

Summary
A prerequisite for antibody secretion and function is the assembly into a defined quaternary structure, composed of two heavy and two light chains for IgG. Unassembled heavy chains are actively retained in the endoplasmic reticulum (ER) until they associate with light chains. Our mechanistic analysis of this critical quality control step revealed that, unlike all other antibody domains studied, the CH1 domain of the murine IgG1 heavy chain is an intrinsically disordered protein in isolation. It adopts the typical immunoglobulin fold only upon interaction with its cognate partner, the CL domain. Structure formation proceeds via a trapped intermediate, can be accelerated by the ER-specific peptidyl-prolyl isomerase cyclophilin B, and is modulated by the molecular chaperone BiP. BiP recognizes incompletely folded states of the CH1 domain and competes for binding to the CL domain. In vivo experiments demonstrate that requirements identified for folding the CH1 domain in vitro, including association with a folded CL domain and isomerization of a conserved proline residue, are essential for antibody assembly and secretion in the cell.
Introduction
In eukaryotic cells, proteins destined for secretion mature within the endoplasmic reticulum and are subject to rigorous quality control prior to their transport to the Golgi (Helenius et al., 1992). This usually involves surveillance of the folding status, the correct posttranslational modifications and proper oligomerization (Helenius et al., 1992; Ellgaard and Helenius, 2003; Christis et al., 2008). A prominent example of this is immunoglobulin G (IgG), the most abundant antibody in the blood. It is a heterotetrameric glycoprotein assembled from two light and two heavy chains that are comprised of two and four compact Ig-domains, respectively, which are structurally almost identical (Huber et al., 1976). Each domain shows a β-barrel topology, a two-layer sandwich structure composed of seven to nine antiparallel β-strands (Huber et al., 1976; Amzel and Poljak, 1979). The fold is stabilized by an internal disulfide bridge (Goto and Hamaguchi, 1979) that is located in the hydrophobic core and lies perpendicular to the β-sheets (Huber et al., 1976). Most of these structural characteristics are shared by the ubiquitous members of the immunoglobulin (Ig) superfamily (Bork et al., 1994), which perform a broad variety of extracellular recognition functions (Williams and Barclay, 1988; Rougon and Hobert, 2003; Aricescu and Jones, 2007). The evolutionary success of the Ig superfamily has fueled a vast scope of investigations on its biophysical properties. In particular, the folding pathways of diverse members of the Ig superfamily have been studied in detail (Goto and Hamaguchi, 1982; Freund et al., 1996; Thies et al., 1999; Cota et al., 2001; Paci et al., 2003; Feige et al., 2004) and have provided insights into determinants of robust folding (Hamill et al., 2000; Feige et al., 2008) as well as potentially harmful misfolding of this class of proteins (Kameda et al., 2005; Jahn et al., 2006; Qin et al., 2007).
To become secreted from the cell and fulfill their biological functions, the individual domains of an antibody not only have to fold into their native tertiary structure, but furthermore must assemble into a defined quaternary structure (Porter, 1973; Huber et al., 1976). While isolated antibody light chains can be secreted from the endoplasmic reticulum (Coffino et al., 1970; Melchers, 1971), unpaired heavy chains are actively and efficiently retained in the ER (Bole et al., 1986; Hendershot et al., 1987). Antibody heavy chain and light chain synthesis occur asynchronously during B cell development (Burrows et al., 1979), and only completely assembled molecules can both bind to antigen and carry out effector functions. Therefore, tight quality control of their assembly prior to secretion is vital. It is known that the first constant domain of the heavy chain, the CH1 domain, plays an important role in this retention process (Hendershot et al., 1987; Kaloff and Haas, 1995). If deleted or replaced with another antibody domain, isolated heavy chains can be secreted, as occurs in the case of the rare heavy chain diseases (Wolfenstein-Todel et al., 1974; Adetugbo, 1978; Hendershot et al., 1987), or naturally in camelid antibodies, which do not contain light chains (Hamers-Casterman et al., 1993). In the context of the whole IgG molecule (Fig. 1A), the CH1 domain is associated with the constant domain of the light chain (CL) and shows the typical immunoglobulin fold (Huber et al., 1976). In vivo, CH1 is the only antibody domain that is stably bound to the molecular chaperone BiP and remains in a reduced form before assembly with light chain (Vanhove et al., 2001). The basis for the unusual behavior of the CH1 domain has remained enigmatic.
Figure 1. Structural characteristics of the CH1 domain and its assembly mechanism with the CL domain.
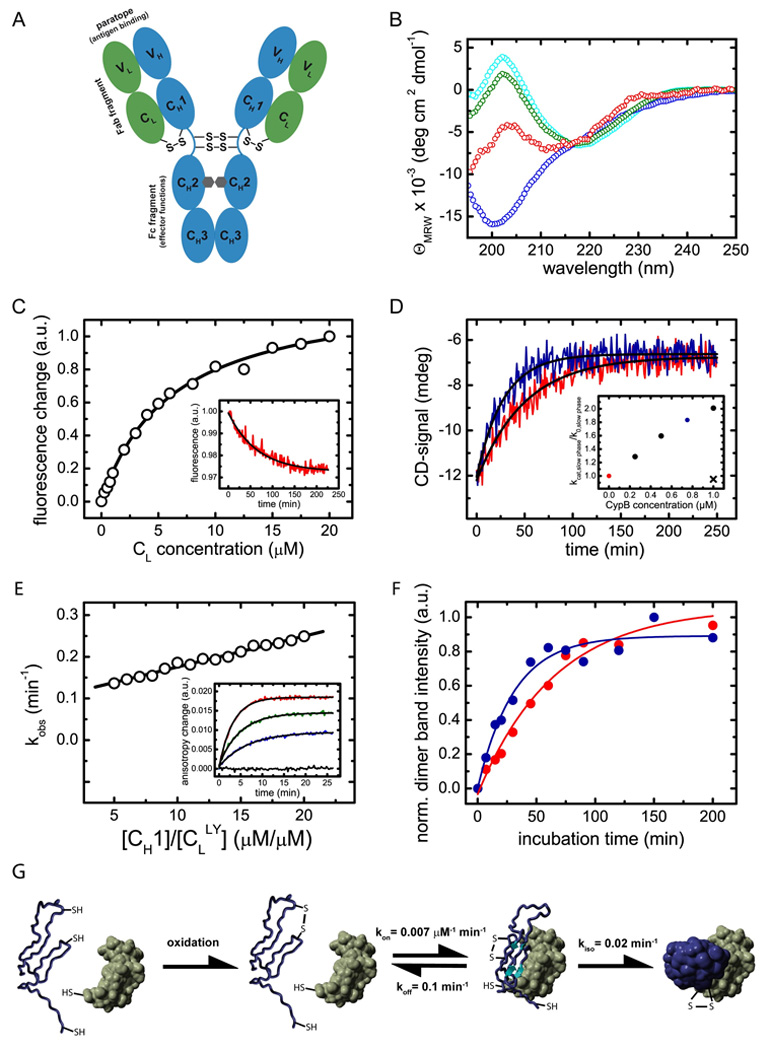
(A) shows a schematic representation of the IgG1 molecule. The IgG molecule is composed of two heavy chains (blue) and two light chains (green). The heavy chain consists of the VH, CH1, CH2 and CH3 domain and the light chain of the VL and the CL domain (listing from N- to C-terminus of each chain). The CH2 domains of the heavy chains are glycosylated with complex biantennary oligosaccharides (depicted in grey). Each domain possesses an internal disulfide bridge (omitted for clarity) and additional disulfide bonds link the two heavy chains in the flexible hinge region. A single disulfide bond covalently connects CH1 with the CL domain. The two identical antigen binding sites (paratopes) are made up by the two variable domains VH and VL. The overall IgG molecule can be divided into two Fab fragments (composed of VH, CH1, VL and CL) and one Fc fragment (composed of two CH2 and two CH3 domains). (B) The isolated CL domain (cyan) displays a typical all-β far-UV CD spectrum whereas the isolated CH1 domain (blue) shows a random coil spectrum. To assess if CL induces structural changes in CH1, the spectrum of co-incubated CL and CH1 was recorded (green). From this spectrum and the far-UV CD spectrum of the isolated CL domain, the spectrum of the CH1 domain in the presence of CL was calculated (red) which shows the characteristics of β-sheet secondary structure. (C) The affinity between CH1 and CL was determined by the change in the intrinsic fluorescence upon CL induced folding of the CH1 domain, recorded before and after a 4 h equilibration step. A one-site binding model was used to fit the data. The inset shows a representative single exponential trace observed after the addition of 1 µM CL to 2 µM CH1. A single exponential reaction with a very similar rate was observed by far-UV CD spectroscopy (D, red trace). The folding reaction could be accelerated by the PPIase CypB (red trace: 10 µM CL and 10 µM CH1 alone, blue trace: in the presence of 0.75 µM CypB). The inset shows the dependence of the slow reaction on CypB concentration. The observed rate in the absence of CypB is denoted as k0, in the presence of CypB as kcat. If 1µM CypB was inhibited by 2 µM cyclosporine A, no acceleration was observed (black cross). (E) Association of the CH1 domain with a lucifer yellow labeled CL domain was followed by the change in the lucifer yellow anisotropy signal. The observed rate constants were fitted with a linear function to yield the kon value and the koff value of 0.007 ±0.0002 µM−1 min−1 respectively 0.1 ±0.01 min−1. The inset shows individual single exponential traces after the addition of 0 µM (black), 5 µM (blue), 10 µM (green) and 20 µM (red) CH1 to 1 µM labeled CL. (F) To assess the formation of the CL/CH1 interchain disulfide bridge, non-reducing SDS-PAGE experiments were carried out and the dimer band intensity was quantified. 25 µM of each domain were used. In the absence of CypB (red), a time constant of τ = 63 ±7 min was observed for the formation of covalent CL/CH1 dimers. In the presence of 5 µM CypB, a time constant of τ = 31 ±5 min was obtained. In (G), the overall CH1/CL assembly mechanism is shown. Only after formation of the internal disulfide bridge in the CH1 domain (blue), the fast formation of a dimeric intermediate with the CL domain (green) is observed. Subsequently, prolyl isomerization limits complete folding and formation of the interchain disulfide bridge. All measurements were carried out at 25°C in PBS.
Here we set out to study the role of the CH1/CL association for correct antibody assembly and secretion. To our surprise we found that CH1 is an unfolded protein in isolation, which gains structure only upon interaction with its cognate partner CL. Based on this finding, we analyzed the association-coupled CH1 folding pathway and its modulation by the chaperone BiP in detail and provide a comprehensive picture for the control of antibody secretion in the cell.
Results
The IgG1 CH1 domain is intrinsically disordered
Antibodies are modular structures composed of a series of structurally highly homologous domains (Fig. 1A). These domains can usually be produced and studied separately as they represent independent structural units (Goto and Hamaguchi, 1982; Lilie et al., 1995). Surprisingly, analysis of the murine IgG1 CH1 domain revealed that, in marked contrast to all antibody domains studied thus far (Goto and Hamaguchi, 1982; Thies et al., 1999; Feige et al., 2004; Rothlisberger et al., 2005), the isolated CH1 domain is an unfolded protein, irrespective of whether its internal disulfide bridge is formed or not (Fig. 1B, supplemental Fig. 1 and supplemental Fig. 2). To further characterize the unfolded state of the CH1 domain under physiological conditions, iodide fluorescence quenching experiments were carried out. The experiments indicate no significant differences in the burial of tryptophan residues between CH1 in PBS and in 3 M GdmCl. Additionally, NMR experiments were recorded on a highly deuterated CH1 sample (data not shown). Deuteration enables detection of long range NOEs even if they belong to only a subset of conformers. In these experiments, no long range NOEs could be determined and consequently no preferential conformation of CH1 seems to persist. Taken together, these data argue against the presence of a significant amount of stable structure in the isolated CH1 domain. However, the pattern changed completely when the CL domain, the cognate association partner of CH1 in the antibody, was added. Only then did we observe folding of the CH1 domain to a well defined β-sheet structure (Fig. 1B). Thus, the CL domain is necessary and sufficient to induce structure formation in CH1. This folding process was observed only if the internal disulfide bridge in the CH1 domain was present (supplemental Fig. 2). Based on these findings, a key role of this association-coupled folding reaction for correct antibody assembly can be anticipated, which we aimed to elucidate in more detail.
The mechanism of induced folding of the CH1 domain
To understand how binding to CL and folding of CH1 are coupled the thermodynamic and kinetic parameters for this reaction were established. The dissociation constant of the two domains was determined to be 6.2 ±0.4 µM (Fig. 1C) by the change in intrinsic fluorescence emission upon CL-induced CH1 folding (Fig. 1C, inset). A moderate affinity is expected since CH1 has to fold upon binding to CL. One should bear in mind that the observed dissociation constant is orders of magnitude lower than the antibody concentration in the ER of plasma cells (Cenci and Sitia, 2007), and thus association will readily occur in vivo. The analysis of the kinetics of secondary, tertiary and quaternary structure formation using far-UV CD spectroscopy (Fig. 1D), near-UV CD spectroscopy and analytical HPLC (supplemental Fig. 1) showed that all three processes occur with virtually identical time constants of τ = 60 ±10 min at 25°C. Hence, all these processes are likely rate-limited by the same slow reaction. This slow folding reaction could be accelerated by the ER-specific peptidylprolyl isomerase cyclophilin B (Fig. 1D). Thus, the slow folding phase can be attributed to the isomerization of peptidyl-prolyl bonds within the CH1 domain, which possesses an unusually high number of three cis prolines in the native state (Augustine et al., 2001). Prior to the slow folding to the native structure, the CH1 domain forms an intermediate with the CL domain in a concentration-dependent reaction (Fig. 1E). As this complex could be detected by fluorescence anisotropy measurements but not by the other techniques outlined above, it is likely a dynamic species with an only marginally folded CH1 domain. In the complete antibody, a disulfide bridge covalently links the CH1 domain with the CL domain (Fig. 1A). If the bridge forming Cys residues were included in the CH1 as well as the CL domain, no change in the folding state of the isolated domains and the CL-induced folding of the CH1 domain was observed (supplemental Fig. 3) but formation of covalent dimers could be readily followed by SDS-PAGE. As covalent dimers were formed with the same rate as the slow CH1 folding reaction and the reaction could be accelerated by cyclophilin B (Fig. 1F), it is clearly limited by proline isomerization and hence complete folding of the CH1 domain.
Taken together, the CL-induced folding of the CH1 domain can be dissected into three reactions: first, oxidation of the internal CH1 disulfide bridge has to take place. Then, a transient heterodimeric intermediate is formed, and subsequently peptidyl-prolyl isomerization is required to allow folding to the native state and covalent assembly with CL (Fig. 1G).
An atomic level description of the CH1 folding pathway
To resolve the specific recognition and the folding pathway of the intrinsically disordered CH1 domain at the level of atomic resolution, NMR experiments were performed. The 15N-1H HSQC spectrum of the isolated CH1 domain is characteristic of an unfolded protein (Fig. 2A, red spectrum) confirming the results described above. In contrast, after induced folding by CL, the CH1 domain shows well-dispersed spectra (Fig. 2A, blue spectrum). The backbone assignment of the CH1 domain in the complex was achieved by a combination of triple resonance experiments and NH residual dipolar couplings (RDCs). All obtained NMR data, the carbon chemical shifts, the NH RDCs and the MEXICO (Gemmecker et al., 1993) water exchange rates (data not shown) agree with an all-β structure for the CH1 domain in the presence of CL, like that observed in the crystal structure of IgG antibodies (Augustine et al., 2001). Because complete folding of the CH1 domain is limited by proline isomerization, and hence associated with a high activation energy, the final folding step is significantly decelerated at low temperatures. This allowed us to characterize the trapped intermediate and resolve the association-coupled folding process using real time 15N-1H HSQC experiments. For each assigned residue, changes of the amplitudes over time could be described by a single exponential function (Fig. 2A, inset). Notably, some residues already exhibit significant intensities in the first spectrum recorded after 20 min (Fig. 2B, red bars). These residues are likely to already adopt a native-like backbone conformation prior to the slow peptidyl-prolyl isomerization reaction. Altogether ten residues, which are part of the β-sheets that form the mature structure, were found to be in a native-like environment in the intermediate. Mapping these residues on the crystal structure of the CH1 domain of a murine IgG1 Fab fragment revealed how the association-coupled folding reaction of this antibody domain might proceed. Residues Thr22, His49, Ser65 and Thr67 in the CH1 domain, which form part of the CL interface, seem to be already correctly positioned in the intermediate (Fig. 2C, left panel). Importantly, His49 and Ser65 are involved in hydrogen bonds with the CL domain in the native state. The interaction with CL apparently initiates the formation of a hydrophobic cluster in the CH1 domain including Val21, Val68, Trp73 and Val78 (Fig. 2C, right panel). Additionally, interaction of Val48 and Val66 might also be involved in this hydrophobic cluster, although this could not be directly addressed due to peak overlap for Val66. Thus, a few key interactions between CL and CH1 establish an interface between the two domains in the intermediate, which allows the formation of a hydrophobic folding nucleus in the CH1 domain, and subsequent prolyl isomerization paves the path to the native state.
Figure 2. NMR spectroscopic characterization of CL induced CH1 folding.
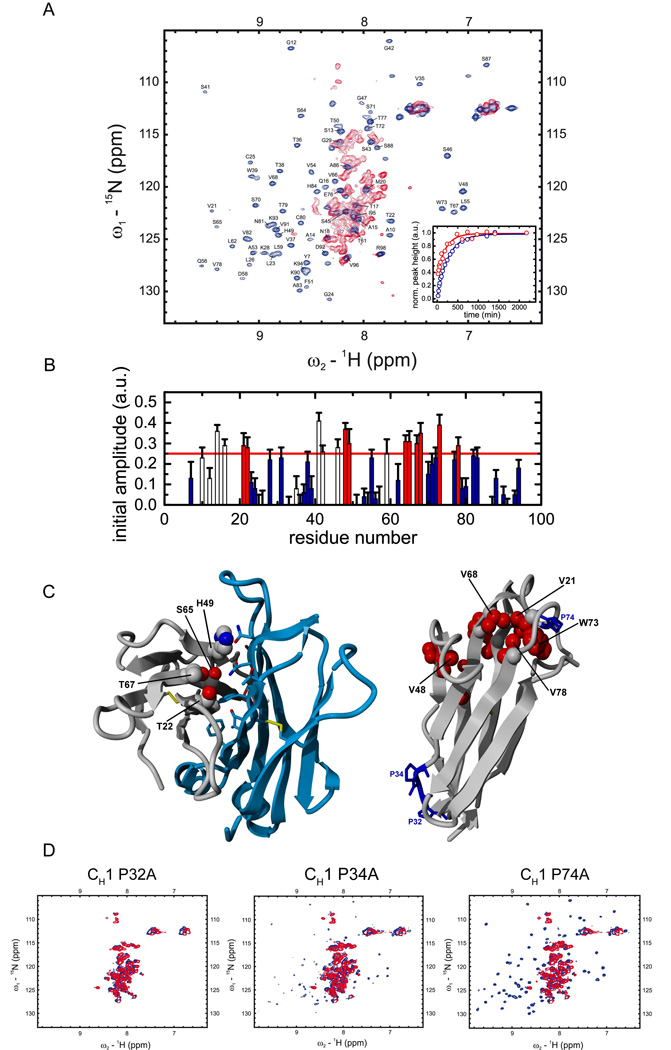
(A) 15N-1H HSQC spectra of the isolated CH1 domain (red) and the assigned CH1 domain in complex with the CL domain (blue) are shown. In order to characterize the folding pathway of the intrinsically disordered CH1 domain, time dependent HSQC intensities upon addition of unlabeled CL to 15N labeled CH1 were measured for each assigned residue at the native chemical shift position and fitted by a single exponential function. Two representative traces for Val68 (red) and Lys90 (blue) are shown in the inset. (B) Initial amplitudes for each assigned CH1 residue were derived from the fitted exponential functions. Residues with an initial HSQC amplitude below a threshold of 25% native intensity are colored in blue and residues above the threshold in red (open bars: residues in loop regions / filled bars: residues in structured regions). Errors indicate standard deviations from single exponential fits. In (C) CH1 residues with intensities above the threshold in the intermediate are mapped on the crystal structure of the CH1/CL dimer (pdb code 1Q9K). The dimerization interface between CH1 (grey) and CL (blue) is shown on the left with only residues of CH1 indicated that are involved in this interaction and above the HSQC amplitude threshold. On the right, internal CH1 residues above the 25% threshold are shown in red, the three cis proline residues in CH1 are depicted and labeled in blue. (D) The HSQC spectra of the CH1 Pro32Ala, Pro34Ala and Pro74Ala mutants show, that only the Pro32Ala mutant is not able to fold in the presence of CL anymore (blue spectrum). For the other two mutants, Pro34Ala and Pro74Ala, well dispersed spectra and hence folding are observed in the presence of CL (blue spectra). In the absence of CL (red spectra), all three mutants show typical HSQC spectra of unfolded proteins. All measurements were carried out in PBS at 25°C except for the folding kinetics which where recorded at 12.5°C.
To identify the residues responsible for the slow folding reaction of the CH1 domain, each of the three proline residues (Pro32, Pro34, and Pro74) that adopt a cis conformation in the native state (Fig. 2C, right panel) was individually mutated to alanine. 15N-1H HSQC spectra were recorded for each of the mutants in the absence and in the presence of CL. All three mutants showed almost indistinguishable spectra compared to the wild type CH1 domain in the absence of CL (Fig. 2D). Importantly, two of the mutants, Pro34Ala and Pro74Ala, displayed well dispersed HSQC spectra in the presence of CL that are very similar to that of wild type CH1 in the presence of CL (Fig. 2D), arguing that isomerization of these two prolines is not essential for CH1 domain folding. However, when the Pro32Ala mutant was similarly examined, identical spectra were obtained in the presence and in the absence of CL (Fig. 2D). Thus, isomerization of Pro32 from trans to cis is a prerequisite for CL-induced folding of the CH1 domain.
The role of the molecular chaperone BiP in CH1/CL assembly
Although association-coupled folding of the CH1 domain is an intrinsic feature of this protein, the folding, assembly and subsequent secretion of IgG molecules in the cell involve additional factors (Meunier et al., 2002). To retain the unassembled heavy chain in the ER, a protein recognizing the unfolded CH1 domain is needed. The molecular chaperone BiP, a member of the Hsp70 chaperone family that is present at high concentrations in the ER, plays a crucial role in this process in vivo (Haas and Wabl, 1983; Lee et al., 1999). After synthesis and prior to assembly with the light chain, the CH1 domain of the heavy chain remains in a reduced state (Vanhove et al., 2001). In line with this finding, BiP formed stable complexes with the reduced CH1 domain in vitro with a dissociation constant of 4.2 ±0.4 µM (Fig. 3A). Oxidation of CH1 only slightly reduced the affinity for BiP to Kd = 12.6 ±0.7 µM (Fig. 3A), which is consistent with this domain remaining unfolded. This is further supported by our finding that reduced CH1 could form its intradomain disulfide bridge while in the BiP bound state (data not shown). Oxidized and reduced CH1 both bind to BiP in a two-state, concentration-dependent reaction guarantying a fast association in the ER (Fig. 3B). To assess whether CL can form stable triple complexes with BiP and CH1, FRET experiments were carried out with ATTO594-labeled BiP and ATTO532-labeled CL to which unlabeled CH1 was added (supplemental Fig. 4). No FRET signal was detected even though BiP readily associates with CH1 under these conditions (supplemental Fig. 4) and CH1 interacts with CL (Fig. 1). Thus, the presence of stable BiP:CH1:CL complexes is very unlikely even though the light chain can trigger the dissociation of BiP:CH1 complexes in vivo (Lee et al., 1999).
Figure 3. Characterization of the interaction between BiP and CH1 in vitro.
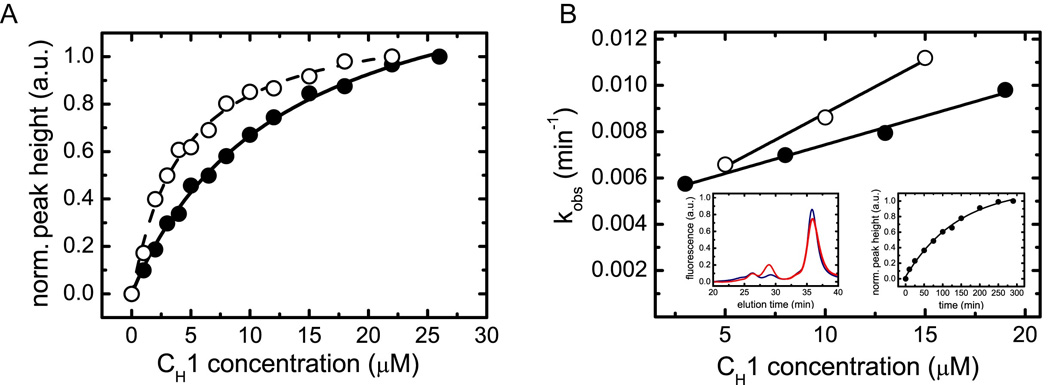
(A) The affinity between BiP and oxidized CH1 (filled circles, straight line) as well as reduced CH1 (open circles, dashed line) was determined by analytical HPLC experiments. The data were fitted to a one-site binding model to determine the Kd. (B) The association kinetics between 1 µM BiP and varying concentrations of oxidized CH1 (filled circles) and reduced CH1 (open circles) were measured to determine the rate constants of the reaction. For oxidized CH1, kon = 0.00026 ±0.00002 µM−1 min−1 and koff = 0.0050 ±0.0002 min−1 were obtained. For the reduced CH1 domain, the corresponding values were kon = 0.00041 ±0.00003 µM−1 min−1 and koff = 0.0047 ±0.0003 min−1. The left inset shows single HPLC runs of 8 µM oxidized CH1 and 1 µM BiP after 10 min (blue) and 200 min (red) co-incubation. The right inset shows the overall observed single exponential association kinetics between 1 µM BiP and 8 µM oxidized CH1.
The antibody domain folding status controls binding to BiP and secretion from the ER in vivo
It has long been appreciated that the CH1 domain is central to correct assembly and transport of IgG molecules and other immunoglobulin isotypes (Wolfenstein-Todel et al., 1974; Adetugbo, 1978; Hendershot et al., 1987; Shaffer and Schlissel, 1997). Deletion of this domain allows secretion or surface expression of free heavy chains and various Ig assembly intermediates (Hendershot et al., 1987), which shows that quality control is focused on this domain. Our data put the unexpected unfolded nature of the CH1 domain at the center of the secretion control mechanism of IgG antibodies. To test this notion in a cellular context, we first expressed the MAK33 κ light chain, which contained the CL domain that was used in the in vitro experiments, in COS-1 cells and performed metabolic labeling and immunoprecipitation assays. As expected, this wild type light chain (LCwt) was detected not only in the cells but also in the medium, indicating that LCwt was secreted efficiently (Fig. 4A, lanes 1 and 3). When we replaced the CL domain of the light chain with the CH1 domain, this light chain (LCCH1) now behaved like a heavy chain in terms of retention in the ER and interaction with BiP as demonstrated by an increase in the amount of the altered light chain that co-precipitated with BiP and its absence in the medium (Fig. 4A, lanes 4–6). This shows that the structural characteristics of the CH1 domain and its role in antibody retention are intrinsic, context-independent features. To more directly address the structural prerequisites for antibody retention, we exchanged the small helical elements of the CL domain, which have been reported to play a crucial role in the folding of this domain (Feige et al., 2008), against the corresponding elements of the CH1 domain. This exchange transformed the CL domain into an unfolded protein in vitro (data not shown). When a light chain containing this altered CL domain (LCCLmut) was expressed in COS-1 cells, it strongly interacted with BiP in vivo and was no longer secreted from the cell (Fig. 4A, lanes 7–9), which argues that the folding status of an antibody domain is key for its retention.
Figure 4. The folding status of an antibody domain controls IgG secretion in vivo.
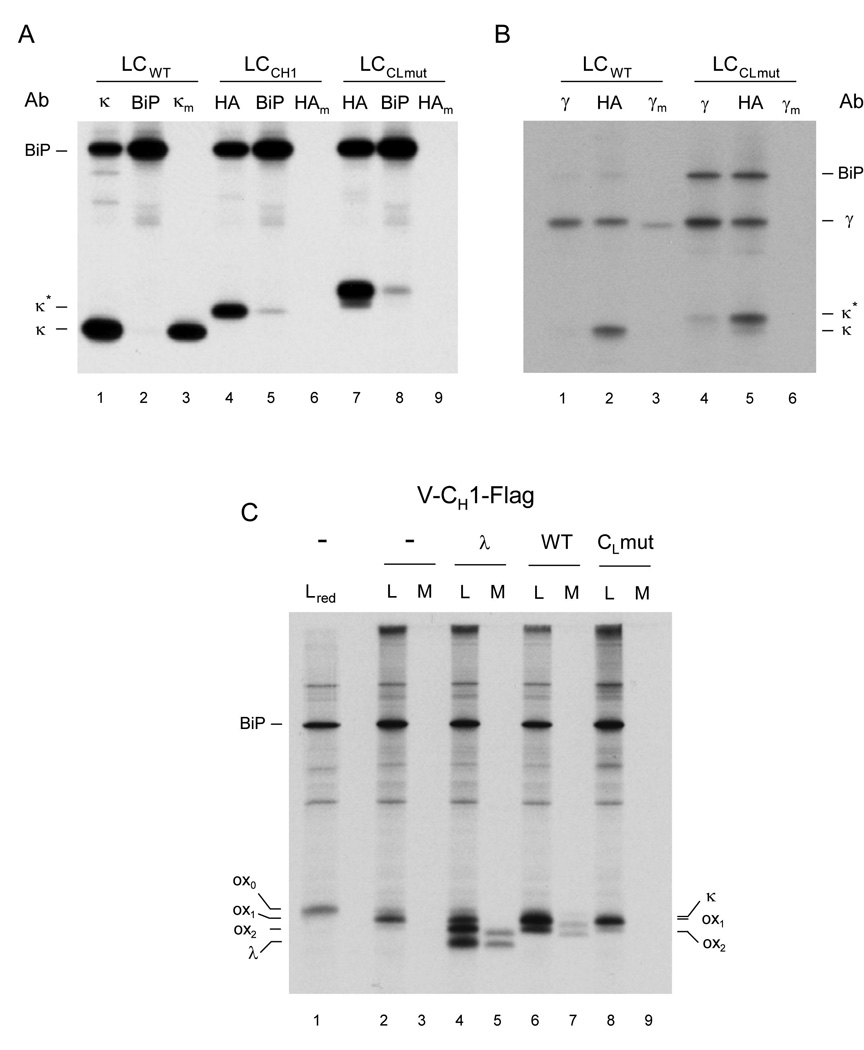
(A) COS-1 cells were co-transfected with vectors encoding BiP and either a wild type light chain (LCwt), a light chain containing the CH1 domain instead of the CL domain (LCCH1), or a light chain containing an unfolded CL domain (LCCLmut). Cells were metabolically labeled for 3 h and both cell lysates (no subscript) and culture supernatants (subscript m) were immunoprecipitated with the indicated antisera (Ab). Precipitated proteins were separated by SDS-PAGE under reducing conditions and visualized by autoradiography. (B) COS-1 cells were co-transfected as in (A) except that a vector encoding a chimeric humanized mouse heavy chain was also included. Cells were labeled and analyzed as in (A). (C) COS-1 cells were co-transfected with vectors encoding BiP, a Flag-tagged truncated heavy chain consisting of only the VH and CH1 domains (Lee et al., 1999), and with the indicated light chain constructs (i.e., λ, wild type κ, or CL mutant κ). Cells were metabolically labeled and both cell lysates (L) and culture supernatants (M) were immunoprecipitated with the anti-Flag antibody. Precipitated proteins were separated by SDS-PAGE under non-reducing condition (except the first lane, which included 2-ME in the sample buffer and is indicated as red) and visualized by autoradiography. Mobilities of completely reduced (ox0), partially oxidized (ox1), and fully oxidized (ox2) forms of truncated heavy chain, as well as those of λ and κ light chains are indicated. The tagged forms of the κ light chain constructs co-migrate with the ox1 form of the truncated heavy chain.
To add support to the key role played specifically by the interactions between CH1 and CL domains, we first performed additional in vivo experiments using two different full length heavy chains (the MAK33 γ1 heavy chain and a humanized mouse IgG γ1 heavy chain (Liu et al., 1987)) in combination with both the wild type (LCwt) and the mutated MAK33 light chain (LCCLmut). The wild type MAK33 light chain assembled with both heavy chains and allowed their secretion from cells, whereas the mutant light chain was unable to assemble or induce secretion of the heavy chains (Fig. 4B and supplemental Fig. 5). This is in keeping with our in vitro data showing that the mutant CL domain was unfolded and therefore unable to induce the folding of the CH1 domain. To determine if this was also the case in vivo, we used a truncated version of the chimeric heavy chain consisting of only the VH and CH1 domains, which allows us to monitor oxidation of the CH1 domain based on an increase in its mobility (Lee et al., 1999). Indeed we found that the wild type MAK33 light chain induced oxidation and secretion comparable to a different λ light chain that was used in previous studies (Lee et al., 1999) (Fig. 4C, compare lanes 4 and 6 and lanes 5 and 7). However the mutant light chain was unable to induce either oxidation or secretion of this truncated heavy chain (Fig. 4C, lanes 8 and 9). These data clearly show that the correct folding of the CL domain is absolutely required to induce the folding and oxidation of the CH1 domain as well as their covalent assembly in vivo, which is in line with our in vitro data. Furthermore, these data demonstrate that the template-assisted folding of CH1 is a general and interchangeable control element functioning in murine and human antibodies.
Isomerization of a conserved proline residue in the CH1 domain is essential for assembly and secretion of IgG molecules in vivo
To directly assess the role of proline isomerization in the association-coupled folding reaction of the CH1 domain in vivo, each single cis proline residue in the CH1 domain was exchanged against alanine in the context of the full length heavy chain and metabolic labeling experiments were conducted. For the MAK33 wild type heavy chain and two of the heavy chains mutated in the CH1 domain, Pro34Ala and Pro74Ala, all Ig assembly intermediates could be detected (Fig. 5A, lanes 1,2,4,5,10 and 11), and importantly, completely assembled Ig molecules were secreted (Fig. 5A, lanes 3,6 and 12), demonstrating that isomerization of neither of these prolines was critical for CH1 domain folding in vivo. In fact, mutation of proline 74 to alanine actually increased the assembly and secretion of the heavy chain. In contrast, when proline 32 in the CH1 domain was mutated to alanine no significant amount of heavy chain and light chain assembly were detected nor was any heavy chain secreted (Fig. 5A, lanes 7–9). This is not due to poor expression of either the light chain or mutant heavy chain in this experiment (Fig. 5B). These results are in excellent agreement with our in vitro data (Fig. 2D) and reveal a key role of the trans to cis isomerization of Pro32 in the CH1 domain for the assembly, interchain disulfide bridge formation and secretion of IgG molecules. It should be noted that this critical proline residue is highly conserved in the various murine Ig isotypes as well as in the immunoglobulins of different species.
Figure 5. The isomerization of a single proline residue controls the assembly and secretion of heavy chains in vivo.
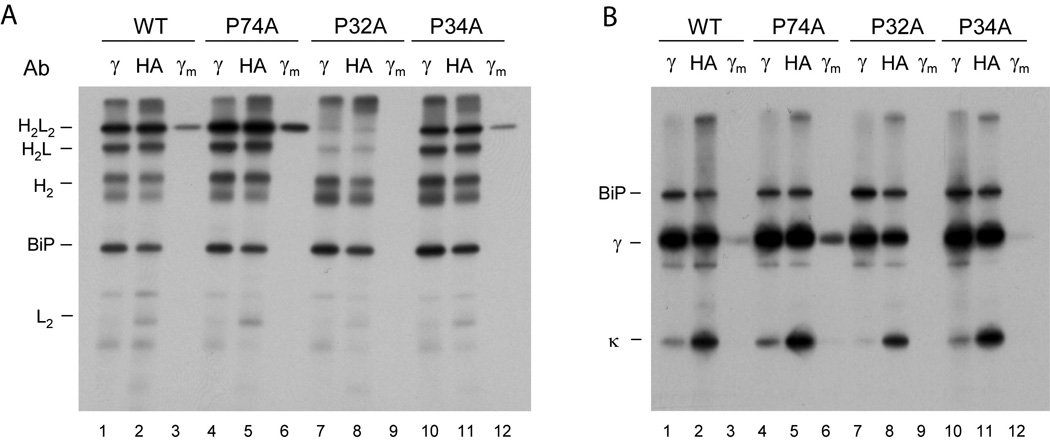
COS-1 cells were co-transfected with vectors encoding wild type MAK33 heavy chain (WT) or one of the three Pro to Ala mutants (P74A, P32A, or P34A) together with wild type light chain and BiP. Cells were metabolically labeled and both cell lysates (no subscript) and culture supernatants (subscript m) were immunoprecipitated with the indicated antisera (Ab). Precipitated proteins were separated by SDS-PAGE under non-reducing conditions and visualized by autoradiography. (B) The cell lysates and culture supernatants from (A) were divided in half, immunoprecipitated with the indicated antibodies, analyzed by SDS-PAGE under reducing conditions, and visualized with autoradiography. These data demonstrate that the failure of the P32A mutant to induce assembly and secretion is not because it is expressed poorly, as the signal for this mutant heavy chain is very similar to that of the wild type heavy chain.
Discussion
In this work we show that the CH1 domain of the murine IgG1 antibody is an intrinsically disordered protein. As CH1 does not possess an unusual number or distribution of charged or hydrophobic residues, it can still form a well defined globular structure once folded and does not show the typical sequence signature usually associated with intrinsically disordered proteins (Ward et al., 2004; Yang et al., 2005; Fink, 2005; Dunker et al., 2008). The large number of proline residues, also in a part of the CH1 domain that was recently identified as forming small helical elements important for antibody domain folding (Feige et al., 2008), might be one determinant contributing to its unfolded nature. Our data thus suggest that the CH1 domain is a representative of a novel class of intrinsically disordered proteins.
We demonstrate that the CL domain, which is the cognate partner of the CH1 domain in the complete IgG molecule, is required to fold CH1 to the structure observed in IgG antibodies. The detailed analysis of the underlying pathway suggests that the reaction is initiated by the recognition of a few key interface residues between CL and CH1, which then promotes the formation of a hydrophobic core in CH1. Both reactions render each other energetically more favorable and thereby allow the entropically demanding structuring of an unfolded polypeptide chain. In vitro, this folding reaction requires the presence of the internal disulfide bridge in CH1. Therefore one might assume that a roughly preformed topology or residual structure, which could not be detected in this study, might play a role in the folding process. Folding of the CH1 domain is rate-limited by proline isomerization as observed for most isolated antibody domains (Goto and Hamaguchi, 1982; Thies et al., 1999; Feige et al., 2004). The presence of several cis proline residues in the native state and the overall very large number of proline residues, however, sets the CH1 domain apart from most other antibody domains and may reflect the special role played by this domain. The key step in CH1 folding, the isomerization of a single highly conserved proline residue in the loop between strand B and C, is likely to be essential for the folding and secretion of most Ig classes and potentially of other Ig superfamily members.
We show that BiP, a major ER chaperone, strongly binds to CH1 in vitro, in agreement with previous in vivo studies (Hendershot et al., 1987). Oxidation of the internal CH1 disulfide bridge was possible in the BiP-bound state and BiP still binds to the oxidized CH1 domain in vitro, although this form has not been detected in vivo. After release from BiP, CH1 can complete folding upon association with CL and, if successful, form an interchain disulfide bridge with CL. Since heavy chains of most isotypes that are devoid of the CH1 domain can be secreted (Coffino et al., 1970; Hendershot et al., 1987), the essential steps controlling IgG assembly described here are likely to be general for all antibodies. Our data allow us to propose a possible order of events for this quality control mechanism in vivo. First, the CH1 domain binds to BiP as it enters the ER cotranslationally in the reduced state. Then, likely triggered by association with light chain (Lee et al., 1999), the oxidation of the internal disulfide bridge between Cys25 and Cys80 takes place, which brings at least two of the residues (Val21 and Val78) that are involved in the formation of the hydrophobic folding nucleus in close proximity to each other. Only after release from BiP can the oxidized CH1 domain complete its folding in association with CL. This scenario is in agreement with the fact that some residues in the CH1 domain found to be involved in the initial interaction with CL were among those identified as putative BiP binding sequences in this domain in a previous study (Knarr et al., 1995). Even though most antibody domains possess BiP binding sequences (Knarr et al., 1995), they interact with BiP in the cell only transiently or not at all due to the competing, rapid folding reaction (Hellman et al., 1999). In contrast, the continued unfolded status of the CH1 domain in the absence of light chain association allows it to permanently expose binding sites for BiP predisposing it for a stable interaction in the ER. In the complete antibody, the CL and the CH1 domain are covalently cross-linked via a disulfide bridge. We found that once this intermolecular disulfide bridge is formed, BiP no longer associates with the CH1 domain in vitro (data not shown), because formation of this interchain disulfide bridge is rate-limited by proline isomerization and hence depends on the complete folding of CH1. Thus, folding-dependent, covalent assembly provides yet another checkpoint for monitoring the proper maturation of Ig molecules in the ER and allows the assembled heavy chain to escape thiol-mediated retention mechanisms in cells (Sitia et al., 1990). It is conceivable that in the ER, association, oxidation and folding of CH1 are tightly coupled by the immunoglobulin assembly machinery, as no oxidized CH1 was found to be bound to BiP in vivo and ATP-induced release of BiP from unassembled heavy chains results in the formation of disulfide linked heavy chain aggregates (Vanhove et al., 2001).
Taken together, our data provide a detailed mechanism by which BiP and an intrinsically disordered antibody domain control the secretion of murine IgG1 antibodies (Fig. 6). The comprehensive model incorporates previous in vitro (Lilie et al., 1995; Mayer et al., 2000) and in vivo findings (Bole et al., 1986; Hendershot et al., 1987; Lilie et al., 1995; Lee et al., 1999; Mayer et al., 2000; Vanhove et al., 2001) and suggests regulatory hubs where additional components may come into play in vivo (Vanhove et al., 2001; Elkabetz et al., 2005) to orchestrate folding, oxidation, assembly and secretion.
Figure 6. A model for the overall IgG secretion control mechanism.
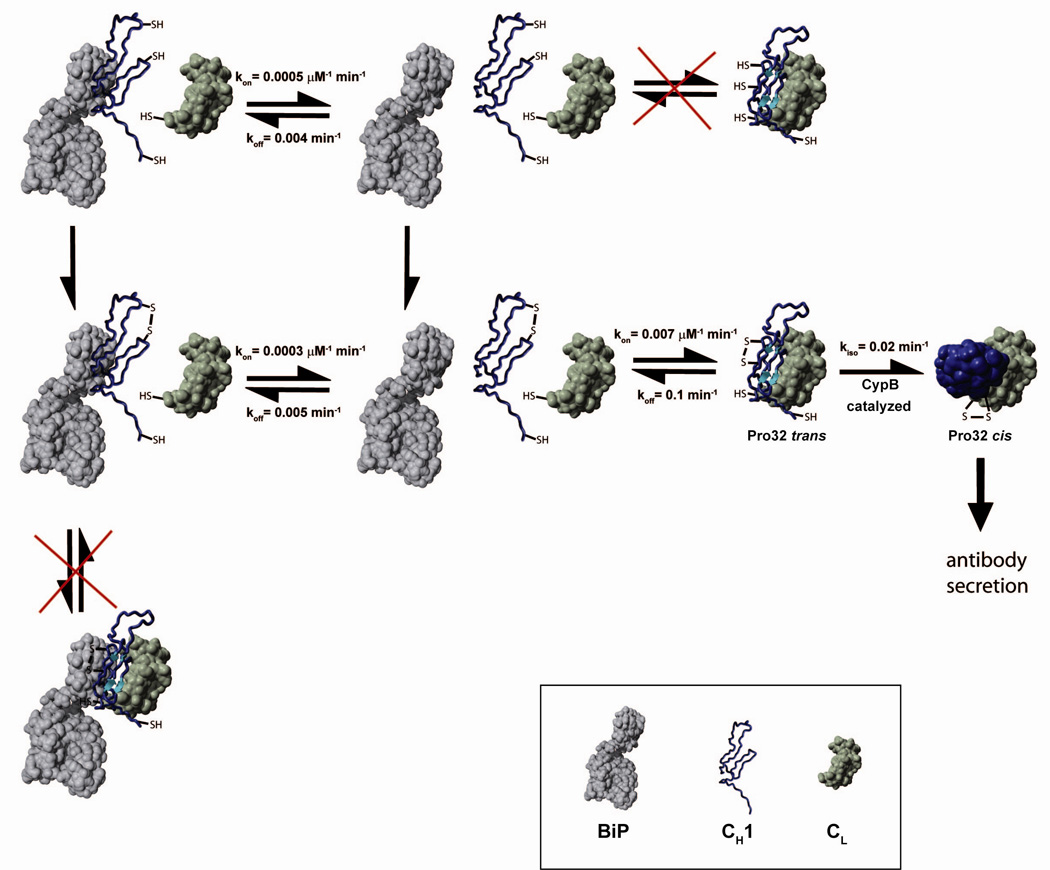
A schematic indicating the possible pathways for the CH1 domain (blue), its folding and assembly in association with CL (green) and BiP (grey) is shown. CH1 has to form its internal disulfide bridge and to be released from BiP before it can associate with CL. In vivo, these processes are tightly coupled and thus cannot be dissected kinetically. Prior to complete folding and irreversible formation of the CL/CH1 interchain disulfide bridge, the proline residue 32 has to isomerize from trans to cis. The isomerization reaction can be accelerated by CyclophilinB. All rate constants were determined at 25°C.
A rigorous assembly control mechanism is particularly important for antibody molecules, which undergo a developmentally asynchronous expression of heavy and light chain genes. In pre-B cells, heavy chain genes are rearranged first and the resultant proteins are largely retained in the cell (Burrows et al., 1979), except for a limited number that assemble with the surrogate light chain (Pillai and Baltimore, 1987). The developmentally more mature B cell expresses light chains, which assemble with heavy chains and allow their transport to the cell surface. Finally, the terminally differentiated plasma cell produces enormous quantities of Ig molecules (Cenci and Sitia, 2007), which must be appropriately assembled to bind specifically to antigens and fulfill their effector functions. Accordingly, the nature of the reactions that govern CH1 folding and assembly with CL allow efficient and accurate assembly of antibodies prior to secretion and might hint towards the co-evolution of substrates and folding factors in the ER as well as a general mechanism of quality control for oligomeric proteins.
Experimental Procedures
Protein production
All antibody domains were produced similar to published protocols (Feige et al., 2004; Feige et al., 2007). Details can be found in the Supplemental Materials section. Numbering of the CH1 domain begins with 1 in this work.
Hamster BiP (Wei and Hendershot, 1995) was mutated by site-directed mutagenesis to the murine sequence. Expression was carried out in HB101 cells for 3 h at 37°C. Following cell disruption, Ni-NTA affinity purification was performed in 50 mM Hepes/KOH, pH 7.5, 400 mM NaCl, 50 mM Imidazole. BiP was eluted with an Imidazole gradient from 0.05 M to 1 M. BiP containing fractions were applied to a Superdex 200pg (26/60) gel filtration column (GE Healthcare, München, Germany) equilibrated in HKM buffer (50 mM Hepes/KOH, pH 7.5, 150 mM KCl, 10 mM MgCl2) and finally to a Superdex 200 10/300GL HPLC column (GE Healthcare, München, Germany) equilibrated in the same buffer. The cyclophilin B (CypB) gene was amplified without the signal sequence from the murine cDNA (imaGenes, Berlin, Germany) and inserted into the pET28a vector. Expression was carried out for 3 h at 37°C in BL21-DE3 cells. The cell pellet was dissolved in 50 mM Hepes/KOH, pH 7.0, 10 mM EDTA and the cleared lysate was applied to a SP-Sepharose column equilibrated in the same buffer. The protein was eluted with a 0–1 M NaCl gradient. Subsequently, CypB containing fractions were applied to a Superdex 75pg (26/60) gel filtration column equilibrated in HKM buffer. All vectors were sequenced and protein masses were verified by mass spectrometry.
Optical spectroscopy
A Jasco J-720 spectropolarimeter was used for all CD measurements (Jasco, Gross-Umstadt, Germany). Far-UV CD spectra were recorded in a 0.2 mm quartz cuvette, far-UV kinetics in a 1 mm quartz cuvette. Spectra of the isolated domains were recorded at 45 µM protein concentration, for the spectrum of the complex 15 µM CH1 in the presence of 45 µM CL was used. Far-UV CD kinetics were recorded at 10 µM protein concentration of each domain and followed at 205 nm. Spectra of the CH1 domain in the complex were calculated by substraction of the spectrum of the isolated CL domain from the spectrum of the complex, measured after a 4 h equilibration step at 25°C. All spectra were averaged 16 times and buffer corrected. Fluorescence measurements were carried out in a Spex FluoroMaxIII spectrofluorimeter (Jobin Yvon, München, Germany) in a stirred 1 cm quartz cuvette. Kinetics and titrations were measured by the change in the intrinsic tryptophan fluorescence, excited at 280 nm and detected at 350 nm. For titrations, varying concentrations of CL were added to 2 µM CH1 and immediately as well as after a 4 h equilibration step at 25°C, the fluorescence of the same samples was determined. The difference between initial and final fluorescence emission was normalized and analyzed according to a one-site binding model. Iodide quenching experiments were performed as published (Feige et al., 2007).
For anisotropy measurements, 1 µM lucifer yellow labeled CL Ala113Cys and varying concentrations of CH1 were used. Lucifer yellow fluorescence was excited at 430 nm and detected at 525 nm. The change in quantum yield of the chromophore was less than 5% upon association of the labeled CL domain with CH1. Individual traces were fitted by single exponential functions. The obtained rate constants were fitted to a linear equation to derive the kon and the koff value.
NMR spectroscopy
Spectra of the CH1 domain in complex with the CL domain were recorded at 25°C on Bruker DMX600 and DMX750 spectrometers (Bruker, Rheinstetten, Germany), whereas spectra for the assignment of the unfolded CH1 domain were measured at 12.5°C on a Bruker AVANCE900 spectrometer (Bruker, Rheinstetten, Germany). Backbone sequential assignment of the isolated CH1 domain was obtained by standard triple resonance experiments implemented with selective proton flip-back techniques for fast pulsing (Diercks et al., 2005). To gain information about any preferential conformations present in the disordered CH1 domain, an NNH-NOESY spectrum and a 15N-HSQC-NOESY spectrum were recorded on a highly deuterated sample with a mixing time of 600 ms in order to detect long range HN-HN NOEs (Mok et al., 1999).
For all measurements of the folded CH1 domain in association with CL, a twofold excess of unlabeled CL was added to 15N or 15N, 13C labeled CH1. Prior to steady state measurements, samples were incubated for at least 6 h at room temperature to ensure complete folding of the CH1 domain. Backbone sequential assignment of the assembled CH1 domain was achieved with standard triple resonance experiments with selective proton flip-back techniques for fast pulsing. The assignment of the carbon chemical shifts was limited to the C’ and Cα chemical shifts due to the relaxation properties of the whole protein complex. To verify the backbone resonance assignment NH residual dipolar couplings (RDCs) were determined. The sample was prepared as described above and aligned with nonionic liquid crystalline media (Ruckert and Otting, 2000). NH RDC values were extracted from IPAP-HSQC spectra using Bruker pulse sequences. The sequential information based on the C’ and Cα chemical shifts as well as the NH RDC values and the crystal structure of the folded CH1 domain (pdb code: 1ORS) served as input for the software MARS (Jung and Zweckstetter, 2004b; Jung and Zweckstetter, 2004a).
In order to characterize the folding pathway of the intrinsically disordered CH1 domain, 15N-HSQC spectra were recorded at 12.5°C every 14 min immediately after adding unlabeled CL to 15N labeled CH1 using selective proton flip-back techniques for fast pulsing. Identical processing of all spectra measured during the folding process was performed with the software TOPSPIN 1.3 (Bruker Biospin). Peak intensities were analyzed using the software SPARKY (www.cgl.ucsf.edu/home/sparky) and normalized to the corresponding intensities in the final spectrum after 36 h. The backbone resonance assignment was transferred from 25°C to 12.5°C recording a HSQC temperature series.
Analytical HPLC experiments
For all experiments, a Shimadzu HPLC system (Shimadzu, München, Germany) was used. Complex formation between BiP and CH1 was analyzed on a Superdex 200 10/300GL column in HKM buffer at a flow rate of 0.5 ml/min. For the determination of the dissociation constant as well as binding kinetics, peak intensities at the retention time of 28.4 min corrected for baseline drifts were plotted against the CH1 concentration respectively the incubation time and normalized. The rate constants kobs of the binding reaction were determined from single exponential fits and evaluated with a linear equation to derive kon and koff. Detection of all proteins was performed by the intrinsic fluorescence excited at 280 nm and monitored at 350 nm. Incubation steps were performed in HKM buffer with 1 mM ADP.
Cell culture experiments
The murine IgG1 MAK33 light chain (LCWT) and heavy chain (HCWT) cDNAs were obtained with an intact signal sequence for expression in mammalian ER. Two LC mutants, one in which the CH1 domain was substituted for the CL domain (LCCH1) and the other where structural features of the CH1 domain were substituted for the corresponding regions of the CL domain (LCCLmut) were produced and all constructs were inserted into the pSVL vector (GE Healthcare, München, Germany). An HA-epitope tag was engineered at the C-terminus of the wild type light chain and the two mutants for immunoprecipitation purposes. Heavy chain proline exchange mutants were generated by site-directed mutagenesis. The cDNA for a chimeric humanized heavy chain was used as published (Liu et al., 1987) and a truncated version of this heavy chain containing only the VH and CH1 domains was produced previously (Lee et al., 1999) as was a mouse lambda light chain cDNA (Hellman et al., 1999). The recombinant plasmids, along with a pMT vector encoding hamster BiP (Lee et al., 1999) were introduced into COS-1 cells (Gluzman, 1981) that were cultured as described (Lee et al., 1999) using FuGENE 6 transfection reagent (Roche, Indianapolis, USA) following the manufacturer’s protocol. Metabolic labeling, cell lysis, immunoprecipitation, and visualization of the proteins were performed as described previously (Lee et al., 1999). Anti-rodent BiP antiserum (Hendershot et al., 1995), a monoclonal anti-HA (12CA5) antibody (kindly provided by Dr. Al Reynolds, Vanderbilt University, USA), and a goat anti-mouse Ig κ and goat anti-mouse γ antibodies (Southern Biotech, Birmingham, AL, USA) and Protein A Sepharose beads were used for immunoprecipitations. For metabolic labeling experiments, cells were cultured in DMEM lacking methionine and cysteine and labeled with 35S Translabel (MP Biomedicals, Irvine, CA, USA) for the indicated times.
Supplementary Material
Acknowledgements
We thank Frank Striebel for preliminary work on the in vivo experiments and Helmut Krause for performing mass spectrometry experiments. Funding of MJF and MM by the Studienstiftung des deutschen Volkes, of LMH by NIH Grant GM54068, the Cancer Center CORE Grant CA21765, and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital and of JB by the SFB749, the Fonds der chemischen Industrie and the Bayerische Forschungsstiftung is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- Adetugbo K. Spontaneous somatic mutations. Structural studies on mutant immunoglobulins. J. Biol. Chem. 1978;253:6076–6080. [PubMed] [Google Scholar]
- Amzel LM, Poljak RJ. 3-Dimensional Structure of Immunoglobulins. Annual Review of Biochemistry. 1979;48:961–997. doi: 10.1146/annurev.bi.48.070179.004525. [DOI] [PubMed] [Google Scholar]
- Aricescu AR, Jones EY. Immunoglobulin superfamily cell adhesion molecules: zippers and signals. Curr. Opin. Cell Biol. 2007;19:543–550. doi: 10.1016/j.ceb.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Augustine JG, de la Calle A, Knarr G, Buchner J, Frederick CA. The crystal structure of the Fab fragment of the monoclonal antibody MAK33 -Implications for folding and interaction with the chaperone BiP. Journal of Biological Chemistry. 2001;276:3287–3294. doi: 10.1074/jbc.M005221200. [DOI] [PubMed] [Google Scholar]
- Bole DG, Hendershot LM, Kearney JF. Posttranslational Association of Immunoglobulin Heavy-Chain Binding-Protein with Nascent Heavy-Chains in Nonsecreting and Secreting Hybridomas. Journal of Cell Biology. 1986;102:1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Holm L, Sander C. The Immunoglobulin Fold - Structural Classification, Sequence Patterns and Common Core. Journal of Molecular Biology. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- Burrows P, LeJeune M, Kearney JF. Evidence that murine pre-B cells synthesise mu heavy chains but no light chains. Nature. 1979;280:838–840. doi: 10.1038/280838a0. [DOI] [PubMed] [Google Scholar]
- Cenci S, Sitia R. Managing and exploiting stress in the antibody factory. FEBS Lett. 2007;581:3652–3657. doi: 10.1016/j.febslet.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Christis C, Lubsen NH, Braakman I. Protein folding includes oligomerization - examples from the endoplasmic reticulum and cytosol. FEBS J. 2008;275:4700–4727. doi: 10.1111/j.1742-4658.2008.06590.x. [DOI] [PubMed] [Google Scholar]
- Coffino P, Laskov R, Scharff MD. Immunoglobulin production: method for quantitatively detecting variant myeloma cells. Science. 1970;167:186–188. doi: 10.1126/science.167.3915.186. [DOI] [PubMed] [Google Scholar]
- Cota E, Steward A, Fowler SB, Clarke J. The folding nucleus of a fibronectin type III domain is composed of core residues of the immunoglobulin-like fold. Journal of Molecular Biology. 2001;305:1185–1194. doi: 10.1006/jmbi.2000.4378. [DOI] [PubMed] [Google Scholar]
- Diercks T, Daniels M, Kaptein R. Extended flip-back schemes for sensitivity enhancement in multidimensional HSQC-type out-and-back experiments. Journal of Biomolecular NMR. 2005;33:243–259. doi: 10.1007/s10858-005-3868-4. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Argon Y, Bar-Nun S. Cysteines in CH1 underlie retention of unassembled ig heavy chains. Journal of Biological Chemistry. 2005;280:14402–14412. doi: 10.1074/jbc.M500161200. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature Reviews Molecular Cell Biology. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Groscurth S, Marcinowski M, Yew ZT, Truffault V, Paci E, Kessler H, Buchner J. The structure of a folding intermediate provides insight into differences in immunoglobulin amyloidogenicity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13373–13378. doi: 10.1073/pnas.0802809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MJ, Hagn F, Esser J, Kessler H, Buchner J. Influence of the internal disulfide bridge on the folding pathway of the C–L antibody domain. Journal of Molecular Biology. 2007;365:1232–1244. doi: 10.1016/j.jmb.2006.10.049. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Walter S, Buchner J. Folding mechanism of the C(H)2 antibody domain. Journal of Molecular Biology. 2004;344:107–118. doi: 10.1016/j.jmb.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Fink AL. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Freund C, Honegger A, Hunziker P, Holak TA, Pluckthun A. Folding nuclei of the scFv fragment of an antibody. Biochemistry. 1996;35:8457–8464. doi: 10.1021/bi952764a. [DOI] [PubMed] [Google Scholar]
- Gemmecker G, Jahnke W, Kessler H. Measurement of Fast Proton-Exchange Rates in Isotopically Labeled Compounds. Journal of the American Chemical Society. 1993;115:11620–11621. [Google Scholar]
- Gluzman Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- Goto Y, Hamaguchi K. Role of the Intrachain Disulfide Bond in the Conformation and Stability of the Constant Fragment of the Immunoglobulin Light Chain. Journal of Biochemistry. 1979;86:1433–1441. doi: 10.1093/oxfordjournals.jbchem.a132661. [DOI] [PubMed] [Google Scholar]
- Goto Y, Hamaguchi K. Unfolding and Refolding of the Constant Fragment of the Immunoglobulin Light Chain. Journal of Molecular Biology. 1982;156:891–910. doi: 10.1016/0022-2836(82)90146-2. [DOI] [PubMed] [Google Scholar]
- Haas IG, Wabl M. Immunoglobulin Heavy-Chain Binding-Protein. Nature. 1983;306:387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally-Occurring Antibodies Devoid of Light-Chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- Hamill SJ, Steward A, Clarke J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. Journal of Molecular Biology. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- Helenius A, Marquardt T, Braakman I. The endoplasmic reticulum as a protein-folding compartment. Trends Cell Biol. 1992;2:227–231. doi: 10.1016/0962-8924(92)90309-b. [DOI] [PubMed] [Google Scholar]
- Hellman R, Vanhove M, Lejeune A, Stevens FJ, Hendershot LM. The in vivo association of BiP with newly synthesized proteins is dependent on the rate and stability of folding and not simply on the presence of sequences that can bind to BiP. J. Cell Biol. 1999;144:21–30. doi: 10.1083/jcb.144.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot L, Bole D, Kohler G, Kearney JF. Assembly and Secretion of Heavy-Chains That do Not Associate Posttranslationally with Immunoglobulin Heavy-Chain Binding-Protein. Journal of Cell Biology. 1987;104:761–767. doi: 10.1083/jcb.104.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM, Wei JY, Gaut JR, Lawson B, Freiden PJ, Murti KG. In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol. Biol. Cell. 1995;6:283–296. doi: 10.1091/mbc.6.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. 1976;264:415–420. doi: 10.1038/264415a0. [DOI] [PubMed] [Google Scholar]
- Jahn TR, Parker MJ, Homans SW, Radford SE. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nature Structural & Molecular Biology. 2006;13:195–201. doi: 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- Jung YS, Zweckstetter M. Backbone assignment of proteins with known structure using residual dipolar couplings. Journal of Biomolecular NMR. 2004a;30:25–35. doi: 10.1023/B:JNMR.0000042955.14647.77. [DOI] [PubMed] [Google Scholar]
- Jung YS, Zweckstetter M. Mars - robust automatic backbone assignment of proteins. Journal of Biomolecular NMR. 2004b;30:11–23. doi: 10.1023/B:JNMR.0000042954.99056.ad. [DOI] [PubMed] [Google Scholar]
- Kaloff CR, Haas IG. Coordination of Immunoglobulin Chain Folding and Immunoglobulin Chain Assembly Is Essential for the Formation of Functional Igg. Immunity. 1995;2:629–637. doi: 10.1016/1074-7613(95)90007-1. [DOI] [PubMed] [Google Scholar]
- Kameda A, Hoshino M, Higurashi T, Takahashi S, Naiki H, Goto Y. Nuclear magnetic resonance characterization of the refolding intermediate of beta(2)-microglobulin trapped by non-native prolyl peptide bond. Journal of Molecular Biology. 2005;348:383–397. doi: 10.1016/j.jmb.2005.02.050. [DOI] [PubMed] [Google Scholar]
- Knarr G, Gething MJ, Modrow S, Buchner J. BiP binding sequences in antibodies. J. Biol. Chem. 1995;270:27589–27594. doi: 10.1074/jbc.270.46.27589. [DOI] [PubMed] [Google Scholar]
- Lee YK, Brewer JW, Hellman R, Hendershot LM. BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Molecular Biology of the Cell. 1999;10:2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilie H, Rudolph R, Buchner J. Association of antibody chains at different stages of folding: prolyl isomerization occurs after formation of quaternary structure. J. Mol. Biol. 1995;248:190–201. doi: 10.1006/jmbi.1995.0211. [DOI] [PubMed] [Google Scholar]
- Liu AY, Robinson RR, Murray ED, Jr, Ledbetter JA, Hellstrom I, Hellstrom KE. Production of a mouse-human chimeric monoclonal antibody to CD20 with potent Fc-dependent biologic activity. J. Immunol. 1987;139:3521–3526. [PubMed] [Google Scholar]
- Mayer M, Kies U, Kammermeier R, Buchner J. BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J. Biol. Chem. 2000;275:29421–29425. doi: 10.1074/jbc.M002655200. [DOI] [PubMed] [Google Scholar]
- Melchers F. The secretion of a Bence-Jones type light chain from a mouse plasmacytoma. Eur. J. Immunol. 1971;1:330–335. doi: 10.1002/eji.1830010505. [DOI] [PubMed] [Google Scholar]
- Meunier L, Usherwood YK, Chung KT, Hendershot L. A sub-set of chaperones and folding enzymes form multi-protein complexes in the ER to bind nascent proteins. Molecular Biology of the Cell. 2002;12:488A. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok YK, Kay CM, Kay LE, Forman-Kay J. NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions. J. Mol. Biol. 1999;289:619–638. doi: 10.1006/jmbi.1999.2769. [DOI] [PubMed] [Google Scholar]
- Paci E, Clarke J, Steward A, Vendruscolo M, Karplus M. Self-consistent determination of the transition state for protein folding: Application to a fibronectin type III domain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:394–399. doi: 10.1073/pnas.232704999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Baltimore D. Formation of disulphide-linked mu 2 omega 2 tetramers in pre-B cells by the 18K omega-immunoglobulin light chain. Nature. 1987;329:172–174. doi: 10.1038/329172a0. [DOI] [PubMed] [Google Scholar]
- Porter RR. Structural studies of immunoglobulins. Science. 1973;180:713–716. doi: 10.1126/science.180.4087.713. [DOI] [PubMed] [Google Scholar]
- Qin ZJ, Hu DM, Zhu M, Fink AL. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry. 2007;46:3521–3531. doi: 10.1021/bi061716v. [DOI] [PubMed] [Google Scholar]
- Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. Journal of Molecular Biology. 2005;347:773–789. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- Rougon G, Hobert O. New insights into the diversity and function of neuronal immunoglobulin superfamily molecules. Annu. Rev. Neurosci. 2003;26:207–238. doi: 10.1146/annurev.neuro.26.041002.131014. [DOI] [PubMed] [Google Scholar]
- Ruckert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. Journal of the American Chemical Society. 2000;122:7793–7797. [Google Scholar]
- Shaffer AL, Schlissel MS. A truncated heavy chain protein relieves the requirement for surrogate light chains in early B cell development. J. Immunol. 1997;159:1265–1275. [PubMed] [Google Scholar]
- Sitia R, Neuberger M, Alberini C, Bet P, Fra A, Valetti C, Williams G, Milstein C. Developmental regulation of IgM secretion: the role of the carboxy-terminal cysteine. Cell. 1990;60:781–790. doi: 10.1016/0092-8674(90)90092-s. [DOI] [PubMed] [Google Scholar]
- Thies MJW, Mayer J, Augustine JG, Frederick CA, Lilie H, Buchner J. Folding and association of the antibody domain C(H)3: Prolyl isomerization preceeds dimerization. Journal of Molecular Biology. 1999;293:67–79. doi: 10.1006/jmbi.1999.3128. [DOI] [PubMed] [Google Scholar]
- Vanhove M, Usherwood YK, Hendershot LM. Unassembled Ig heavy chains do not cycle from BiP in vivo but require light chains to trigger their release. Immunity. 2001;15:105–114. doi: 10.1016/s1074-7613(01)00163-7. [DOI] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Wei JY, Hendershot LM. Characterization of the Nucleotide-Binding Properties and Atpase Activity of Recombinant Hamster Bip Purified from Bacteria. Journal of Biological Chemistry. 1995;270:26670–26676. doi: 10.1074/jbc.270.44.26670. [DOI] [PubMed] [Google Scholar]
- Williams AF, Barclay AN. The immunoglobulin superfamily--domains for cell surface recognition. Annu. Rev. Immunol. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- Wolfenstein-Todel C, Mihaesco E, Frangione B. "Alpha chain disease" protein def: internal deletion of a human immunoglobulin A1 heavy chain. Proc. Natl. Acad. Sci. U. S. A. 1974;71:974–978. doi: 10.1073/pnas.71.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.