigate this possibility.
In the review we consider the strong possibility that the cumulative effects of potentially preventable “silent strokes” may have measurable functional sequelae primarily as noted in decreased executive function. Such processes synergistically lead to progressive dementia in concert with other pathophysiologic causes such as hypertension, diabetes and Alzheimer’s disease. The presentation of stroke often takes place due to the presence of atherosclerotic disease which may be embolizing even when total brain flow rate is maintained by collateral vessels. The possibility that a component of the pathophysiology such as unstable atherosclerotic plaque may be noninvasively detectable calls for further study of brain changes, plaque structure, and the frequency of emboli in patients at risk. Stabilizing such plaques, whether medically with antioxidants, dietary, or surgically by structural stabilization by stenting or excision of the plaque by endarterectomy, all may be testable hypotheses as we increase our understanding of the scope and breadth of cerebrovascular disease.
Carotid Atherosclerosis
Carotid artery atherosclerosis and its relationship to stroke have clinically been an area of considerable research focus due to the devastating effects of artery to artery emboli and the potential for diagnostic and therapeutic advances. Indeed, the accessibility beneath the skin of the neck of this source of stroke to noninvasive study has allowed the opportunity for far greater understanding of the pathophysiology of stroke disease processes. A major endpoint of most carotid atherosclerosis studies has been major clinical stroke3 manifested generally by motor, sensory, speech and vision deficits. Due to the eloquence of individual regions of the brain, this has allowed us to understand the focal presentation of major carotid embolic disease. It, however, has not lent itself to increased understanding of the role of carotid atherosclerosis in microembolic disease which may present as a diffuse load of difficult to detect microemboli.
Several lines of evidence suggest that in carotid atherosclerotic disease, brain atrophy and vascular cognitive impairment may be related, suggesting that symptomatology may exist far beyond the clinically recognized visual, sensory, motor or speech deficits of transient ischemic attacks (TIA) and fixed stroke4,5. Doppler studies searching for carotid emboli distal to symptomatic carotid plaques, suggest that at the time of presentation with a motor TIA, multiple other clinically unrecognized emboli are passing to the brain4. Studies of coronary surgery patients have measured a significant number of cerebral emboli occurring up the carotids during these procedures and have documented multi-factorial cognitive decline in the post-operative patients as compared to preoperative cognition5. Leukoariosis white matter changes are particularly frequent in patients with dementia and stroke6 and are associated with a particularly bad outcome in patients with symptomatic carotid stenosis6.
It is clear that ischemic cerebrovascular disease is a complex process with contributions from macro and microemboli, large and small vessel occlusion and small vessel regeneration within the target organ. These processes are likely to be related by the basic promoters of vessel degeneration which include inflammation, atherogenesis, vascular recruitment of macrophages, lipid deposition and neo vascularity of the walls of these vessels. We have previously published studies suggesting that understanding of the presence and importance of these processes can be aided by a careful analysis of the genetic up-regulation which heralds the development of symptomatology within atherosclerotic plaques7,8. While the processes of atherogenesis in vascular degeneration are systemic throughout the body, the carotid bifurcation provides a particularly accessible site in vivo in which we may noninvasively examine the plaque, recover the specimen in life through carotid endarterectomy and perform genetic and immunochemical and histological examination of the disease process to use these results as predictors for future pathophysiology.
Silent Strokes and Vascular Cognition Decline
What causes silent strokes? The logic behind the study of vascular cognitive impairment, carotid plaque instability, microemboli, and brain imaging is an attempt to increase our understanding of the pathophysiology of one important cause of silent strokes. In 1998 it was estimated that in the United States there were 770,000 clinical cerebrovascular accidents. At the same time, imaging studies suggested 9,440,000 silent strokes and 1,940,000 micro hemorrhages2. Other studies have suggested that for every one clinical stroke recognized, five silent strokes take place1. The importance of these 11 million silent strokes and their cumulative effect on patients is only now being recognized. Indeed, only thirteen percent of strokes are preceded by a clinical warning, such as classical TIAs. The need for improved risk profiling and early intervention is obvious. Accumulating evidence suggests that these silent strokes have a risk profile similar to those of the major strokes; therefore, many may have the same embolic, macro and microvascular characteristics. The data suggests that their presentation may be best understood by examination of cognitive decline, especially regarding executive function. The relationship between cognitive impairment and stroke is of vital importance. Sixty-four percent of stroke patients over the age of 65 have cognitive impairment, not dementia but loss of executive function. One-fourth of cognitively impaired patients over 65 have had a stroke9. One in three of people over 65 go on to stroke and/or dementia and evidence suggests these two processes may be linked10,11,12.
Functional loss after stroke is of vital importance for the patient, family and the quality of life. Pathological studies suggest an equivalence even of classic Alzheimer’s and stroke suggesting an interaction between stroke and dementia. This is present not only in executive function decline, but also dementia. In the classic Nun study13,14,15, the pathological changes of dementia were more common than the actual clinical presentation of Alzheimer’s disease. That is, post-mortem changes were more common than pre-mortem dementia. Only ½ of Nuns with Alzheimer’s pathology had dementia; however, if their MRI suggested a structural lesion of a stroke, dementia was far more common and if they had had a clinical stroke, 93% of this subgroup presented with cognitive decline12,13,15. Animal studies have further suggested that unilateral stroke may affect bilateral function, especially regarding cognition. These clinical studies suggest that stroke may be a trigger in a patient susceptible to Alzheimer’s. Elias’ studies16 suggest that the examination of cognitive testing, especially decline in executive function greater than memory, is a predictor of stroke risk over the next 10 years. This suggests that an ongoing process, such as subclinical emboli, may be existent for some time in such patients. Steps such as ultrasound and Doppler may recognize early on, not only the presence of the emboli, but also a potential source such as unstable carotid atherosclerotic plaque. This may greatly increase our understanding of the pathophysiology and also suggest future therapeutic interventions16.
Carotid Atherosclerosis Plaques and Emboli
Cerebrovascular disease is a major public health problem, which is only expected to increase in future years as our population ages at an accelerated pace.
One important and quite plausible cause of both the imaged silent strokes and vascular cognitive decline, is the loss of structural stability and breaking up of atherosclerotic plaque, which over time may initiate the cerebral decline. We studied one important site which is accessible for noninvasive examination that is carotid atherosclerotic plaque and tested the hypothesis that the physical structural stability of plaque is measurable. When this stability is disturbed, we hypothesize that it is related through increased emboli to a decline in cerebral function. If this relationship is established, it opens wide frontiers for further investigation of the pathophysiology and potential future prevention of such a devastating disorder as silent stroke and vascular cognitive decline.
Imaging criteria for treatment until now has focused primarily on percent stenosis and ulceration of the carotid vessel. However, the literature suggests that the process of developing symptoms of carotid plaques mostly involves factors beyond the geometry of such lesions. Thus, we must consider the biochemical, genetic and physical structural properties of the plaques, which may predispose to clinical decline.
A series of studies have suggested that cerebral emboli as measured by transcranial Doppler (TCD) may predict vascular cognitive impairment. In studies17,18, in patients with carotid stenotic disease transcranial Doppler placed distal to the lesion and measured for a period of one hour showed a bimodal distribution. The presence of measurable microemboli during that time correlated with functional cognitive impairment suggesting a functional deficit beyond that of traditional markers of stroke symptomatology.
Considerable debate remains regarding the cause of vascular cognitive impairment and brain atrophy. Multiple cortical infarcts are believed to cause dementia19 and cerebrovascular disease may increase the severity of classic Alzheimer’s disease19. However, studies have suggested that this vascular component may be a result of cumulative microvascular changes with white, gray and hippocampal volume loss over time being more important than specific subcortical lacunes19. The primary microvascular process may be worsened by chronic micro emboli. The suspected relationship of significant atherosclerotic carotid disease to cognition suggests that the pathophysiology of cognitive decline may be influenced by a microembolic process taking place in select carotid plaques above and beyond those processes which produce large clinically recognizable strokes and TIAs. The need to look at the pathophysiology of carotid atherosclerotic disease beyond simple parameters of degree of stenosis or irregularity of surface has led us to look at the structural stability of these plaques as a potential marker for risk of both clinically evident emboli as well as subclinical microemboli.
Carotid artery atherosclerosis is a particular presentation of the larger systemic disease of atherosclerosis which affects many organs. Although stroke is a major cause of death and disability in the USA, many studies have suggested that symptomatically sensitive areas such as the coronary arteries, the carotid arteries and the descending aorta have a stereotypic pattern of plaque development, which is likely to be influenced by a wide variety of modifiers20,21,22,23,24,25.
In humans, carotid artery atherosclerosis is far more common than the major strokes, i.e. sudden motor, sensory, visual and speech deficits, which clinicians recognize from carotid embolic disease26,27. Proliferation of smooth muscle cells, formation of connective tissue, cholesterol deposition, calcification and extravasation of inflammatory cells are all thought to promote atherosclerotic plaque formation. Less is known about the factors that predispose a given atherosclerosis plaque to become symptomatic. At the systemic level, smoking, dietary lipid intake, diabetes, hypertension and infection might promote plaque maturation and rupture28,29,30,31,32.
The clinically evident neurological deficits that follow stroke (i.e. motor, sensory, speech and vision) have been used to classify the stroke and provide some information about the prognosis and pathophysiology. Studies have suggested that a consistent increase in atherosclerosis is seen with age and it may be studied by such noninvasive testing as ultrasonic analysis of plaque at the carotid arteries3,33,34,35,36,37,38,39. The disease process may be further modified by smoking, dietary lipid intake, diabetes, hypertension, and possibly by other factors such as infection, inflammation, and flow characteristics22,26,40,41,42,43,44. It is the latter, the distribution of local flow, which suggests a particular vulnerability of the carotid bifurcation in the neck to atherosclerotic development. The carotid arteries are extremely high flow vessels, as 20% of the cardiac output is delivered to the brain mainly through these 4-5 mm diameter vessels. Flow is therefore robust but remarkably laminar. This flow is disrupted considerably at the first major flow divider, the bifurcation of the internal and external carotids of the neck. Long-standing flow studies have suggested that turbulence here creates a back wall injury on the internal carotid at its origin, at which point a constant process of repair and cellular activity may be ongoing45,46,47,48. Local events significantly affect the progress of atherosclerosis. Theories of atherogenesis suggest that plaque may form at this area and may be enhanced by processes which increase local oxidation in the presence of abnormal circulating cholesterol moieties49,50,51,52. We have studied these events for signature genetic markers7,8.
The clinical pathophysiology of carotid atherosclerosis had originally emphasized flow stenosis. However, since the pioneering work of C. Miller Fisher, it has been suggested that a second mechanism present in both the coronaries and carotids may be of great importance, the mechanism of artery-to-artery emboli53,54. Because of the extreme eloquence of critical regions of the cerebral vasculature, even moderate emboli may produce devastating and clinically relevant consequences when arriving at the brain. At the same time multiple small emboli may cause atrophy and multiple small infarcts that are not recognized as a single event but rather as a progressive decline in function and cognition. Clinical trials of treatments for carotid atherosclerotic disease such as, ACAS, the asymptomatic carotid artery plaque study have taken the practical NASCET step of categorizing carotid atherosclerosis by the degree of narrowing or stenosis of flow38,55,56,57,58. This has been necessary to allow variable methods of measuring carotid atherosclerosis, angiography, etc. to make meaningful comparisons of measurements. Nevertheless, each study has emphasized the importance of emboli in the pathophysiology of clinical symptoms. Such studies have led to the logical conclusion that it would be clinically important if physicians could identify not only the presence of atherosclerotic plaques, but also those plaques which are biochemically or functionally more likely to produce symptoms, generally by the creation of emboli59. That is to say, can we clinically differentiate carotid atherosclerotic plaques at greater risk of causing symptoms and then direct therapies towards those aspects which predispose a plaque to becoming symptomatic?
Structural Stability of Carotid Plaques
Structural stability of a carotid plaque is a result of its chemical composition, cellular material and new vessel formation. The main components of atherosclerotic plaque are connective tissue extracellular matrix, including collagen, proteoglycans, and fibronectin elastic fibers; crystalline cholesterol, cholesterol esters, and phospholipids; and cells such as monocytederived macrophages, lymphocytes, smooth muscle cells and new endothelial lined vessels (Fig. 1). Atherosclerosis develops as lipid is laid down within the inner layers of the artery wall. As the lipid pool enlarges, fibrin and calcium are incorporated within the plaque. Due to the cellular components of the plaque it is not inert, but has a genetic signature, metabolic needs and cellular representation which require its own microvasculature. The structure of the plaque itself may be fissured by the neovascular channels forming within this growing and metabolically active lesion. Rupture of the new blood vessels within the plaque wall or blood entering from the lumen, secondary to fissuring of the plaque, may result in hemorrhage and embolization of contents, platelets and thrombus into the cerebrovascular circulation. All of these events may result in a measurable loss of structural stability of the plaque60,61.
Figure 1.
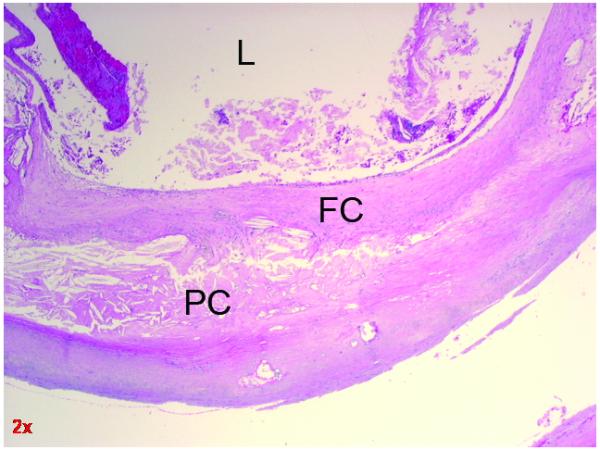
Cross section of carotid atherosclerosis showing fibrous cap adjacent to the lumen. L = Lumen FC = Fibrous Cap PC = Plaque
Elastic Properties of Plaque
Plaque vulnerability is determined primarily by the mechanical (elastic) properties of the vessel wall and the plaque composition62,63,64. Various studies have indicated that the pulsatile pressure induced due to blood flow may rupture the thin fibrous cap overlying fatty tissue (or lipid rich lesions) on the plaque, which may lead to subsequent thrombosis62. Techniques that are able to characterize the elastic properties of plaque may therefore provide clinical information that may have a significant impact on patient care. Imaging of tissue elastic properties using ultrasound has shown great promise in the detection of cancers in the breast and prostate65,66,67,68,69,70,71,72,73, with several investigators using this modality to examine vascular tissue properties73,74,75,76. Elastography is one of the methods that have been used in imaging the elastic properties of tissue68,69,72,73. Characterization of the plaque composition in vascular tissue may significantly help in the selection of appropriate interventional techniques.
Features Leading to Embolization
Randomized clinical trials have attempted to address the problem of symptomatic vs. asymptomatic plaques by utilizing tools such as measurements of ulceration by angiography, magnetic resonance angiography, or duplex ultrasound Doppler scanning35,77,78. These studies have not developed sufficient power to identify useful clinically predictive markers for future plaque symptomatology other than the presence of a significant stenosis with or without a prior ischemic event in the same vascular distribution.
It should be understood that the inability to make such correlations in the past may be due to our restriction of symptomatology to classic stroke or death. It may also be due to the lack of innovative ways to assess the plaque’s structural stability and histopathological characteristics.
It has been shown that macroscopic intraplaque hemorrhage was significantly more common in plaques removed from symptomatic patients21,79,80,81. This classified carotid plaques as “hard” (predominantly composed of collagen or calcium) or “soft” (containing atheromatous debris or intraplaque hemorrhage). In these studies, soft plaques were significantly more common in symptomatic carotid lesions21,23. Some studies report gross macroscopic findings, while others examine the tissue microscopically. Most of the previous studies have noted the presence or absence of specific features, but not in quantifiable terms. Intraplaque hemorrhage may lead to the development of clinical neurological symptoms if this process then ruptures into the luminal region of the artery24. Thinning of the fibrous cap, which usually functions to stabilize the luminal surface of the plaque, foam cell infiltration of the fibrous cap, and an inflammatory infiltrate all predispose the fibrous cap to rupture, for which there are strong correlations with symptomatic carotid artery disease59,82,83. It appears that plaque instability is a major factor in the development of thromboembolic events but a noninvasive method of measuring this has not been well established.
Other theories such as examining excised plaques for ulceration, inflammatory changes, age or lipid composition, have not clearly identified those factors which predispose plaque to symptomatic embolization20,48,84,85,86,87. However, these prior studies may not have had the power to arrive at these conclusions. A further explanation may be that molecular and biochemical mediators, as yet unidentified, are the factors which are predisposed to such changes.
Gene Expression in Symptomatic Carotid Atherosclerosis
A series of experiments have been done in our laboratories to establish the methodologies and the logic behind the hypothesis relating measurable carotid atherosclerotic plaque stability to embolization. In the first series of experiments we looked for differential gene expression in symptomatic carotid atherosclerotic plaques that presented with clinically recognizable symptoms as opposed to those patients who had not shown such major symptomatology and therefore, were classically considered asymptomatic as in the asymptomatic carotid stenosis study (ACAS).
There were ten patients in the genetic study. These patients are typical of the entire plaque study and representative. Six were symptomatic (3 males and 3 females) and four were asymptomatic (2 males and 2 females). The average age was 60 +/− 15 of the symptomatic group and 63 +/− 10 of the asymptomatic group. All six of the symptomatic group had presented with a stroke and none of the asymptomatic had symptoms. The mean degree of stenosis was 73% in the symptomatic and 72% in the asymptomatic group. None of the patients in either group had a cardiac infarction. In the symptomatic group, 4 were hypertensive and 3 in the asymptomatic group were hypertensive. Both groups contain one patient who was a diabetic. The only major clinical difference between the two groups was the presence of symptomatology. Neuropathologic examination did not show a difference in the typical characteristics of calcification, gross lipid hemorrhage or ulceration. Therefore genetic differences between these groups suggest the presence of factors expressed in a fashion beyond classic neuropathological examination.
Patients underwent preoperative evaluation imaging, which estimated the degree of stenosis. Using Affymetrix human GeneChip microarray set we evaluated the plaque tissue from the symptomatic and asymptomatic patients for the mRNA expression profiles8. __Of the 44,860 probe sets representing various mRNAs, 236 transcripts were expressed more abundantly in the symptomatic plaques compared to asymptomatic plaques. Of these 213 (~90%) transcripts were those that code for proteins which control cell growth, cell maintenance, cell adhesion and motility, signal transduction, organogenesis, nucleotide metabolism, amino acid and protein metabolism indicating an active cell proliferation process (Fig. 2). This indicates that carotid atherosclerotic plaques from symptomatic stroke patients share the molecular fingerprints to develop in a neoplastic fashion7. The other groups of transcripts expressed more abundantly in the symptomatic plaques are those that control ionic homeostasis and those that participate in the progression of degenerative neurological diseases (Alzheimer’s disease, ALS and Huntington’s disease) and epilepsy. This indicates that symptomatic plaques are molecularly and biochemically more active than the asymptomatic plaques. It further suggests a possible gene expression relationship between carotid atherosclerosis and later diagnosed degenerative neurological disorders including cognitive impairment8.
Figure 2.

Gene ontological analysis of the biological functions of transcripts more abundantly expressed in the symptomatic plaques showed a neoplastic phenotype.
Neovascularity with Carotid Atherosclerosis
Gene expression analysis also showed increased expression of many transcripts that are the putative promoters of cell division and angiogenesis in the symptomatic plaque (Fig. 3). This supports a strong possibility of increased microvascular formation in symptomatic plaques over the asymptomatic plaques. In a series of patients we measured the density of new vessels in the plaque of classically symptomatic and asymptomatic patients. The median neo-vessel density (number of vessels/area) within the fibrous cap and the plaque proper for plaques from symptomatic patients are significantly higher than asymptomatic patients. This suggests that symptomatic plaques may be more prone to rupture and embolization due to the changes in the vessels which fissure the fibrous cap88.
Figure 3.

Increased expression of genes that control angiogenesis in symptomatic plaque samples.
Plaque Strain distribution
Having established a potential mechanism by which a plaque may become physically unstable based on our genetic analysis and the histopathophysiology of new vessel formation, we then began to establish a reliable non-invasive measurement to study the local strain distribution within the plaque itself, both ex vivo89,90 and in vivo61,91. By having the opportunity to measure these parameters from endarterectomy patients we were able to characterize this technology by comparison of noninvasively obtained preoperative data followed by ex vivo analysis of the same plaque after its removal at surgery and histopathological analysis with point by point comparisons of identical regions.
Elastograms of ex-vivo Plaque Tissue
Apparatus and Method
Surgically excised carotid plaque tissue was encased in a gelatin cube mold with 80 mm sides and studied ex vivo using a 7.5 MHz linear array transducer window length of 3 mm with a 75% overlap between data segments. Axial strains in plaque tissue were evaluated over 114 Regions of Interests (ROI) from 44 patients were plotted versus the ultrasound attenuation coefficient. This exhibits a linear relationship. This data demonstrated ex vivo the ability to define regions of interest of friable plaque material with ultrasound elasticity measurements60,89,90.
NEUROPSYCHOLOGICAL ASSESSMENT
Baseline Screening Prior to Surgery
A consecutive series of 25 preoperative carotid endarterectomy patients were administered a mental status screening measure (Repeatable Battery for the Assessment of Neuropsychology Status [RBANS]) that assesses cognitive status and executive functions, immediate and delayed memory, language function, attention, spatial/construction skills, and delayed memory. As a group, these patients showed substantial cognitive pathology prior to surgery. It is likely that this profile of cognitive impairment underrepresents the degree of pathology that would be revealed by more traditional cognitive assessment (Fig. 4).
Figure 4.

Age adjusted standard scores demonstrating a pattern of generally depressed mental status across tested cognitive domains. Relationships were in the similar direction, but weaker, across tests of visual perception, language and attention r’s from −.37 to −.31).
Having obtained data for the maximum accumulated axial strain value61 in a series of pilot patients, we then studied the neuropsychological status in a subgroup of patients undergoing in vivo elastographical imaging for structural strain of their carotid plaques. These were all patients with significant carotid atherosclerotic plaques as measured by NASCET and ACAS criteria that presented both with and without major motor, sensory, vision or speech clinically recognizable symptoms and were scheduled to undergo carotid endarterectomy. All patients underwent strain imaging analysis60,61 and neuro-psychological assessment.
In-vivo Strain Imaging
The maximum accumulated axial strain in plaque was estimated as an index of plaque vulnerability to rupture. The presence of the high strain region (brighter region) corresponds to plaque from the B-mode and color Doppler image in Fig, 5. For any given location, variations in the frame strain and accumulated strain will be seen over the cardiac cycle60. Greatest accumulated strain suggests greatest instability of plaque60.
Figure 5.
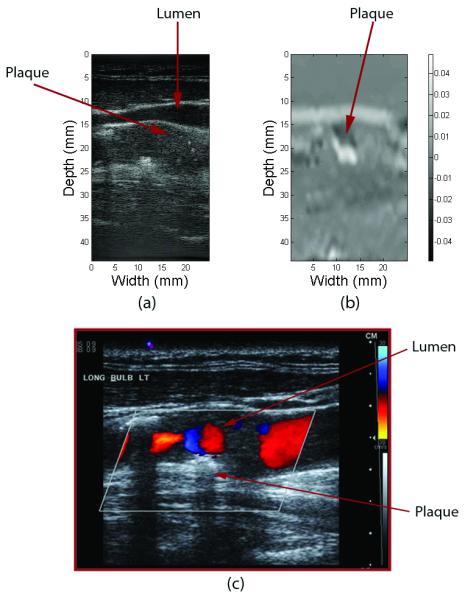
(a) B-mode gray scale ultrasound image, (b) 2-D elastogram, (c) B-mode with colorflow of an in-vivo carotid artery with plaque visible near the wall of the vessel close to the bifurcation. Note also the presence of turbulent flow around this region. Elastographically strain here appears as a more friable or more likely to fracture region and is depicted as the brighter region.
In the context of the above pattern of general mental status decline described above, there was a considerable degree of individual variability. To determine whether vessel strain was associated with variability in neuropsychological status, the RBANS total score was regressed on the maximum accumulated axial strain index for the 10 patients who had undergone assessment of both cognition and strain. There was a significant correlation between strain and RBANS total performance (p=.03), with higher strain associated with poorer cognitive performance, the relationship depicted in Fig. 6. Inspection of additional RBANS scales showed similar significant associations between strain and immediate (p=.03) and delayed memory (p=0.3). Relationships were in the similar direction, but weaker across tests of visuoperception, language and attention r’s from −.37 to −.31.
Figure 6.

Plot of the RBANS total score versus maximum accumulated frame strain (single observer measurement) over a cardiac cycle.
Discussion
The present state of our understanding raises several important pathophysiology questions of regarding the circumstances under which carotid plaques become unstable. Several forces are probably working in concert. The first is understanding that the plaque itself has multiple components with active metabolism, cell division, and nutritional needs. This is especially true of endothelium itself, which is serving to protect circulating blood components from contact with the thrombogenic core of the plaque. Progressive increase in plaque development will elevate that endothelium further and further from its vas vasorum. In light of the genetic material presented here, it is likely that an angiogenic stimulus occurs resulting in the new vessel formation which fissure the plaque itself. These in turn may structurally serve to destabilize the integrity of the plaque as it pulsates. They additionally can be a source of microhemorrhage within the plaque leading to sudden failure and ulceration of the endothelium and instability of the plaque. Speculation exists as to the relative contributions of brittle and soft plaque, especially if they result in an interface creating instability with pulsation. At the same time, flow dynamics must be considered which change dramatically as the plaque protrudes into the lumen. Velocity and turbulence of flow also serve to denude vulnerable endothelium, exposing more thrombogenic material to circulating blood components. This process is enhanced by the local inflammatory process and migration of inflammatory cells to the lesion. Our local gene data suggests a local alteration in immune function. The process of symptomatology, therefore, is likely to be active with vascular and cellular components as well as the physical forces of pulsation and vessel jetting. Treatment contributions therefore should also be multiple affecting the physics of flow, the rheology of blood components, inflammation of the vessel wall, genetics of vessel wall and thrombotic potential. These forces, as demonstrated in our work, are regulated by definable genetic processes and local upregulation of genes which orchestrate the process.
While this review concentrates on a source of microembolic damage to the brain, it is important to note that the processes of neovascularity of the carotid plaque including lipid deposition, angiogenesis and new vessel formation are very similar to that which is seen in brain parenchyma itself. It is unlikely that macro and microvascular forces work separately but rather much more likely, that they represent a continuum of presentations driven by the genetically determined response of the microvasculature to injury, inflammation, lipid deposition and new vascular permeability. It is intriguing to speculate that future research may systemically target both processes by understanding that the macrovascular changes seen in a carotid plaque are themselves determined by microvessel changes within the large vessel walls themselves. These commonalities of vascular biology may be most important for future diagnostic and therapeutic endeavors of cerebrovascular disease.
CONCLUSION
These preliminary results suggest a direct relationship between increasing axial strain within atherosclerotic plaque and decreased cognitive function. This is present in classically symptomatic and asymptomatic patients. The relationship suggests that the processes by which clinically recognizable major TIA or stroke symptoms of motor, vision, speech or sensation are seen, may be inadequate to recognize those processes by which cognitive decline take place. Hence, these high strain deficits suggest cognitive impairment whether the patients presented with major motor symptomatology or not. If proven, this may suggest that subclinical emboli from structurally unstable plaques are taking place for a much greater time that clinically recognizable strokes and TIAs. These subclinical microemboli may cause cumulative decline, which may in the future be measured by loss or atrophy of white matter and hippocampal volume, as well as, by a clinical measure of cognitive defects. The clear need is for ongoing carotid clinical studies of the role of unstable plaque and microemboli in the pathophysiology of vascular cognitive decline. The recognition of such pathophysiology in the establishment of a useful noninvasive diagnostic tool could direct our medical and surgical treatments at an earlier stage in the disease by recognizing patients at risk based on their abnormal structural elasticity strain measurements within their vulnerable plaques.
Article Summary.
In this article, the authors review the recent work regarding the symptomology of carotid atherosclerosis and the diagnosis of vascular cognitive decline. Increasing emphasis is being placed on the importance of the quality of life, especially regarding the cognition and executive function of patients. With additional testing, it has become clear that some aspects of this decline may be related to atherosclerotic disease, and therefore may be amenable to preventive therapy. In this paper the authors discuss those aspects which may cause silent carotid emboli to result from destabilized atherosclerotic plaques. Looking at the genetic, anatomic, and physical features of atherosclerotic plaque suggest that mechanically unstable areas of the carotid artery plaques may be related to vascular cognitive decline. This suggests that further investigation of potentially treatable embolic sources for multiple, small, or previously considered “silent strokes” would be a fruitful area of future investigation.
Footnotes
There are no disclosures from any of the authors listed above.
Contributor Information
Robert J Dempsey, University of Wisconsin School of Medicine and Public Health Department of Neurological Surgery.
Raghu Vemuganti, University of Wisconsin School of Medicine and Public Health Department of Neurological Surgery.
Tomy Varghese, University of Wisconsin School of Medicine and Public Health Department of Medical Physics Electrical and Computer Engineering.
Bruce P. Hermann, University of Wisconsin School of Medicine and Public Health Department of Neurology.
References
- 1.Vermeer SE, Prins ND, Heijer HA, et al. Silent Brain Infarcts and the Risk of Dementia and Cognitive Decline. NE Jrnl of Med. 2003;348:1215–21. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 2.Leary M, Saver J. Annual incidence of first silent stroke in the United States: A Preliminary Estimate. Cerebrovascular Diseases. 2003;16(3):280–285. doi: 10.1159/000071128. [DOI] [PubMed] [Google Scholar]
- 3.Fisher M, Martin A, Cosgrove M, Norris JW. The NASCET-ACAS plaque project. North American Symptomatic Carotid Endarterectomy xTrial. Asymptomatic Carotid Atherosclerosis Study. Stroke. 1993 Dec;24(12 Suppl):124–5. (discussion:131-2) [PubMed] [Google Scholar]
- 4.Droste DW, Decker W, Siemens HJ, Kaps M, Schulte-Altedorneburg G. Variability in occurrence of embolic signals in long term transcranial Doppler recordings. Neurol Res. 1996 Feb;18(1):25–30. doi: 10.1080/01616412.1996.11740372. [DOI] [PubMed] [Google Scholar]
- 5.Siebler M, Sitzer M, Rose G, Steinmetz H. Microembolism in Carotid Artery Disease. Echocardiography. 1996 Sep;13(5):529–536. doi: 10.1111/j.1540-8175.1996.tb00931.x. [DOI] [PubMed] [Google Scholar]
- 6.Streifler J, Eliasziw M, Benavente O, et al. Prognostic Importance of Leukoaraiosis in Patients With Symptomatic Internal Carotid Artery Stenosis. Stroke. 2002;33:1651. doi: 10.1161/01.str.0000018010.38749.08. [DOI] [PubMed] [Google Scholar]
- 7.Vemuganti R, Dempsey RJ. Carotid atherosclerotic plaques from symptomatic stroke patients share the molecular fingerprints to develop in a neoplastic fashion: a microarray analysis study. Neuroscience. 2005;131(2):359–74. doi: 10.1016/j.neuroscience.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 8.Vemuganti R, Dempsey RJ. Increased expression of genes that control ionic homeostasis, second messenger signaling and metabolism in the carotid plaques from patients from symptomatic plaques. J. Neurochem. 2006;97:92–96. doi: 10.1111/j.1471-4159.2005.03516.x. [DOI] [PubMed] [Google Scholar]
- 9.Cordoliani-Mackowiak M, Henon H, Pruvo J, Pasquier Fl, Leys D. Poststroke Dementia. Archives of Neurology. 2003;60:585–590. doi: 10.1001/archneur.60.4.585. [DOI] [PubMed] [Google Scholar]
- 10.Ivan C, Seshadri S, Beiser A, et al. Dementia after Stroke: The Framingham Study. Stroke. 2003;60:585–590. doi: 10.1161/01.STR.0000127810.92616.78. [DOI] [PubMed] [Google Scholar]
- 11.Tang WK, Chan S, Chiu H, et al. Frequency and Determinants of Post stroke Dementia in Chinese. Stroke. 2004;35:930–935. doi: 10.1161/01.STR.0000119752.74880.5B. [DOI] [PubMed] [Google Scholar]
- 12.Seshadri S, Beiser A, Kelly-Hayes M, et al. The Lifetime Risk of Stroke: Estimates From the Framingham Study. Stroke. 2006;37:345–350. doi: 10.1161/01.STR.0000199613.38911.b2. [DOI] [PubMed] [Google Scholar]
- 13.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease: The Nun Study Journal of the American Medical Association. JAMA. 1997;277(10):813–817. [PubMed] [Google Scholar]
- 14.Smith CS, Snowdon DA, Wang H, Markesbery WR. White matter volumes and periventricular white matter hyperintensities in aging and dementia. Neurology. 2000;54(4):838–842. doi: 10.1212/wnl.54.4.838. [DOI] [PubMed] [Google Scholar]
- 15.Greiner PA, Snowdon DA, Greiner LG. Self-rated function, self-rated health and postmortem evidence of brain infarcts: Findings from the Nun Study. Journal of Gerontology: Social Sciences. 1999;54B(3):S219–222. doi: 10.1093/geronb/54b.4.s219. [DOI] [PubMed] [Google Scholar]
- 16.Elias M, Sullivan L, D’Agostino R, et al. Stroke Risk Profile and Lowered Cognitive Performance. Stroke. 2004;35:404–409. doi: 10.1161/01.STR.0000103141.82869.77. [DOI] [PubMed] [Google Scholar]
- 17.Purandare N, Welsh S, Hutchinson S, Riding G, Burns A, McCollum C. Cerebral emboli and paradoxical embolisation in dementia: a pilot study. Int J Geriatr Psychiatry. 2005;20:12–16. doi: 10.1002/gps.1202. [DOI] [PubMed] [Google Scholar]
- 18.Purandare N, Voshaar RC Oude, Morris J, et al. Asymptomatic Spontaneous Cerebral Emboli Predict Cognitive and Functional Decline in Dementia. Biol Psychiatry. 2007 doi: 10.1016/j.biopsych.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Mungas D, Jagust WJ, Reed BR, et al. MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology. 2001;57:2229–2235. doi: 10.1212/wnl.57.12.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulz UG, Rothwell PM. Association between arterial bifurcation anatomy and angiographic plaque ulceration among 4,627 carotid stenoses. Cerebrovasc Dis. 2003;15(4):244–51. doi: 10.1159/000069491. [DOI] [PubMed] [Google Scholar]
- 21.Avril G, Batt M, Guidoin R, et al. Carotid endarterectomy plaques: correlations of clinical and anatomic findings. Ann Vasc Surg. 1991 Jan;5(1):50–4. doi: 10.1007/BF02021778. [DOI] [PubMed] [Google Scholar]
- 22.van Swijndregt AD Montauban, Elbers HR, Moll FL, de Letter J, Ackerstaff RG. Cerebral ischemic disease and morphometric analyses of carotid plaques. Ann Vasc Surg. 1999 Sep;13(5):468–74. doi: 10.1007/s100169900285. [DOI] [PubMed] [Google Scholar]
- 23.Park AE, McCarthy WJ, Pearce WH, Matsumura JS, Yao JS. Carotid plaque morphology correlates with presenting symptomatology. J Vasc Surg. 1998 May;27(5):872–8. doi: 10.1016/s0741-5214(98)70267-8. (discussion:878-9) [DOI] [PubMed] [Google Scholar]
- 24.Persson AV, Robichaux WT, Silverman M. The natural history of carotid plaque development. Arch Surg. 1983 Sep;118(9):1048–52. doi: 10.1001/archsurg.1983.01390090038008. [DOI] [PubMed] [Google Scholar]
- 25.Galili O, Herrmann J, Woodrum J, Sattler KJ, Lerman LO, Lerman A. Adventitial vasa vasorum heterogeneity among different vascular beds. J Vasc Surg. 2004 Sep;40(3):529–35. doi: 10.1016/j.jvs.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 26.Rothwell PM, Gibson R, Warlow CP. Interrelation between plaque surface morphology and degree of stenosis on carotid angiograms and the risk of ischemic stroke in patients with symptomatic carotid stenosis. On behalf of the European Carotid Surgery Trialists’ Collaborative Group. Stroke. 2000 Mar;31(3):615–21. doi: 10.1161/01.str.31.3.615. [DOI] [PubMed] [Google Scholar]
- 27.Rothwell PM, Villagra R, Gibson R, Donders RC, Warlow CP. Evidence of a chronic systemic cause of instability of atherosclerotic plaques. Lancet. 2000 Jan 1;355(9197):19–24. doi: 10.1016/s0140-6736(99)04470-0. [DOI] [PubMed] [Google Scholar]
- 28.Burns DM. Epidemiology of smoking induced cardiovascular disease. Porg Cariodvasc Dis. 2003 Jul-Aug;46(1):11–29. doi: 10.1016/s0033-0620(03)00079-3. (Review) [DOI] [PubMed] [Google Scholar]
- 29.Linton MS, Fazio S. Macrophages, inflammation and atherosclerosis. International Journal of Obesity. 2003;27(3):S35–40. doi: 10.1038/sj.ijo.0802498. [DOI] [PubMed] [Google Scholar]
- 30.Droste DW, Ritter MA, Dittrick R, et al. Arterial hypertension and ischemic stroke. ACTA Neurological Scandinavic. 2003;107(4):241–251. doi: 10.1034/j.1600-0404.2003.00098.x. [DOI] [PubMed] [Google Scholar]
- 31.Emsley HC, Tyrrell PJ. Inflammation and infection in clinical stroke. Jrnl of Cer Blood Flow and Met. 2002;22(12):1399–1419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- 32.Feigin VL, Anderson CS, Mhurchu CN. Systemic inflammation, endothelial dysfunction, dietary fatty acids and micronutrients as risk factors for stroke: a selective review. Cerebrovasc Dis. 2002;3(4):219–24. doi: 10.1159/000057846. [DOI] [PubMed] [Google Scholar]
- 33.Nicolaides A, Sabetai M, Kakkos SK, et al. The Asymptomatic Carotid Stenosis and Risk of Stroke (ACSRS) study. Aims and results of quality control. Int Angiol. 2003 Sep;22(3):263–72. [PubMed] [Google Scholar]
- 34.Brook RH, Park RE, Chassin MR, Kosecoff J, Keesey J, Solomon DH. Carotid endarterectomy for elderly patients: predicting complications. Ann Intern Med. 1990 Nov 15;113(10):747–53. doi: 10.7326/0003-4819-113-10-747. [DOI] [PubMed] [Google Scholar]
- 35.Tegos TJ, Kalomiris KJ, Sabetai MM, Kalodiki E, Nicolaides AN. Significance of sonographic tissue and surface characteristics of carotid plaques. AJNR Am J Neuroradiol. 2001 Sep;22(8):1605–12. [PMC free article] [PubMed] [Google Scholar]
- 36.Grant EG, Benson CB, Moneta GL, et al. Carotid artery stenosis: grayscale and Doppler ultrasound diagnosis-Society of Radiologists in Ultrasound consensus conference. Ultrasound Q. 2003 Dec;19(4):190–8. doi: 10.1097/00013644-200312000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Dempsey RJ, Diana AL, Moore RW. Thickness of carotid artery atherosclerotic plaque and ischemic risk. Neurosurgery. 1990 Sep;27(3):343–8. doi: 10.1097/00006123-199009000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Riley WA, Barnes RW, Evans GW, Burke GL. Ultrasonic measurement of the elastic modulus of the common carotid artery. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke. 1992 Jul;23(7):952–6. doi: 10.1161/01.str.23.7.952. [DOI] [PubMed] [Google Scholar]
- 39.Dempsey RJ, Moore RW. Amount of smoking independently predicts carotid artery atherosclerosis severity. Stroke. 1992;23:693–696. doi: 10.1161/01.str.23.5.693. [DOI] [PubMed] [Google Scholar]
- 40.Katsenis C, Kouskouni E, Kolokotronis L, Rizos D, Dimakakos P. The significance of Chlamydia pneumoniae in symptomatic carotid stenosis. Angiology. 2001 Sep;52(9):615–9. doi: 10.1177/000331970105200905. [DOI] [PubMed] [Google Scholar]
- 41.Vainas T, Kurvers HA, Mess WH, et al. Chlamydia pneumoniae serology is associated with thrombosis-related but not with plaque-related microembolization during carotid endarterectomy. Stroke. 2002 May;33(5):1249–54. doi: 10.1161/01.str.0000014508.65367.8f. [DOI] [PubMed] [Google Scholar]
- 42.Alvarez GB, Ruiz C, Chacon P, Sabin JA, Matas M. High-sensitivity C-reactive protein in high-grade carotid stenosis: risk marker for unstable carotid plaque. J Vasc Surg. 2003 Nov;38(5):1018–24. doi: 10.1016/s0741-5214(03)00709-2. [DOI] [PubMed] [Google Scholar]
- 43.Dempsey RJ, Moore RW, Cordero S. Factors leading to early recurrence of carotid plaque after carotid endarterectomy. Surg Neurol. 1995 Mar;43(3):278–82. doi: 10.1016/0090-3019(95)80014-8. (discussion:282-3) [DOI] [PubMed] [Google Scholar]
- 44.Matetzky S, Tani S, Kangavari S, et al. Smoking increases tissue factor expression in atherosclerotic plaques: implications for plaque thrombogenicity. Circulation. 2000 Aug 8;102(6):602–4. doi: 10.1161/01.cir.102.6.602. [DOI] [PubMed] [Google Scholar]
- 45.Kim C, Cervos-Navarro J, Patzold C, Tokuriki Y, Takebe Y, Hori K. In vivo study of flow pattern at human carotid bifurcation with regard to aneurysm development. Acta Neurochir (Wien) 1992;115(3-4):112–7. doi: 10.1007/BF01406368. [DOI] [PubMed] [Google Scholar]
- 46.Tang D, Yang C, Kobayashi S, Ku DN. Steady flow and wall compression in stenotic arteries: a three-dimensional thick-wall model with fluid-wall interactions. J Biomech Eng. 2001 Dec;123(6):548–57. doi: 10.1115/1.1406036. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi K, Akishita M, Yu W, Hashimoto M, Ohni M, Toba K. Interrelationship between non-invasive measurements of atherosclerosis: flow-mediated dilation of brachial artery, carotid intima-media thickness and pulse wave velocity. Atherosclerosis. 2004 Mar;173(1):13–8. doi: 10.1016/j.atherosclerosis.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 48.Lovett JK, Rothwell PM. Site of carotid plaque ulceration in relation to direction of blood flow: an angiographic and pathological study. Cerebrovasc Dis. 2003;16(4):369–75. doi: 10.1159/000072559. [DOI] [PubMed] [Google Scholar]
- 49.Waddington EL, Croft KD, Sienuarine K, Latham B, Puddey IB. Fatty acid oxidation products in human atherosclerotic plaque: an analysis of clinical and histopathological correlates. Atherosclerosis. 2003 Mar;167(1):111–20. doi: 10.1016/s0021-9150(02)00391-x. [DOI] [PubMed] [Google Scholar]
- 50.Nishi K, Itabe H, Uno M, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol. 2002 Oct 1;22(10):1649–54. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 51.Waddington E, Sienuarine K, Puddey I, Croft K. Identification and quantitation of unique fatty acid oxidation products in human atherosclerotic plaque using high-performance liquid chromatography. Anal Biochem. 2001 May 15;292(2):234–44. doi: 10.1006/abio.2001.5075. [DOI] [PubMed] [Google Scholar]
- 52.Saba PS, Roman MJ, Longhini C, et al. Carotid intimal-medial thickness and stiffness are not affected by hypercholesterolemia in uncomplicated essential hypertension. Arterioscler Thromb Vasc Biol. 1999 Nov;19(11):2788–94. doi: 10.1161/01.atv.19.11.2788. [DOI] [PubMed] [Google Scholar]
- 53.Fisher CM, Ojemann RG. A clinico-pathologic study of carotid endarterectomy plaques. Rev Neurol (Paris) 1986;142(6-7):573–89. [PubMed] [Google Scholar]
- 54.Fisher CM. Clinical picture of cerebral arteriosclerosis. Minn Med. 1955 Dec;38(12):839–51. [PubMed] [Google Scholar]
- 55.Mukherjee D, Yadav JS. Effect of contralateral occlusion on long-term efficacy of endarterectomy in the Asymptomatic Carotid Atherosclerosis Study (ACAS) Stroke. 2001 Jun;32(6):1443–8. doi: 10.1161/01.str.32.6.1443-d. [DOI] [PubMed] [Google Scholar]
- 56.Baker WH, Howard VJ, Howard G, Toole JF. Effect of contralateral occlusion on longterm efficacy of endarterectomy in the asymptomatic carotid atherosclerosis study (ACAS) Stroke. 2000 Oct;31(10):2330–4. doi: 10.1161/01.str.31.10.2330. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein LB, Samsa GP, Matchar DB, Oddone EZ. Multicenter review of preoperative risk factors for endarterectomy for asymptomatic carotid artery stenosis. Stroke. 1998 Apr;29(4):750–3. doi: 10.1161/01.str.29.4.750. [DOI] [PubMed] [Google Scholar]
- 58.North American Symptomatic Carotid Endarterectomy Trial Collaborators (NASCET) Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991;325:445–453. doi: 10.1056/NEJM199108153250701. [DOI] [PubMed] [Google Scholar]
- 59.Golledge J, Greenhalgh RM, Davies AH. The symptomatic carotid plaque. Stroke. 2000 Mar;31(3):774–81. doi: 10.1161/01.str.31.3.774. [DOI] [PubMed] [Google Scholar]
- 60.Shi H. Ph.D dissertation, Medical Physics. University of Wisconsin-Madison; 2007. Atherosclerotic Carotid Plaque Characterization using Ultrasound and Elastography. [Google Scholar]
- 61.Shi H, Mitchell CC, McCormick M, Kliewer MA, Dempsey RJ, Varghese T. Preliminary in vivo atherosclerotic carotid plaque characterization using the accumulated axial strain and relative lateral shift strain indices. Phys Med Biol. 2008;53(22):6377–94. doi: 10.1088/0031-9155/53/22/008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richardson PD, Davies MJ, Born GV. Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet. 1989;2(8669):941–944. doi: 10.1016/s0140-6736(89)90953-7. [DOI] [PubMed] [Google Scholar]
- 63.Fuster V, Stein B, Ambrose JA, Badimon L, Badimon JJ, Chesebro JH. Atherosclerotic plaque rupture and thrombosis. Evolving Concepts. 1990;82(3 Suppl):1147–59. [PubMed] [Google Scholar]
- 64.Gronholdt ML. Ultrasound and lipoproteins as predictors of lipid-rich, rupture-prone plaques in the carotid artery. Arterioscler Thromb Vasc Biol. 1999;19(1):2–13. doi: 10.1161/01.atv.19.1.2. [DOI] [PubMed] [Google Scholar]
- 65.Wilson LS, Robinson DE. Ultrasonic measurement of small displacements and deformations of tissue. Ultrasonic Imaging. 1982;4:71–82. doi: 10.1177/016173468200400105. [DOI] [PubMed] [Google Scholar]
- 66.Bertrand M, Meunier M, Doucet M, Ferland G. Ultrasonic biomechanical strain gauge based on speckle tracking. IEEE Ultrasonics Symposium. 1989:859–864. [Google Scholar]
- 67.Parker KJ, Huang SR, Musulin RA, Lerner RM. Tissue response to mechanical vibrations for sonoelasticity imaging. Ultrasound in Medicine & Biology. 1990;16(3):241–246. doi: 10.1016/0301-5629(90)90003-u. [DOI] [PubMed] [Google Scholar]
- 68.Ophir J, Cespedes I, Ponnekanti H, Yazdi Y, Li X. Elastography: a quantitative method for imaging the elasticity of biological tissues. Ultrasonic Imaging. 1991;13(2):111–134. doi: 10.1177/016173469101300201. [DOI] [PubMed] [Google Scholar]
- 69.Cespedes EI. Ph.D. Dissertation. University of Houston; 1993. Elastography: Imaging of Biological Tissue Elasticity. [Google Scholar]
- 70.O’Donnell M, Skovoroda AR, Shapo BM, Emelianov SY. Internal displacement and strain imaging using ultrasonic speckle tracking. IEEE Transactions on Ultrasonics, Ferroelectrics and Frequency Control. 1994;41(3):314–325. [Google Scholar]
- 71.Bamber JC, Bush NL. Freehand elasticity imaging using speckle decorrelation rate. Acoustic Imaging. 1996;22:285–292. [Google Scholar]
- 72.Varghese T, Ophir J, Konofagou E, Kallel F, Righetti R. Tradeoffs in elastographic Imaging. Ultrasonic Imaging. 2001;23(4):216–248. doi: 10.1177/016173460102300402. [DOI] [PubMed] [Google Scholar]
- 73.de Korte CL, vanderSteen AF. Intravascular ultrasound elastography: an overview. Ultrasonics. 2002;40(1-8):859–865. doi: 10.1016/s0041-624x(02)00227-5. [DOI] [PubMed] [Google Scholar]
- 74.O’Donnell M, Skovoroda AR, Shapo BM. Measurement of arterial wall motion using Fourier based speckle tracking algorithms. Proc. IEEE Ultras. Symp. 1991:1101–1104. [Google Scholar]
- 75.Shapo BM, Crowe JR, Skovoroda AR, Eberle MJ, Cohn NA, M OD. Displacement and strain imaging of coronary arteries with intraluminal ultrasound. IEEE Transactions on Ultrasonics, Ferroelectrics and Frequency Control. 1996;43(2):234–246. [Google Scholar]
- 76.Ryan LK, Foster FS. Ultrasonic measurement of differential displacement and strain in a vascular model. Ultrasonic Imaging. 1997;19(1):19–38. doi: 10.1177/016173469701900102. [DOI] [PubMed] [Google Scholar]
- 77.Moody AR, Murphy RE, Morgan PS, et al. Characterization of complicated carotid plaque with magnetic resonance direct thrombus imaging in patients with cerebral ischemia. Circulation. 2003 Jun 24;107(24):3047–52. doi: 10.1161/01.CIR.0000074222.61572.44. [DOI] [PubMed] [Google Scholar]
- 78.Murphy RE, Moody AR, Morgan PS, et al. Prevalence of complicated carotid atheroma as detected by magnetic resonance direct thrombus imaging in patients with suspected carotid artery stenosis and previous acute cerebral ischemia. Circulation. 2003 Jun 24;107(24):3053–8. doi: 10.1161/01.CIR.0000074204.92443.37. [DOI] [PubMed] [Google Scholar]
- 79.Imparato AM, Riles TS, Mintzer R, Baumann FG. The importance of hemorrhage in the relationship between gross morphologic characteristics and cerebral symptoms in 376 carotid artery plaques. Ann Surg. 1983 Feb;197(2):195–203. doi: 10.1097/00000658-198302000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lusby RJ, Ferrell LD, Ehrenfeld WK, Stoney RJ, Wylie EJ. Carotid plaque hemorrhage. Its role in production of cerebral ischemia. Arch Surg. 1982 Nov;117(11):1479–88. doi: 10.1001/archsurg.1982.01380350069010. [DOI] [PubMed] [Google Scholar]
- 81.Sillesen H, Nielsen T. Clinical significance of intraplaque hemorrhage in carotid artery disease. J Neuroimaging. 1998 Jan;8(1):15–9. doi: 10.1111/jon19988115. [DOI] [PubMed] [Google Scholar]
- 82.Carr S, Farb A, Pearce WH, Virmani R, Yao JS. Atherosclerotic plaque rupture in symptomatic carotid artery stenosis. J Vasc Surg. 1996 May;23(5):755–65. doi: 10.1016/s0741-5214(96)70237-9. (discussion):765-6. [DOI] [PubMed] [Google Scholar]
- 83.Bassiouny HS, Sakaguchi Y, Mikucki SA, et al. Juxtalumenal location of plaque necrosis and neoformation in symptomatic carotid stenosis. J Vasc Surg. 1997 Oct;26(4):585–94. doi: 10.1016/s0741-5214(97)70056-9. [DOI] [PubMed] [Google Scholar]
- 84.Schminke U, Motsch L, Hilker L, Kessler C. Three-dimensional ultrasound observation of carotid artery plaque ulceration. Stroke. 2000 Jul;31(7):1651–5. doi: 10.1161/01.str.31.7.1651. [DOI] [PubMed] [Google Scholar]
- 85.Dogan A, Dempsey RJ. Diagnostic modalities for carotid artery disease. Neurosurg Clin N Am. 2000 Apr;11(2):205–20. [PubMed] [Google Scholar]
- 86.McCarthy MJ, Loftus IM, Thompson MM, et al. Angiogenesis and the atherosclerotic carotid plaque: an association between symptomatology and plaque morphology. J Vasc Surg. 1999 Aug;30(2):261–8. doi: 10.1016/s0741-5214(99)70136-9. [DOI] [PubMed] [Google Scholar]
- 87.Mofidi R, Crotty TB, McCarthy P, Sheehan SJ, Mehigan D, Keaveny TV. Association between plaque instability, angiogenesis and symptomatic carotid occlusive disease. Br J Surg. 2001 Jul;88(7):945–50. doi: 10.1046/j.0007-1323.2001.01823.x. [DOI] [PubMed] [Google Scholar]
- 88.Tureyen K, Vemuganti R, Salamat MS, Dempsey RJ. Increased angiogenesis and angiogenic gene expression in carotid artery plaques from symptomatic stroke patients. Neurosurgery. 2006 May;58(5):971–7. doi: 10.1227/01.NEU.0000210246.61817.FE. (discussion:971-7) [DOI] [PubMed] [Google Scholar]
- 89.Shi H, Tu H, Dempsey RJ, Varghese T. Ultrasonic attenuation estimation in small plaque samples using a power difference method. Ultrason Imaging. 2007 Jan;29(1):15–30. doi: 10.1177/016173460702900102. [DOI] [PubMed] [Google Scholar]
- 90.Shi H, Varghese T, Dempsey RJ, Salamat MS, Zagzebski JA. Relationship between ultrasonic attenuation, size and axial strain parameters for ex vivo atherosclerotic carotid plaque. Ultrasound Med Biol. 2008;34(10):1666–77. doi: 10.1016/j.ultrasmedbio.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi H, Varghese T. Two-dimensional multi-level strain estimation for discontinuous tissue. Phys Med Biol. 2007;52(2):389–401. doi: 10.1088/0031-9155/52/2/006. [DOI] [PubMed] [Google Scholar]