

Abstract
Heterogeneous heparan sulfate and chondroitin sulfate glycosaminoglycan (GAG) polysaccharides are important components of blood circulation. Changes in GAG quantity and structure in blood have been indicated in cancers and other human diseases. However, GAG quantities and structures have not been fully characterized due to lack of robust and sensitive analytical tools. To develop such tools, we isolated GAGs from serum and plasma. We employed liquid chromatography (LC) for GAG quantification and LC/mass spectrometry (MS) for GAG structural analysis. Twenty-four heparan and chondroitin sulfate motifs were identified, including linkage hexasaccharides, repeating disaccharide compositions, reducing, and non-reducing end mono-, di-, tri-, and tetrasaccharide structures. Disaccharides were detectable at picomolar level without radiolabeling or derivitization, so only a few ml of human and fetal bovine serum was required for this study. The detection of different reducing end structures distinct from GAG linkage hexasaccharides revealed that free GAG chains generated by GAG degradation enzymes co-existed with proteoglycans in serum. In addition, a novel sialic acid-modified linkage hexasaccharide was found conjugated to bikunin, the most abundant serum proteoglycan.
Keywords: human and fetal bovine serum, heparan sulfate, chondroitin sulfate, mass spectrometry
Introduction
Glycosaminoglycans (GAGs) are linear polysaccharides comprised of glucosamine-/galactosamine-containing repeating disaccharides. Two major types of GAGs, heparan sulfate (HS) and chondroitin sulfate (CS), are produced by most animal cells.1 Because of their differential sulfation, these linear polysaccharides are the most information dense biopolymers found in nature. GAGs are generated during the biosynthesis of protein/GAG conjugates known as proteoglycans. The biological functions of proteoglycans are largely determined by their GAG chains. Rapid turnover2 and structure diversity generated by both biosynthesis and post biosynthesis editing allow GAGs to regulate a myriad of signaling pathways.3 Many proteoglycans, such as syndecan, endocan, and glypican, and GAG structure-editing enzymes, such as heparanase, sulfatases, and hyaluronidases, are regarded as potential cancer diagnosis/prognosis markers or treatment targets4–11 GAGs interact with hundreds of plasma proteins,12 including growth factors, cytokines, chemokines, proteases, protease inhibitors, coagulant and anticoagulant proteins, complement proteins, lipoproteins, and lipolytic enzymes.13,14 More than 95% of plasma GAGs form complexes with plasma proteins.15 The ability to quantify changes in blood GAG structures will advance the understanding and diagnosis of human diseases.
Knowledge of blood circulating GAGs has accumulated over the last 50 years.16 CS represents the major GAG in blood circulation. Other serum GAGs include HS, keratan sulfate, and hyaluronan. The relative amount of HS and keratan sulfates varies greatly in patients.17,18 Each of these GAGs is heterogeneous with respect to molecular size, disaccharide composition and sulfate content. Numerous analytical methods such as cellulose acetate membrane electrophoresis, paper chromatography, gas chromatography, affinity chromatography, HPLC, capillary electrophoretic analysis have been developed and applied to quantitative, qualitative, and structural analysis of GAGs.19,20 However, plasma GAG structures have not yet been fully characterized due to a general lack of robust and sensitive analytical tools. There are no standardized methods for serum/plasma GAG isolation and quantification. Moreover, when GAGs are co-purified with their protein binding partners, their quantification, biological activity, and structural analysis can be compromised or impaired.17
GAG fine structure is traditionally assessed by analyzing repeating disaccharide compositions. GAG disaccharides are obtained by either chemical or enzymatic depolymerization of GAG chains followed by gel filtration chromatography to separate disaccharides from salt, mono-, tri-, and other oligosaccharides and undigested materials. GAG disaccharides have been separated based on charge, position of sulfate groups and uronic acid composition by anion exchange HPLC, reverse phase HPLC, capillary electrophoresis, and fluorophore-assisted carbohydrate electrophoresis (FACE).21–24 Thus far, twenty-three HS disaccharides3 and twenty-five CS disaccharides,16 derived from a variety of biological sources, have been described. GAG disaccharide composition analysis can quantify GAGs and provide structural information.22,25 However, these methods are critically dependent on a comparison of retention times relative to authentic standards which are sometimes difficult to obtain. Moreover, interpretation of the chromatograms can be problematic due to contaminants and/or the presence of unknown saccharide peaks.
Mass spectrometry (MS) has emerged as the tool of choice for GAG disaccharide, oligosaccharide, and GAG sequence analysis.26–39 Electrospray ionization mass spectrometry (ESI MS) utilizes a flowing stream containing the analyte, provides for very gentle ionization, and detects all ions simultaneously. Reverse phase HPLC coupled with ESIMS greatly benefits GAG disaccharide analysis. The unique elution times of the disaccharides from capillary HPLC plus the mass/charge (m/z) values obtained from MS provide enough information to assign each disaccharide unambiguously without the use of standards.36,37
In this study we isolated serum and plasma GAGs from a few ml of human and fetal bovine serum (FBS) and employed LC/MS for GAG structural analysis. We detected twenty-four HS and CS structural motifs, including linkage hexasaccharides, repeating disaccharide compositions, reducing, and non-reducing end mono-, di-, tri-, and tetrasaccharide structures. In addition, a novel sialic acid-modified linkage hexasaccharide was found conjugated to bikunin, the most abundant serum proteoglycan.
Material and Methods
Materials
DEAE-Sephacel (Sigma I-6505) was washed three times in 5 volumes of 2.0 M NaCl, 0.01% Triton X-100, 20 mM NaAc pH 6.0, five times in 5 volumes of water, and then suspended 1:1 in water before use. FBS was purchased from Sigma, Hyclone, and Mediatech, Normal human whole blood samples were collected into either regular tubes for serum preparation or two 4 ml vacutainers containing K3EDTA (1.6 mg EDTA per ml blood) for plasma preparation, and centrifuged at 1500 g for 10 min. Serum and plasma were immediately stored at −80 °C before use.
Preparation of metabolically-labeled35 S-sulfate HS
Nine CHO cell lines, including CHO K1, CHOF17, CHOF17.3, CHOK1.5, CHOK1.N7, CHOPPP6, 3OST-1-, 3OST3A-, and 3OST-5-expressing CHO lines established in Dr. Esko,40,41 Dr. Rosenberg,42–44 and Dr. Liu’s45 laboratories, were metabolically labeled with35 S-sulfate using the published protocols. 46,47 GAGs were purified and pooled from the 9 CHO cell lines. CS was removed by chondroitinase ABC digestion and HS was used in some experiments to assess the recovery of GAGs from serum and plasma samples. CHO HS has ~0.8 sulfate per disaccharide, which is comparable to the sulfation degree of plasma GAGs. We used the 9 CHO cell lines because together they might represent a variety of GAG structures, which allow a fair estimation of the recovery of GAGs from serum and plasma samples.
Isolation of proteoglycan core protein peptide-containing GAGs
We isolated serum GAG chains from proteoglycans and GAG-binding proteins by exhaustive digestion with Streptomyces griseus protease (Sigma P5147): A protease stock solution (2 ml), containing 1 mg/ml protease, 1.92 M NaCl, and 0.24 M NaAc, pH 6.5, was added to 10 ml serum solution containing 5 ml FBS or human serum plus 5 ml of water. The digestion was performed overnight at 37 °C. The sample was filtered (0.2 μm). The filtrate was diluted with 20 ml of water and the pH and Triton X-100 concentration were adjusted to 6 and 0.01%, respectively. The sample was loaded onto a 0.5 ml DEAE-Sephacel column pre-equilibrated with 12 ml of 0.25 M NaCl, 0.01% Triton X-100, 20 mM NaAc, pH 6.0 and the column was washed with 30 ml of the same equilibrating buffer. GAGs were eluted with 2.5 ml 1 M NaCl in 0.01% Triton X-100, 20 mM NaAc, pH 6.0, precipitated with 10 ml of ethanol, and incubated at 4 °C overnight. The GAG ethanol precipitate was collected by centrifugation (16000 g for 10 min), washed with 1 ml of 75% ethanol, collected by centrifugation (16000 g; 10 min), and suspended in 200 μl water and stored at 4 °C for further analysis. The proteoglycan GAG chains collected in this manner were each expected to be covalently attached to a residual peptide that derived from the core protein after the protease digestion.
Isolation of proteoglycan core peptide-free GAGs
GAGs prepared as above were freed of their residual core peptide by a β-elimination reaction followed by DEAE-Sephacel purification. In brief, 100 μl of 1M NaBH4 in 0.5M NaOH was added to 100 μl GAG sample and incubated at 4 °C for 24 h (This treatment cleaves GAG chains from proteoglycan core protein peptides). The borohydride was then destroyed by adding 20 μl of glacial HAc to the sample. One ml of water and 5 μl of 1 mg/ml phenol red were added to the sample to monitor pH. The acidic sample (yellow) was neutralized with 2.0 M NaOH until the sample turned pinkish. The pH was checked by applying 1 μl of sample to pH paper allowing adjustment to pH 6 by addition of 1 M HAc. DEAE-Sephacel slurry (100 μl, beads to water 1:1) was added to the β-eliminated GAG samples. The maximum sample and DEAE-Sephacel interaction was achieved by inversion for at least 5 min on a rocker platform (Using radiolabeled GAGs as a tracer, we found that complete absorption of GAGs to DEAE-Sephacel beads took less than 2 min). The sample was microcentrifuged at 16000 g for 1 min. The supernatant was aspirated and the beads were washed three times with 1 ml 0.25 M NaCl, 0.01% Triton X-100, 20 mM NaAc pH 6.0 by re-suspension and microcentrifugation (1 min). The GAGs were pooled from the supernatants by four successive 100 μl 1.0 M NaCl, 0.01% Triton X-100, 20 mM NaAc pH 6.0 elution steps. The pooled sample was microcentrifuged at 16000 g for 1 min to pellet any carryover beads and the supernatant was transferred to a new 1.5-ml tube. The GAGs were precipitated by adding 10 μl of 20 mg/ml glycogen and 1.1 ml of 100% ethanol to the supernatant for at least 2 h at 4 °C. The sample was microcentrifuged at 16000g for 15 min and the supernatant was aspirated. The pellet was washed with 0.5 ml of 75% ethanol by a quick vortex/spin cycle. The GAGs in the dried pellet were dissolved in 100 μl of water in a siliconized tube and stored at 4 °C for further analysis.
GAG quantification
This assay has been described in detail previously.48 The steps are acid hydrolysis, sodium borohydride reduction, precolumn derivatization with O-phthaldialdehyde (OPA) and 3-mercaptopropionic acid (3 MPA), and reversed phase HPLC separation with fluorescence detection of the isoindole derivatives. GAG aliquots containing 6 nmol of norleucine as an internal standard are dried in pyrolized glass vials (Agilent, Palo Alto, CA, part 5181-8872) before hydrolysis with HCl vapor in N2 gas at 100 °C for 3 hours. The samples are rehydrated in 45 μl of 0.56% NaBH4 to reduce the glucosamine and galactosamine liberated by acid hydrolysis into glucosaminitol and galactosaminitol, respectively. After 3 hours or overnight at room temperature, the reaction is terminated by adding 5 μl of 2N acetic acid to each vial. A 5-μl aliquot of each hydrolyzed and reduced sample is transferred to a fresh autosampler insert vial for precolumn derivatization with 35 μl of 7.5 mM OPA, 375 mM 3 MPA, in 0.4 N borate adjusted to pH 9.3 with NaOH. Half of this reaction mixture is injected onto a 4.6 × 250 mm C-12 column, a Synergi 4 μMAX-RP 80 Å (Phenomenex, Torrance, CA, part 00G-4337-E0), heated to 35 °C. The column was equilibrated with Buffer A, consisting of 0.05 M (monobasic and dibasic) sodium phosphate, pH 7.2 in 25% methanol, at a flow rate of 0.8 ml/min. Buffer B consisted of methanol/water/tetrahydrofuran at 70:30:3 volume ratios. After injection, Buffer B is increased from 0 to 8% by a linear gradient between 0 and 3 min, is maintained at 8% between 3 and 18 min, at 55% between18 and 30.5 min, at 100% between 30.5 and 32.5 min, and at 0% between 32.5 and 35 min. A 5 min post-run interval at 0% B precedes the initiation of the next precolumn derivatization injection sequence. The fluorescent derivatives of glucosaminitol, galactosaminitol, and the amino acids contained in the GAG preparations were excited at 337 nm and detected at 454 nm. Quantification of the GlcN and GalN peaks was based on calibration curves derived from 10 external standards ranging from 2.5 pmol to 1280 pmol (y = 2.085 × −22.59, r2 = 0.997 for GlcN and y = 2.085 × −22.59, r2 = 0.9958 for GalN).
Enzymatic degradation of GAGs
Heparitinase I (EC 4.2.2.8), heparitinase II (EC 4.2.2.8), heparinase (EC 4.2.2.7), chondroitinase ABC (EC 4.2.2.4), chondroitinase ACII (EC 4.2.2.5), CS 4-O-sulfatase (EC 3.1.6.9), and CS 6-O-sulfatase (EC 3.1.6.10) were purchased from Seikagaku. Heparitinase IV was a kind gift from Dr. Yoshida, Seikagaku Corporation. The enzymes 2-O-sulfatase (S1062), Δ4, 5-glycuronidase (G9028), and α (2 → 3,6,8,9) sialidase (N2876) were purchased from Sigma-Aldrich. HS and CS disaccharide standards were purchased from Sigma-Aldrich and Seikagaku. The disaccharides were dissolved in water to make 1 mM stock solutions and the concentrations were confirmed by GAG quantification.48 All enzymatic GAG digestions were performed at 37 °C. For HS digestions, GAGs were incubated in 100 μl of digestion solution containing 5 mM NH4Ac pH 7.0, 0.125 mM CaCl2, and 0.33 mU each of heparin lyase I, II, III and IV. For CS digestions, GAGs were incubated in 100 μl of digestion solution containing 5 mM ammonium acetate pH 7.0 and 1 mU chondroitinase ABC. Combined HS and CS digestions were in 100 μl of digestion solution containing 0.33 mU of each heparin lyase, plus 1 mU chondroitinase ABC. Digestion was monitored by increased absorbance at 232 nm by using a Spectra MAX M2 plate-reading spectrophotometer (Molecular Devices, Sunnyvale, CA) with the temperature of the plate chamber set at 37 °C. For exhaustive digestions, the digests were incubated at 37 °C overnight after adding another 100 μl of digestion buffer and enzymes. The same heparin lyase buffer was used for all sulfatases and Δ4, 5-glycuronidase digestions. The sialidase digestion was carried out overnight in a buffer containing 25 mM NH4Ac pH 5.0 with 10 mU of sialidase. The digests were lyophilized and reconstituted in 10 μl water. Unless otherwise indicated, 0.5 μl of digested GAG sample was analyzed by LC/MS as described below.
LC/MS
An Agilent 1100 series capillary HPLC workstation (Agilent, Palo Alto, CA) with Chemstation software was used for data acquisition, analysis, and management. HPLC separations were performed on a 0.3 × 250 mm C18 column (Zorbax 300SB, 5 μm, Agilent) using a binary solvent system composed of 5% methanol (eluent A) and 90% methanol in water (eluent B), both contained 3.5 mM dibutylamine and adjusted to pH 5.5 with 2 M HAc. While tetrabutylammonium is more commonly used as a reverse phase HPLC ion-pairing reagent to achieve baseline separation of all disaccharides, tetrabutylammonium would interfere with MS detection and so we employed dibutylamine for this purpose. After injection of 0.5 μl sample, the elution profile was 0% B for 7 min, 15% B for 9 min, 40% B for 11 minutes, and 100% B for 23 minutes. The flow rate was 5 μl per minute, and absorbencies at 232, 260, and 280 nm were monitored during each run. After each run the column was washed with 90% B for 15 minutes, and equilibrated with 100% A for 40 minutes. The capillary HPLC was directly coupled to the mass spectrometer. Mass spectra were acquired on a Mariner BioSpectrometry Workstation ESI time-of-flight mass spectrometer (PerSeptive Biosystems; Framingham, MA) in the negative-ion mode. Nitrogen was used as a desolvation gas as well as a nebulizer. Conditions for ESI-MS were as follows: nebulizer flow, 1 liter/min; nozzle temperature, 140 °C; N2 flow, 0.1 liters/min; spray tip potential, 2.8 kV; nozzle potential, 70 V; and skimmer potential, 9V. Negative ion spectra were collected by scanning the m/z range 150–1000. During analyses, the vacuum was 2.1 × 10−6 Torr. TIC chromatograms and mass spectra were processed with Data Explorer software version 3.0.
Results
Isolation and quantification of GAGs from serum
Our goal was the development of straightforward methods that allow quantification and structural analysis of serum/plasma GAGs by LC/MS. Our application is the discovery and characterization of plasma GAG-based disease biomarkers. In preliminary experiments, we analyzed commercially available heparin, HS, and several structural varieties of CS by LC/MS. All disaccharides were easily detected and quantified and the disaccharide compositions obtained were consistent with literature values based on either UV or MS signals (data not shown). In contrast, these same methods proved unsatisfactory for the analysis of GAGs isolated from animal cells and tissues, including FBS. Non-sulfated disaccharides could not be detected by either UV or MS. The UV signal was obscured by a 232 nm signal produced by contaminants in the enzymatically digested GAGs. The contaminants also made the non-sulfated disaccharide undetectable by MS (data not shown). These results are consistent with previous publications that non-sulfated disaccharides were not detected in digested GAGs isolated from different biological sources,44,49,50 which indicates that GAGs from small biological samples need to be further purified to accommodate disaccharide composition analysis by LC/MS.
We developed a serum GAG purification procedure using FBS as a starting material (see Experimental Procedures). We added 35S-labeled HS mixture purified from 9 CHO cell lines to the serum samples prior to GAG isolation to monitor GAG recovery in each step. Briefly, we subjected FBS to exhaustive protease treatment in order to free GAGs from proteoglycans and GAG-binding proteins. We then isolated the GAGs by column chromatography. Following ethanol precipitation, the purified GAGs were subjected to β-elimination reaction with NaBH4/NaOH in order to free them from residual peptide remaining from the core protein. Peptide-free GAGs were isolated by affinity to DEAE-Sephacel beads, eluted from the beads, ethanol precipitated and resuspended. We recovered more than 95% of the 35S-HS in our final GAG preparations.
We used the modified procedure to purify GAGs from 5 ml samples of FBS, human serum, and EDTA-anticoagulated human plasma that had been prepared from the same blood donor, a portion (10%) of each GAG preparation was assayed for GlcN and GalN GAG composition using an established reverse phase HPLC method with series of GlcN and GalN standards48 (Fig. 1). These experiments yielded the following values: 36.8 μmol of GalN and 6.23 μmol of GlcN per ml human serum, 22.6 μmol of GalN and 3.20 μmol of GlcN per ml human plasma, and 24.4 μmol of GalN and 7.97 μmol of GlcN per ml FBS. Using the observed GAG average disaccharide M.W. of 460 for our calculations, we estimate that human serum had 17.0 μg/ml CS and 2.86 μg/ml of the GlcN-containing GAGs (HS, HA, and KS), human plasma had 10.4 μg/ml CS and 1.47 μg/ml GlcN GAGs, and FBS had 10.2 μg/ml CS and 3.67 μg/ml GlcN-containing GAGs.
Figure 1.

Quantification of glucosamine (GlcN) and galactosamine (GalN) content of GAGs isolated from FBS (red), human serum (black), and human plasma (blue). GAGs were purified and then digested by acid hydrolysis, reduced with NaBH4, and reacted with OPA (O-phthaldialdehyde)/3MPA (3-mercaptopropionic acid); a reagent to form fluorescent isoindole derivatives of glucosaminitol or galactosaminitol prior to HPLC separation and detection.
Disaccharide analysis of HS and CS isolated from FBS
We next conducted a structural analysis of GAGs derived from FBS by LC/MS. Proteoglycan core peptide-free GAGs were isolated from FBS as described in material and methods. A portion (10%) of the core peptide-free GAGs was digested with a mixture of heparin lyases and a portion of the digested (5%) of GAGs was analyzed by LC/MS. The resulting UV and MS chromatograms are shown in Figure 2. Peak 1 detected by UV was defined as ΔUA-GlcNAc (z1 378.1) by MS. Peak 2 detected by UV had two disaccharides, ΔUA-GlcNAcS (z1 458.03, z2 228.53) and ΔUA-GlcNS (z1 416.07, z2 207.52) based on MS analysis. Peak 3 detected by UV were indicated to be disulfated disaccharides, ΔUA-GlcNS+1S (z1 496.1, z2 247.50) by MS. Peak 4 detected by UV was a trisulfated disaccharide, ΔUA2S-GlcNS6S (z1 576.0, z2 287.49) based on MS analysis.
Figure 2.
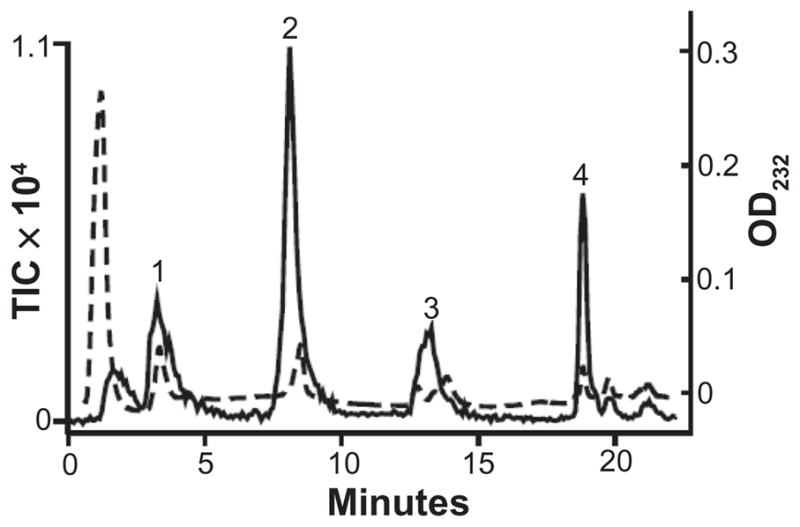
UV absorbance (dashed line) and TIC (solid line) of a heparin lyase digest of FBS GAGs. Peak 1 contains ΔUA-GlcNAc (z1 378.1). Peak 2 contains ΔUA-GlcNAc+S1 (z1 458.03, z2 228.53) and ΔUA-GlcNS (z1 416.07, z2 207.52). Peak 3 contains ΔUA2S-GlcNS (z1 496.1) and ΔUA-GlcNS 6S (z1 496.1). Peak 4 contains ΔUA2S-GlcNS 6S (z1 576.0).
To distinguish ΔUA2S-GlcNS from ΔUA-GlcNS6S in peak 3, we treated the heparin lyase digested mixture further with Δ4, 5-glycuronidase prior to HPLC-MS analysis. The Δ4, 5-glycuronidase enzyme removes ΔUA from the disaccharides that do not have 2-O-sulfated residues.51 In this case we observed a decrease in the UV and MS signals in peak 3 (a mixture of ΔUA2S-GlcNS and ΔUA-GlcNS6S), but not peak 4 (ΔUA2S-GlcNS6S) (not shown), which allows us to estimate the relative abundance of ΔUA2S-GlcNS vs. ΔUA-GlcNS6S in the peak 3.
The CS disaccharide composition of the FBS GAGs was determined by treatment of the proteoglycan core peptide-free GAGs isolated above with chondroitinase ABC prior to analysis by LC/MS. The UV and MS chromatograms are shown in Figure 3. Several species were detected: Nonsulfated disaccharide (ΔUA-GalNAc, z1 378.1), monosulfated disaccharide (ΔUA-GalNAc+1S, z1 458.1), and di-sulfated z1 538.1 disaccharides (ΔUA-GalNAc+2S, z1 538.1), as well as a z2 585.6 corresponding to the linkage hexasaccharide ΔUA-GalNAc-GlcA-Gal-Gal-Xylol (plus 2 sulfates or 1 sulfate and 1 phosphate).
Figure 3.
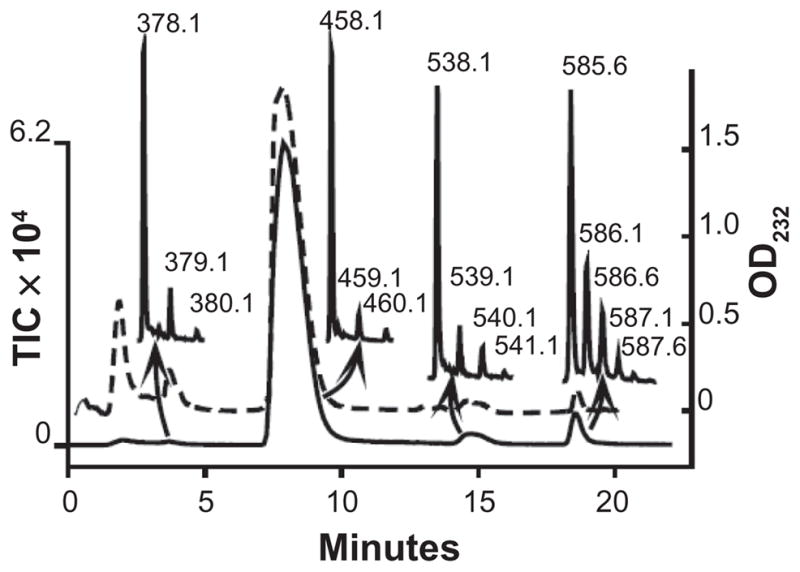
UV absorbance (dashed line) and TIC (solid line) of chondroitinase ABC digested FBS GAGs. The most abundant ions found in each peak are shown. Peak 1 contains ΔUA-GalNAc (z1 378.1). Peak 2 contains ΔUA-GalNAc+ S1 (z1 458.0, z2 228.5). Peak 3 contains ΔUA-GalNAc+S2 (z1 538.1). Peak 4 contains ΔUA-GalNAc-GlcA-Gal-Gal-Xylol+S2 (z2 585.6).
The sulfation positions of the CS disaccharides were then determined by treatment of the chondroitinase- treated material with a variety of sulfatases prior to HPLC/MS analysis. Unlike the newly cloned heparan sulfate 6-Osulfatase,52 chondroitin 4-O- and 6-O-sulfatases hydrolyze unsaturated sulfated disaccharides and oligosaccharides produced by chondroitinase digestion.53,54 Indeed, we observed that chondroitin lyases 4-O-sulfatase digestion decreased the sulfated disaccharide UV and MS signals while it increased nonsulfated disaccharide UV and MS signals (data not shown). These results indicate that the monosulfated disaccharides were mostly 4-O-sulfated at GalNAc residues. The 4-O-sulfate-digested material was further treated with CS 6-O-sulfatase and then 2-O-sulfatase. Each successive treatment reduced the monosulfated z1 458.1 signal further, indicating the presence of both 2-O- and 6-O-sulfated disaccharides in addition to 4-O-sulfated disaccharides in the original FBS GAG sample. Disaccharide composition was calculated from the observed changes in signal intensity, as described later.
The reducing and non-reducing end mono-, di-, tri-, and tetrasaccharide structures in FBS
Several non-reducing end species were identified in the chondroitinase-treated fractions: Non-reducing end mono-sulfated GalNAc (z1 300.05, IC 6374) eluted at 3.8 min. Non-reducing end di-sulfated GalNAc (z1 380.02 and z2 189.50, IC 1408) eluted at 9.0 min. Non-reducing end mono-sulfated disaccharide, GlcA-GalNAc+ 1S (z1 476.10), eluted at 7.2 min (IC 677). The elution position and m/z values for the mono- and di-saccharides were in agreement with the non-reducing end structures of CS identified by Midura et al.55
Extracted ion current (XIC) of m/z 556 and 636 is shown in Figure 4. The two peaks represented two reducing end trisaccharides, ΔUA-GalNAc-UA-ol (z1 556.07, IC 2658) and ΔUA-GalNAc4S-UA-ol (z1 636.15, z2 317.56, IC 2311). ΔUA-GalNAc-UA-ol eluted at 7.3 min and ΔUA-GalNAc-UA-ol+1S eluted at 13.5 min. Chondroitin sulfate 4-O-sulfatase treatment eliminated ΔUA-GalNAc-UA-ol+1S signals (z1 636.15, z2 317.56) and resulted an increase in non-sulfated trisaccharide ΔUA-GalNAc-UA-ol signal (z1 556.07, data not shown), which indicated that there was a 4-O-sulfated GalNAc residue in the monosulfated trisaccharide, ΔUA-GalNAc4S-UA-ol. The ICs of the trisaccharides were about 21% that of the linkage hexasaccharide IC. Thus, a significant amount of free CS chains ending in uronic acid co-existed with proteoglycans in FBS.
Figure 4.
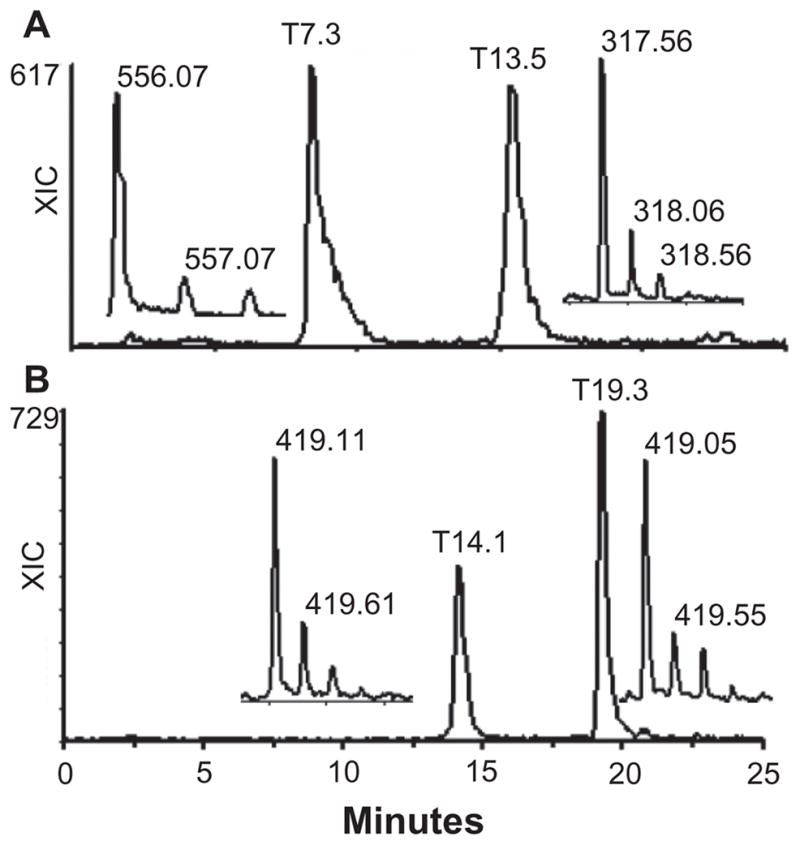
XIC of FBS CS reducing and non-reducing end structures from the data presented in Figure 3. A) The XIC of the reducing end trisaccharides, ΔUA-GalNAc-UA-ol and ΔUA-GalNAc4S-UA-ol. The inserts show the z1 for ΔUA-GalNAc-UA-ol and z2 for ΔUA-GalNAc4S-UA-ol. B) The XIC of the reducing end tetrasaccharide, ΔUA-GalNAc-UA-Gal-NAc-ol+S1 (14.1 min peak) and the non-reducing end trisaccharide, GalNAc-UA-GalNAc+S3 (19.3 min peak). The inserts show the z2 ions observed for each peak.
We also observed another reducing end tetrasaccharide ΔUA-GalNAc-UA-GalNAc-ol+1S, and a non-reducing end trisaccharide, GalNAc-UA-GalNAc+ 3S, in that both had the same mass but different elution time and m/z values. The XIC of m/z 419 is shown in Figure 4b. The 14.1 min peak had z1 839.26 (IC 63) and z2 419.11 (IC 708) ions, whereas the 19.3 min peak had z2 419.05 (IC 1090) and z3 279.03 (IC 514) ions. No z3 ion was observed at 14.1 min and no z1 ion was observed at 19.3 min. The m/z values, the elution position, and the charge status suggested that the 14.1 min peak was the reducing end monosulfated tetrasaccharide, ΔUA-GalNAc-UA-GalNAc-ol+1 sulfate, and the 19.3 min peak was the non-reducing end, tri-sulfated trisaccharide, GalNAc-UA-GalNAc+3 sulfates. To confirm this, FBS GAGs were treated with NaBD4 instead of NaBH4 to label the reducing end saccharides in the β-elimination reaction and digested with chondroitinase ABC. In that case the 14.1 min peak had a z2 419.61 instead of z2 419.05, whereas the m/z of the ions in 19.3 min peak remained the same (data not shown). While no internal tetrasaccharides were observed after one round of chondroitinase ABC digestion, the 19.3 min peak was still seen after 4 rounds of chondroitinase ABC digestion, indicating that the trisaccharide was resistant to enzymatic depolymerization. However, its ion intensity was reduced with repeated chondroitinase ABC digestions, and its ion reduction was accompanied by an increase in TIC of di-sulfated GalNAc (z1 380.02 and z2 189.50) (data not shown). Together these observations suggest that the 19.3 min peak represents a trisaccharide that had 2 sulfates at its non-reducing end GalNAc residue.
CS Linkage hexasaccharides
After chondroitinase ABC digestion of FBS GAGs, the following known linkage hexasaccharides were observed: 1. Non-sulfated hexasaccharide (z2 505.6, IC 599), eluted at 7.1 min. 2. Mono-sulfated hexasaccharide (z2 545.6, IC 2366), eluted at 13.0 min. 3. Di-sulfated hexasaccharide (z2 585.6, IC 1.3 × 104), eluted at 18.6 min. 4. Tri-sulfated hexasaccharide (z2 690.2 = 625.2 + 1 dibutylamine (DBA), z3 416.74, IC 327), eluted at 20.2 min. The structure of the most abundant di-sulfated linkage hexasaccharide (z2 585.6, IC 1.3 × 104) was confirmed by the following experiment: The GAG sample was first digested with chondroitinase ACII, a treatment which cleaves linkage hexasaccharides into di- and tetrasaccharides. Indeed, the hexasaccharide peak disappeared and a new z1 713.1 peak appeared (data not shown). The z1 713.1 ion was likely the tetrasaccharide ΔUA-Gal-Gal-Xylol, plus one sulfate or phosphate. Alkaline phosphatase treatment did not affect the z1 713.1 ion. However, further digestion with CS 4-O-sulfatase led to the appearance of a new species, z1 633.1 and partial disappearance of z1 713.1 ion. Z1 633.1 ion corresponds to a non-sulfated linkage tetrasaccharide (ΔUA-Gal-Gal-Xylol). These results indicate that the most abundant linkage hexasaccharide was a di-sulfated hexasaccharide (ΔUA-GalNAc)+1S-(GlcAc-Gal-Gal-Xylol)+1S. To further confirm the identity of the linkage hexasaccharides, FBS GAGs were treated with NaBD4 instead of NaBH4 in β-elimination reaction prior to chondroitinase ABC digestion and LC/MS analysis, all the putative linkage hexasaccharides underwent a +1 mass increase (data not shown), indicating that they all derived from the reducing end of CS chains.
Theoretically, every non-reducing end uronic acid double bond in both repeating disaccharides and linkage hexasaccharides should produce the same UV absorbance. The summed UV signal of all the disaccharides divided by the summed UV signal of the linkage hexasaccharide gave an estimated average chain length of 12 disaccharides for FBS CS.
Identification of sialic acid-containing CS linkage hexasaccharides
The most striking difference between FBS and human serum CS was a 20-fold higher level in human serum of an ion with a z2 731.2 (IC 110 vs. 2672) and z3 487.1 (IC 98 vs. 1605) (Fig. 5). This ion eluted at 20.1 min, between di- and tri-sulfated linkage hexasaccharides. It was not observed in commercial CS-A, CS-B, or CS-C reduced with NaBH4 and digested with chondroitinase ABC, which indicates that it is not generated during CS degradation. To determine whether this novel ion species was derived from a serum proteoglycan, we exhaustively digested human serum with protease and isolated the GAGs. Half of the GAG sample was treated with NaBH4 to remove the core protein peptides. Both the NaBH4-treated and unreduced GAG fractions were then digested with chondroitinase ABC and analyzed by capillary HPLC coupled MS. TIC chromatograms for both GAGs are shown in Figure 6. The major peak in both chromatograms eluting at 8.3 min represents the mono-sulfated repeating disaccharides (z1 458.03 and z2 228.54). In the chromatograph of the reduced GAGs, the T1 peak was identified as the di-sulfated linkage hexasaccharide which has a natural molecular weight of (585.6228 × 2 + 2) − 2 = 1171.25 and the T2 peak is the novel ion with a molecular weight 1464.36 (731.1787 × 2 + 2). Neither T1 nor T2 were present in the chromatograph of the non-reduced GAGs. Instead we observed a major peak, T3 (molecular weight 2001.54 (499.3855 × 4 + 4), and a minor peak, T4 (molecular weight 2292.65 (457.5291 × 5 + 5).
Figure 5.

Significant increase of a novel ion with a z2 731.2 in human serum. The XIC of the ions, z2 731.2 and other linkage hexasaccharides (z2 505.6, 545.6, 585.6, 690.2 = 625.6 + 1DBA) in and FBS (upper panel) and human serum (lower panel. The inserts show the most abundant ions observed for each peak.
Figure 6.

Bikunin, the most abundant CS PG in blood circulation, was covalently linked to linkage hexasaccharide and the novel ion. TIC of chondroitinase ABC digested human serum GAGs before (black line) and after β-elimination reactions (red line). Inserts show the most abundant ions for T1, T2, T3, and T4 peaks. The peptide molecular weight (830.29+18.01) was derived based on the molecular weight changes from (T3 peak) to (T1peak) and from (T4 peak) to (T2 peak). Therefore, the same bikunin peptide, QEEEGS-GGG, with a molecular weight of 830.29+18.01 was removed by the β-elimination reaction from both linkage hexasaccharide and the novel ion.
Assuming the novel ion T2 is derived from a GAG linkage saccharide, with a natural molecular weight of 1464.36 − 2 = 1462.36, then the molecular difference between peak T3 and the linkage hexasaccharide T1 (2001.54−1171.25 = 830.29) is equal to the molecular difference between peak T4 and the novel ion T2 (2292.65−1462.36 = 830.29). This suggests the two unreduced ions, T3 and T4, could be attached to a common peptide of molecular weight 830.29. We searched the MS protein databank (http://www.proteomeconsortium.org) for appropriately sized peptides bearing at least one Ser-Gly consensus sequence, and identified a logical candidate peptide, QEEEGSGGG (830.29 + 18.01), derived from bikunin, the most abundant CS proteoglycan in normal human serum. Together these observations indicate that large fractions of the CS species we have resolved, including the novel ion, are derived from the bikunin proteoglycan.
The mass difference between the novel ion and the disulfated linkage hexasaccharide ion, 291.1 amu, immediately suggested that the novel ion could be a disulfated linkage hexasaccharide modified by the addition of a sialic acid residue. Moreover, the elution of the novel ion, between di- and tri-sulfated linkage hexasaccharides is consistent with the extra negative charge which sialic acid would provide. While sialic acid modified linkage hexasaccharides have never been reported, Wakabayashi et al, have identified a proteoglycan linkage-like tetrasaccharides with 3-Osulfated GlcA and sialic acid modified Gal residue.56 Thus, we treated chondroitinase ABC digested human serum GAGs with sialidase and compared the results to non-sialidase treated controls by LC/MS. Neither z2 731.2 nor z3 487.1 ions survived the treatment, while all other linkage hexasaccharide ions remained (Fig. 7). This result confirmed that the novel ion is derived from a linkage hexasaccharide modified with a sialic acid moiety.
Figure 7.

Sialidase treatment eliminated the novel ion. XIC of z2 585.6, z2690.2, and the novel ion, z2 731.2, before (black line) and after (red line) sialidase treatment. The novel ion, z2 731.2, was no longer detectable after sialidase treatment whereas the trisulfated hexasaccharide ion (690.2) was unaffected and the percentage of di-sulfated hexasaccharide ion was increased.
FBS and human serum HS and CS disaccharide compositions
We next sought to use the LC/MS protocol to quantify the disaccharide composition of FBS and human serum GAGs. However, the TIC signals derived from the LC/MS system cannot be used directly for disaccharide composition analysis: Sulfated disaccharides are eluted from the HPLC at a higher level of methanol than the non-sulfated disaccharides, and the greater desolvation that accompanies the higher methanol levels results in higher sensitivity for MS detection. We circumvented this problem by employing a series of HS and CS standard disaccharides.
To this end, UV and MS signals from a mixture of five different HS disaccharides were analyzed over a 20-fold concentration range (from 50 to 1000 pmol per injection) (Fig. 8). Similar experiments were also conducted by using a mixture of five different CS disaccharides (data not shown). For all volumes and all HS and CS disaccharides, the average UV area/pmol disaccharide was 9.06 ± 1.4 mAU (mean ± standard deviation) irrespective of their structures and elution position. The MS signals for equimolar amount of disaccharides were much higher for sulfated than for nonsulfated disaccharides. However, the ratio of UV to MS signal intensity for each disaccharide was constant over the range of concentrations analyzed. These ratios, derived from pure disaccharide standards, are shown in Table 1. The formula, IC multiplied by UV/IC equalizing factor, was used to normalize the MS signal for disaccharide quantification purposes. In our system, the MS detection limit was ~1 pmol for -GlcNAc/ΔUA-GalNAc and ~100 fmol for ΔUA2S-GlcNS6S. In contrast, the UV detection limit, defined as peak heights at 3 times baseline noise levels, was 10 pmol for all disaccharides.
Figure 8.

Quantification of HS disaccharide standards by LC/MS. Volumes of 0.05, 0.10, 0.50, or 1.0 μl of a mixture of disaccharide standards were analyzed. Both the HPLC UV absorbance (dashed lines) and MS TIC (solid lines) increase with increased analyte. Peak 1 is ΔUA-GlcNAc (z1 [mass − 1 proton] = 378.1). Peak 2 is ΔUA-GlcNS (z1 416.1). Peak 3 contains both ΔUA2S-GlcNS (z1 496.1) and ΔUA-GlcNS 6S (z1 496.1). Peak 4 is ΔUA2S-GlcNS 6S (z1 576.0). The quantitative data are summarized in Table 1.
Table 1.
MS signal equalizing factors (the ratio of TIC/pmol to UV area/pmol) for HS and CS disaccharides.
| Disaccharide standards | Mean TIC/UV area* |
|---|---|
| ΔUA-GlcNAc | 0.761 ± 0.076 |
| ΔUA-GlcNS | 2.019 ± 0.066 |
| ΔUA-GlcNS6S/ΔUA2S-GlcNS | 3.659 ± 0.543 |
| ΔUA2S-GlcNS6S | 4.431 ± 0.426 |
| ΔUA-GalNAc/ΔUA-GlcNAcβ | 0.841 ± 0.071 |
| ΔUA-GalNAc4S/ΔUA-GalNAc6S | 2.017 ± 0.125 |
| ΔUA2S-GalNAc4S | 4.128 ± 0.224 |
Average results from injection volumes of 0.05, 0.1, 0.5, and 1.0 μl. For all volumes and all disaccharides, the average UV area/pmol disaccharide was 9.06 ± 1.4 mAU (mean ± standard deviation). MS signals are higher for sulfated disaccharides than for equimolar amounts of nonsulfated disaccharides. This is due to an increased percentage of organic solvent in the elution at later times and the increased ionizability of more highly charged sulfated disaccharides.
Based on the equalizing factors, HS and CS disaccharide compositions were calculated for FBS and human serum (Table 2). Typical blank TIC background was 10 ± 5. In principle, any GAG ions with a TIC signal above 50 can be identified by the LC/MS method from its stable isotope distribution pattern and unique elution position. In all we have detected twenty-four distinct heparan sulfate and chondroitin sulfate components from FBS and human serum (summarized in the Table 2). There were still a few minor unidentified ions in both FBS and human serum GAGs that are under current investigation.
Table 2.
Twenty four FBS and human serum HS and CS mono-, di-, tri-, tetra-, and hexasaccharides.
| Heparan sulfate | ||||
|---|---|---|---|---|
| (HS Disaccharide %. B:FBS, H:human serum) | Observed ions | Elution time | TIC and % of TIC |
|
| FBS | Human | |||
| ΔUA-GlcNAc (B46.6%, H37.5%) | z1 378.12 | 3.9 | 1822 18.80% |
990 14.10% |
| ΔUA-GlcNS (B20.9%, H25.5%) | z1 416.08, z2 207.53 | 9 | 2128 22.00% |
1787 25.40% |
| ΔUA-GlcNAc6S (B8.5%, H9.6%) | z1 458.09, z2 228.54 | 8.7 | 852 8.80% |
673 9.60% |
| ΔUA2S-GlcNAc (B1.2%, H1.6%) | z1 458.09, z2 228.54 | 8.7 | 135 1.40% |
112 1.60% |
| ΔUA-GlcNS6S (B5.4%, H9.6%) | z1 496.04, z2 247.51 | 14.2 | 1048 10.80% |
1214 17.30% |
| ΔUA2S-GlcNS (B8.2%, H8.5%) | z1 496.04, z2 247.51 | 14.2 | 1610 16.50% |
1081 15.40% |
| ΔUA2S-GlcNS6S (B9.2%, H7.6%) | z1 576.01, z2 287.49 z3 191.32 | 19.3 | 2105 21.70% |
1167 16.60% |
| Chondroitin sulfate | ||||
| (CS Disaccharide %. B:FBS, H:human serum) | ||||
| ΔUA-GalNAc (B6.3%, H2.8%) | z1 378.12 | 3.7 | 13906 2.50% |
54000 5.50% |
| ΔUA-GalNAc6S (B7.2%, H10.0%) | z1 458.03, z2 228.53 | 7.8 | 4154 6.80% |
81663 8.30% |
| ΔUA-GalNAc4S (B83.7%, H85.0%) | z1 458.03, z2 228.53 | 7.8 | 48992 80.20% |
693647 70.50% |
| ΔUA2S-GalNAc (B1.5%, H2.2%) | z1 458.03, z2 228.53 | 7.8 | 855 1.40% |
17710 1.80% |
| ΔUA-GlcNAc+S2 (B1.3%, H0.02%) | z1 538.06, z2 268.51,z3 178.67 | 14.9 | 14428 2.60% |
1239 0.13% |
| GalNAc+S1 | z1 300.05 | 3.8 | 6674 1.20% |
2165 0.22% |
| GalNAc+S2 | z1 380.02, z2 189.50 | 9 | 2750 0.46% |
186 0.02% |
| GlcA/IdoA-GalNAc+S1 | z1 476.10, z2 237.53 | 7.2 | 677 0.12% |
520 0.05% |
| ΔUA-GalNAc-UA-ol | z1 556.07 | 7.3 | 2658 0.48% |
259 0.26% |
| ΔUA-GalNAc4S-UA-ol | z1 636.15, z2 317.56 | 13.5 | 2311 0.42% |
319 0.03% |
| ΔUA-GalNAc-UA-GalNAc-ol | z1 839.26, z2 419.11 | 14.1 | 1190 0.21% |
88 0.01% |
| GalNAc-UA-GalNAc+S3 | z2 419.05, z3 279.03 | 19.3 | 1604 0.29% |
149 0.02% |
| ΔUA-GalNAc-GlcA-Gal-Gal-Xylol | z2 505.67 | 7.1 | 550 0.10% |
0 0 |
| ΔUA-GalNAc-GlcA-Gal-Gal-Xylol+S1 | z2 545.63, z3 363.42 | 13 | 2539 0.46% |
371 0.04% |
| ΔUA-GalNAc-GlcA-Gal-Gal-Xylol+S2 | z2 585.63, z3 390.08 | 18.6 | 14007 2.52% |
125000 12.70% |
| ΔUA-GalNAc-GlcA-Gal-Gal-Xylol+S3 | z2 690.2, z3 416.74 | 20.2 | 282 0.05% |
1817 0.18% |
| ΔUA-GalNAc-GlcA-Gal-Gal-Xylol+S2+Sialic acid | z2 731.22, z3 487.12, z4 365.08 | 20.1 | 208 0.04% |
4277 0.44% |
Discussion
This report describes a LC/MS method for the analysis of GAGs isolated from serum and plasma. Samples undergo extensive proteolysis to free GAGs from their respective proteoglycans and GAG-binding proteins, treatment with NaBH4 or NaBD4 to eliminate residual core protein peptides and to tag reducing ends of free GAGs, and digestion with a variety of GAG-specific enzymes to identify mono-, di, tri-, tetrasaccharides, linkage hexasaccharides, and other oligosaccharides. Reverse phase capillary HPLC is used to separate GAG mono-, di-, tri-, and oligosaccharides according to charge and structure and to remove contaminating salts and undigested material, and the resulting fractions are simultaneously analyzed by both UV and MS. GAG structures are identified on the basis of UV absorbance, m/z values, and HPLC retention times. Sulfatases are used to distinguish among disaccharide isomers. Twenty-four distinct HS and CS components including reducing end structures and a novel sialic acid-modified chondroitin sulfate linkage hexasaccharide were identified from FBS, human serum, and human plasma. The purified serum/plasma GAGs were quantified by a previously-reported reversed-phase HPLC method that quantifies GlcN and GalN content simultaneously48 (Fig. 1). It was reported that CS content is higher in serum because CS is released from platelets during coagulation process.57 Indeed, CS levels were substantially greater in the human serum sample (17.0 μg/ml) than in plasma (10.4 μg/ml) derived from the same blood sample. We also observed greater GlcN-containing GAGs in the human serum (2.86 μg/ml) than in the plasma (1.47 μg/ml), which suggests that GlcN-containing GAGs might also be released during the coagulation process. There is a wide disparity in previous estimates of human serum and plasma GAG concentrations18,58 which may reflect differences in the methods used for GAG isolation and quantification. The values we observed in human serum (20 μg/ml) and plasma (12 μg/ml) are at the high end of literature values, likely due to the exhaustive protease digestion we employed to dissociate GAGs from their respective proteoglycans and GAG-binding proteins.
Previous use of LC/MS for the analysis of GAGs in biological samples failed to detect non-sulfated disaccharides and underestimated mono-sulfated disaccharides.44,50,59 The modifications we have introduced in the GAG isolation steps allowed us to detect non-sulfated disaccharides and other oligosaccharides via LC/MS. The capillary HPLC step is particularly important for the removal of salts that interfere with MS-based GAG analysis. Since the UV and MS signals we obtained were linear over a wide range (Fig. 8), we calculated GAG disaccharide composition using equalizing factors, a technique previously established for tandem ESI-MSn 27–29 and LC/MS.36,37 However, there are limitations to such quantifications. The estimation of quantities of 24 detected components in Table 2 relied on “equalizing factors” that were derived from dose curves for pure disaccharide standards. Because these standards were assayed pure and not mixed in with complex serum/plasma GAG samples that contain contaminants affecting ionization thus MS detection, these equalizing factors do not account for possible quenching of analyte ionization. In addition, MS detection sensitivity could change during a run and between different runs in the same day, which made this method a more qualitative than quantitative one.
Using this method, we have identified a novel sialic acid-modified CS linkage hexasaccharide, primarily in GAGs derived from human serum. We have also observed small amounts of sialic acid-modified linkage hexasaccharides in GAGs derived from mouse tissues and mouse growth plates (Zhang, L., unpublished results). Previously, CS linkage hexasaccharides from different tissues and cell lines were characterized via both NMR and enzymatic sequencing approaches by Dr. Sugahara’s group,60 and CS linkage hexasaccharides from rat chondrosarcoma cells were analyzed using a combination of Progel-TSK G2500 and CarboPac PA1 chromatography steps in conjunction with monosaccharide analyses by Shibata et al.61 Neither of those studies identified a sialic acid modified linkage hexasaccharide. It is possible that sialic acid-modified GAGs have not been detected before because they occur in low abundance. Proteins that bind to sialic acid are involved in regulating many biological processes.62 Thus, sialic acid-modified CS may provide GAGs with unique biological functions.
The estimation of GAG molecular weights has long been difficult, complicated by the effects of chain length and sulfation heterogeneity and conformational differences among the GAGs. Number-average mean molecular weight values have been obtained by osmometry, and weight average mean molecular weight values have been obtained by viscometry and light scattering, but these methods require relatively large amounts of material. Several groups have used heparin, dermatan sulfate, and CS to estimate their molecular weights by intrinsic viscosity and light scattering intensity, and subsequently these characterized GAGs were used as molecular weight standards for gel electrophoresis or gel filtration chromatography-based biological GAG chain length estimation. However, the hydrodynamic size of GAGs was found to be influenced by the sulfation isomer composition of the chains.63 Moreover, heparin and dermatan sulfate samples with the same molecular weight values as observed by gel filtration chromatography had different intrinsic viscosity and different radius of gyration when right-angle laser light scattering, refractometer and viscometer were used for triple on-line detections.64 Thus, the molecular weight measured by these approaches has limited accuracy.
Number-average molecular weight can be estimated if the ratio of GAG reducing or non-reducing end saccharide to repeating disaccharides is obtained. Desai and Linhardt have used 13C NMR to estimate the molecular weight of low molecular weight heparin based on the signal strength of reducing end GlcA vs. repeating disaccharides.65,66 However, most biological GAGs have GlcA-Gal-Gal-Xyl-linkage tetrasaccharides rather than GlcA at the reducing end. Plaas et al67 have estimated CS average chain length by quantifying the non-reducing end mono- and disaccharides vs. repeating disaccharides. However, an unknown late eluting peak, which might be a non-reduced trisaccharide, was observed but was not included in calculations. 55 We have used LC/MS to detect reducing end, non-reducing end, and repeating disaccharides simultaneously. In principle, CS chain length can be estimated by comparing either reducing end or non-reducing end groups to repeating disaccharide. Since we do not have mono- di-, and trisaccharide standards to generate proper equalizing factors for quantifying non-reducing ends, we compared the ratio of UV signal intensities of total disaccharides to reducing end linkage hexasaccharides. We calculated an average of 12 disaccharides per CS chain in FBS and 14 disaccharides per CS chain in human serum. These values are quite similar to 15 ± 3 disaccharides per bikunin CS chain based on three independent studies using several different approaches.68,69 Interestingly, bikunin CS chains derived from a patient with septic shock and measured by MALDI-MS were 6 disaccharides longer than those derived from a normal patient.70 Thus, serum GAG chain length could vary with respect to physiological conditions. We could not analyze the average HS chain length: The heparin lyase III used in the isolation procedure cleaves HS linkage hexasaccharides into di- and tetra-saccharides.71 However, no FBS or human serum HS linkage tetrasaccharides were observed based on LC/MS analysis. This could indicate that serum HS chains are much longer than the CS chains, and/or the MS signals emitted by the linkage tetrasaccharides are weak due to a lack of sulfation in the HS linkage tetrasaccharides.72
The reduction of free GAGs by either NaBH4 or NaBD4 terminates the ends of free GAGs with an alcohol group while further enzymatic digestion leaves a residual aldehyde at the repeating disaccharides. This allows the detection of reduced and un-reduced GAG structures simultaneously by the LC/MS method (Table 2). Such detection showed the coexistence of free GAGs and proteoglycans in serum. Free GAGs would likely be produced in vivo. Heparanase (endo-β-glucuronidase) cleaves HS at GlcA-GlcNAc/SO3 bonds5 and also degrades CS in vitro (Zhang, L, unpublished observation). Free GAGs terminating with GlcA (Indicated with a GlcA-OH end in Fig. 4a) may have been generated from proteoglycans by extracellular heparanase. Hyaluronidase (endo-β-Nacetylhexosaminidase) cleaves hyaluronan,73 as well as CS and DS in vitro.74 A CS specific hyaluronidase has been characterized recently.75 Free GAG chains with GalNAc at reducing end (Indicated by a Gal-NAc-OH end in Fig. 4b) may have been generated from proteoglycans by such hyaluronidases.
The GAGs present in the serum are derived from many different cells and tissues including the liver,76 platelets,57 endothelial cells,77 and malignant cells.78 Specific GAG structures are the results of modifications that occur during proteoglycan biosynthesis, in the extracellular matrix, and in the plasma by the GAG editing enzymes. Thus, the spectrum of serum GAG quantity and structures is a reflection of physiological state, and is therefore a potentially rich source of biomarkers for the assessment of human health and disease. To overcome intrinsic difficulties in quantifying GAG di- and oligosaccharide changes by the LC/MS method for biomarker discoveries, we recently adapted the Glycan Reductive Isotope Labeling (GRIL) technique.38 The technique involves enzymatic digestion of purified GAGs using chondroitinase and/or heparin lyases, followed by reductive amination of the reducing ends of the disaccharides with aniline for one sample and deuterated (D5)-aniline for the other sample. The addition of the regular aniline group adds 77 amu to the mass of the free molecular ion ([M-H]−1), whereas D5-aniline group adds 82 amu. Thus, normal and diseased biological samples tagged with different isotopically labeled anilines can be mixed and analyzed simultaneously to yield quantitative or ratiometric comparisons. This analysis overcomes the problematic run-to-run and sample-to-sample variation of LC/MS sensitivity and has pinpointed specific GAG structural changes in adducted thumb-clubfoot syndrome patient fibroblasts.79 We expect that serum/plasma GAG-based biomarker discovery will be enabled by this method.
Acknowledgments
Funding
This work is supported in part by National Institutes of Health Grant ROIGM069968 to LZ.
Abbreviations
- GAG
glycosaminoglycan
- HS
heparan sulfate
- CS
chondroitin sulfate
- MS
mass spectrometry
- LC
liquid chromatography
- ESI
electrospray ionization
- ΔUA
unsaturated uronic aid
- GlcA
glucuronic acid
- IdoA
Iduronic acid
- GlcNAc
N-acetylglucosamine
- GlcNS
N-sulfated glucosamine
- GalNAc
N-acetylgalactosamine, GlcN, glucosamine
- GalN
galactosamine
- Gal
galactose
- Xyl
xylose
- -ol
reduced terminal sugar
- S1
one sulfate
- S2
two sulfates
- S3
three sulfates
- OPA
o-phthaldialdehyde
- 3MPA
DBA, dibutylamine, 3-mercaptopropionic acid
- m/z
mass/charge
- IC
ion current
- TIC
total ion current
- XIC
extracted ion current
- FBS
fetal bovine serum
Footnotes
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The views expressed in this article do not necessarily reflect those of Libertas Academica. The authors report no conflicts of interest.
This is an open access article. Unrestricted non-commercial use is permitted provided the original work is properly cited.
References
- 1.Esko JD, Zhang L. Influence of core protein sequence on glycosaminoglycan assembly. Curr Opin Struct Biol. 1996;6:663–70. doi: 10.1016/s0959-440x(96)80034-0. [DOI] [PubMed] [Google Scholar]
- 2.Yanagishita M, Hascall VC. Cell surface heparan sulfate proteoglycans. J Biol Chem. 1992;267:9451–9454. [PubMed] [Google Scholar]
- 3.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, MacLeod V, Dai Y, et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood. 2007;110:2041–8. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and Characterization of Two Extracellular Heparin-degrading Endosulfatases in Mice and Humans. J Biol Chem. 2002;277:49175–85. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lokeshwar VB, Rubinowicz D, Schroeder GL, et al. Stromal and Epithelial Expression of Tumor Markers Hyaluronic Acid and HYAL1 Hyaluronidase in Prostate Cancer. J Biol Chem. 2001;276:11922–32. doi: 10.1074/jbc.M008432200. [DOI] [PubMed] [Google Scholar]
- 8.Kim H, Xu GL, Borczuk AC, et al. The Heparan Sulfate Proteoglycan GPC3 Is a Potential Lung Tumor Suppressor. Am J Respir Cell Mol Biol. 2003;29:694–701. doi: 10.1165/rcmb.2003-0061OC. [DOI] [PubMed] [Google Scholar]
- 9.Grigoriu BD, Depontieu F, Scherpereel A, et al. Endocan Expression and Relationship with Survival in Human Non-Small Cell Lung Cancer. Clin Cancer Res. 2006;12:4575–82. doi: 10.1158/1078-0432.CCR-06-0185. [DOI] [PubMed] [Google Scholar]
- 10.Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–42. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 11.Anttonen A, Leppa S, Ruotsalainen T, Alfthan H, Mattson K, Joensuu H. Pretreatment serum syndecan-1 levels and outcome in small cell lung cancer patients treated with platinum-based chemotherapy. Lung Cancer. 2003;41:171–7. doi: 10.1016/s0169-5002(03)00196-x. [DOI] [PubMed] [Google Scholar]
- 12.Saito A, Munakata H. Analysis of plasma proteins that bind to glycosaminoglycans. Biochim Biophys Acta. 2007;1770:241–6. doi: 10.1016/j.bbagen.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Conrad HE. Heparin-Binding Proteins. San Diego: Academic Press; 1998. [Google Scholar]
- 14.Yu H, Munoz EM, Edens RE, Linhardt RJ. Kinetic studies on the interactions of heparin and complement proteins using surface plasmon resonance. Biochim Biophys Acta. 2005;1726:168–76. doi: 10.1016/j.bbagen.2005.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calatroni A, Vinci R, Ferlazzo AM. Characteristics of the interactions between acid glycosaminoglycans and proteins in normal human plasma as revealed by the behaviour of the protein-polysaccharide complexes in ultrafiltration and chromatographic procedures. Clin Chim Acta. 1992;206:167–80. doi: 10.1016/0009-8981(92)90086-6. [DOI] [PubMed] [Google Scholar]
- 16.Lamari FN, Theocharis AD, Asimakopoulou AP, Malavaki CJ, Karamanos NK. Metabolism and biochemical/physiological roles of chondroitin sulfates: analysis of endogenous and supplemental chondroitin sulfates in blood circulation. Biomed Chromatogr. 2006;20:539–50. doi: 10.1002/bmc.669. [DOI] [PubMed] [Google Scholar]
- 17.Volpi N, Cusmano M, Venturelli T. Qualitative and quantitative studies of heparin and chondroitin sulfates in normal human plasma. Biochim Biophys Acta. 1995;1243:49–58. doi: 10.1016/0304-4165(94)00123-f. [DOI] [PubMed] [Google Scholar]
- 18.Contini M, Pacini S, Ibba-Manneschi L, et al. Modification of plasma glycosaminoglycans in long distance runners. Br J Sports Med. 2004;38:134–7. doi: 10.1136/bjsm.2002.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beaty NB, Mello RJ. Extracellular mammalian polysaccharides: glycosaminoglycans and proteoglycans. J Chromatogr. 1987;418:187–222. doi: 10.1016/0378-4347(87)80009-9. [DOI] [PubMed] [Google Scholar]
- 20.Imanari T, Toida T, Koshiishi I, Toyoda H. High-performance liquid chromatographic analysis of glycosaminoglycan-derived oligosaccharides. J Chromatogr A. 1996;720:275–93. doi: 10.1016/0021-9673(95)00338-x. [DOI] [PubMed] [Google Scholar]
- 21.Guo YC, Conrad HE. Analysis of oligosaccharides from heparin by reversed-phase ion-pairing high-performance liquid chromatography. Anal Biochem. 1988;168:54–62. doi: 10.1016/0003-2697(88)90009-7. [DOI] [PubMed] [Google Scholar]
- 22.Toyoda H, Kinoshita-Toyoda A, Fox B, Selleck SB. Structural analysis of glycosaminoglycans in animals bearing mutations in sugarless, sulfateless, and tout-velu. Drosophila homologues of vertebrate genes encoding glycosaminoglycan biosynthetic enzymes. J Biol Chem. 2000;275:21856–61. doi: 10.1074/jbc.M003540200. [DOI] [PubMed] [Google Scholar]
- 23.Ampofo SA, Wang HM, Linhardt RJ. Disaccharide compositional analysis of heparin and heparan sulfate using capillary zone electrophoresis. Anal Biochem. 1991;199:249–55. doi: 10.1016/0003-2697(91)90098-e. [DOI] [PubMed] [Google Scholar]
- 24.Calabro A, Midura R, Wang A, West L, Plaas A, Hascall VC. Fluorophore-assisted carbohydrate electrophoresis (FACE) of glycosaminoglycans. Osteoarthritis Cartilage. 2001;9 (Suppl A):S16–22. doi: 10.1053/joca.2001.0439. [DOI] [PubMed] [Google Scholar]
- 25.Toyoda H, Kinoshita-Toyoda A, Selleck SB. Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J Biol Chem. 2000;275:2269–75. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- 26.Zaia J, Li XQ, Chan SY, Costello CE. Tandem mass spectrometric strategies for determination of sulfation positions and uronic acid epimerization in chondroitin sulfate oligosaccharides. J Am Soc Mass Spectrom. 2003;14:1270–81. doi: 10.1016/S1044-0305(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 27.Saad OM, Leary JA. Compositional analysis and quantification of heparin and heparan sulfate by electrospray ionization ion trap mass spectrometry. Anal Chem. 2003;75:2985–95. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- 28.Saad OM, Ebel H, Uchimura K, Rosen SD, Bertozzi CR, Leary JA. Compositional profiling of heparin/heparan sulfate using mass spectrometry: Assay for specificity of a novel extracellular human endosulfatase. Glycobiology. 2005;15:818–26. doi: 10.1093/glycob/cwi064. [DOI] [PubMed] [Google Scholar]
- 29.Behr JR, Matsumoto Y, White FM, Sasisekharan R. Quantification of isomers from a mixture of twelve heparin and heparan sulfate disaccharides using tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:2553–62. doi: 10.1002/rcm.2079. [DOI] [PubMed] [Google Scholar]
- 30.Chai W, Luo J, Lim CK, Lawson AM. Characterization of heparin oligosaccharide mixtures as ammonium salts using electrospray mass spectrometry. Anal Chem. 1998;70:2060–6. doi: 10.1021/ac9712761. [DOI] [PubMed] [Google Scholar]
- 31.Zaia J, Costello CE. Compositional analysis of glycosaminoglycans by electrospray mass spectrometry. Anal Chem. 2001;73:233–9. doi: 10.1021/ac000777a. [DOI] [PubMed] [Google Scholar]
- 32.Zaia J, McClellan JE, Costello CE. Tandem mass spectrometric determination of the 4S/6S sulfation sequence in chondroitin sulfate oligosaccharides. Anal Chem. 2001;73:6030–9. doi: 10.1021/ac015577t. [DOI] [PubMed] [Google Scholar]
- 33.Venkataraman G, Shriver Z, Raman R, Sasisekharan R. Sequencing complex polysaccharides. Science. 1999;286:537–42. doi: 10.1126/science.286.5439.537. [DOI] [PubMed] [Google Scholar]
- 34.Pope RM, Raska CS, Thorp SC, Liu J. Analysis of heparan sulfate oligosaccharides by nano-electrospray ionization mass spectrometry. Glycobiology. 2001;11:505–13. doi: 10.1093/glycob/11.6.505. [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock AM, Costello CE, Zaia J. Glycoform quantification of chondroitin/dermatan sulfate using a liquid chromatography-tandem mass spectrometry platform. Biochemistry. 2006;45:2350–61. doi: 10.1021/bi052100t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solakyildirim K, Zhang Z, Linhardt RJ. Ultraperformance liquid chromatography with electrospray ionization ion trap mass spectrometry for chondroitin disaccharide analysis. Anal Biochem. 2009 doi: 10.1016/j.ab.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Xie J, Liu H, Liu J, Linhardt RJ. Quantification of heparan sulfate disaccharides using ion-pairing reversed-phase microflow high-performance liquid chromatography with electrospray ionization trap mass spectrometry. Anal Chem. 2009;81:4349–55. doi: 10.1021/ac9001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrence R, Olson SK, Steele RE, et al. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem. 2008;283:33674–84. doi: 10.1074/jbc.M804288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Studelska DR, Mandik-Nayak L, Zhou X, et al. High affinity glycosaminoglycan and autoantigen interaction explains joint specificity in a mouse model of rheumatoid arthritis. J Biol Chem. 2009;284:2354–62. doi: 10.1074/jbc.M806458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai X, Esko JD. An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J Biol Chem. 1996;271:17711–7. doi: 10.1074/jbc.271.30.17711. [DOI] [PubMed] [Google Scholar]
- 41.Bame KJ, Esko JD. Undersulfated heparan sulfate in a Chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J Biol Chem. 1989;264:8059–65. [PubMed] [Google Scholar]
- 42.Zhang L, Lawrence R, Schwartz JJ, et al. The effect of precursor structures on the action of 3-OST-1 and the biosynthesis of anticoagulant heparan sulfate. J Biol Chem. 2001;276:28806–13. doi: 10.1074/jbc.M100204200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L, Yoshida K, Liu J, Rosenberg RD. Anticoagulant heparan sulfate precursor structures in F9 embryonal carcinoma cells. J Biol Chem. 1999;274:5681–91. doi: 10.1074/jbc.274.9.5681. [DOI] [PubMed] [Google Scholar]
- 44.Lawrence R, Kuberan B, Lech M, Beeler DL, Rosenberg RD. Mapping critical biological motifs and biosynthetic pathways of heparan sulfate. Glycobiology. 2004;14:467–79. doi: 10.1093/glycob/cwh057. [DOI] [PubMed] [Google Scholar]
- 45.Xu D, Tiwari V, Xia G, Clement C, Shukla D, Liu J. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem J. 2005;385:451–9. doi: 10.1042/BJ20040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, David G, Esko JD. Repetitive Ser-Gly sequences enhance heparan sulfate assembly in proteoglycans. J Biol Chem. 1995;270:27127–35. doi: 10.1074/jbc.270.45.27127. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Esko JD. Amino acid determinants that drive heparan sulfate assembly in a proteoglycan. J Biol Chem. 1994;269:19295–9. [PubMed] [Google Scholar]
- 48.Studelska DR, Giljum K, McDowell LM, Zhang L. Quantification of glycosaminoglycans by reversed-phase HPLC separation of fluorescent isoindole derivatives. Glycobiology. 2006;16:65–72. doi: 10.1093/glycob/cwj037. [DOI] [PubMed] [Google Scholar]
- 49.Kuberan B, Lech M, Zhang L, Wu ZL, Beeler DL, Rosenberg RD. Analysis of heparan sulfate oligosaccharides with ion pair-reverse phase capillary high performance liquid chromatography-microelectrospray ionization time-of-flight mass spectrometry. J Am Chem Soc. 2002;124:8707–18. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
- 50.Kuberan B, Lech M, Borjigin J, Rosenberg RD. Light-induced 3-O-sulfotransferase expression alters pineal heparan sulfate fine structure. A surprising link to circadian rhythm. J Biol Chem. 2004;279:5053–4. doi: 10.1074/jbc.C300492200. [DOI] [PubMed] [Google Scholar]
- 51.Myette JR, Shriver Z, Claycamp C, McLean MW, Venkataraman G, Sasisekharan R. The heparin/heparan sulfate 2-O-sulfatase from Flavobacterium heparinum. Molecular cloning, recombinant expression, and biochemical characterization. J Biol Chem. 2003;278:12157–66. doi: 10.1074/jbc.M211420200. [DOI] [PubMed] [Google Scholar]
- 52.Myette JR, Soundararajan V, Shriver Z, Raman R, Sasisekharan R. Heparin/heparan sulfate 6-O-sulfatase from Flavobacterium heparinum: Integrated structural and biochemical investigation of enzyme active site and substrate specificity. J Biol Chem. 2009;284:35177–88. doi: 10.1074/jbc.M109.053801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugahara K, Kojima T. Specificity studies of bacterial sulfatases by means of structurally defined sulfated oligosaccharides isolated from shark cartilage chondroitin sulfate D. Eur J Biochem. 1996;239:865–70. doi: 10.1111/j.1432-1033.1996.0865u.x. [DOI] [PubMed] [Google Scholar]
- 54.Yamagata T, Saito H, Habuchi O, Suzuki S. Purification and properties of bacterial chondroitinases and chondrosulfatases. J Biol Chem. 1968;243:1523–35. [PubMed] [Google Scholar]
- 55.Midura RJ, Calabro A, Yanagishita M, Hascall VC. Nonreducing end structures of chondroitin sulfate chains on aggrecan isolated from Swarm rat chondrosarcoma cultures. J Biol Chem. 1995;270:8009–15. doi: 10.1074/jbc.270.14.8009. [DOI] [PubMed] [Google Scholar]
- 56.Wakabayashi H, Natsuka S, Mega T, et al. Novel proteoglycan linkage tetrasaccharides of human urinary soluble thrombomodulin, SO4–3GlcAbeta1-3Galbeta1-3(+/−Siaalpha2-6)Galbeta1-4Xyl. J Biol Chem. 1999;274:5436–42. doi: 10.1074/jbc.274.9.5436. [DOI] [PubMed] [Google Scholar]
- 57.Imanari T, Toyoda H, Yamanashi S, Shinomiya K, Koshiishi I, Oguma T. Study of the measurement of chondroitin sulphates in rabbit plasma and serum. J Chromatogr. 1992;574:142–5. doi: 10.1016/0378-4347(92)80109-4. [DOI] [PubMed] [Google Scholar]
- 58.Calabro L, Musolino C, Spatari G, Vinci R, Calatroni A. Increased concentration of circulating acid glycosaminoglycans in chronic lymphocytic leukaemia and essential thrombocythaemia. Clin Chim Acta. 1998;269:185–99. doi: 10.1016/s0009-8981(97)00200-3. [DOI] [PubMed] [Google Scholar]
- 59.Sugumaran G, Elliott-Bryant R, Phung N, Vitseva O, Kuberan B, Lech M. Characterization of splenic glycosaminoglycans accumulated in vivo in experimentally induced amyloid-susceptible and amyloid-resistant mice. Scand J Immunol. 2004;60:574–83. doi: 10.1111/j.0300-9475.2004.01516.x. [DOI] [PubMed] [Google Scholar]
- 60.Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612–20. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 61.Shibata S, Midura RJ, Hascall VC. Structural analysis of the linkage region oligosaccharides and unsaturated disaccharides from chondroitin sulfate using CarboPac PA1. J Biol Chem. 1992;267:6548–55. [PubMed] [Google Scholar]
- 62.Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–9. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 63.Deutsch AJ, Midura RJ, Plaas AH. Structure of chondroitin sulfate on aggrecan isolated from bovine tibial and costochondral growth plates. J Orthop Res. 1995;13:230–9. doi: 10.1002/jor.1100130211. [DOI] [PubMed] [Google Scholar]
- 64.Bertini S, Bisio A, Torri G, Bensi D, Terbojevich M. Molecular weight determination of heparin and dermatan sulfate by size exclusion chromatography with a triple detector array. Biomacromolecules. 2005;6:168–73. doi: 10.1021/bm049693s. [DOI] [PubMed] [Google Scholar]
- 65.Desai UR, Linhardt RJ. Molecular weight of heparin using 13C nuclear magnetic resonance spectroscopy. J Pharm Sci. 1995;84:212–5. doi: 10.1002/jps.2600840218. [DOI] [PubMed] [Google Scholar]
- 66.Desai UR, Linhardt RJ. Molecular weight of low molecular weight heparins by 13C nuclear magnetic resonance spectroscopy. Carbohydr Res. 1994;255:193–212. doi: 10.1016/s0008-6215(00)90979-0. [DOI] [PubMed] [Google Scholar]
- 67.Plaas AH, Wong-Palms S, Roughley PJ, Midura RJ, Hascall VC. Chemical and immunological assay of the nonreducing terminal residues of chondroitin sulfate from human aggrecan. J Biol Chem. 1997;272:20603–10. doi: 10.1074/jbc.272.33.20603. [DOI] [PubMed] [Google Scholar]
- 68.Enghild JJ, Thogersen IB, Cheng F, Fransson LA, Roepstorff P, Rahbek-Nielsen H. Organization of the inter-alpha-inhibitor heavy chains on the chondroitin sulfate originating from Ser(10) of bikunin: posttranslational modification of IalphaI-derived bikunin. Biochemistry. 1999;38:11804–13. doi: 10.1021/bi9908540. [DOI] [PubMed] [Google Scholar]
- 69.Toyoda H, Kobayashi S, Sakamoto S, Toida T, Imanari T. Structural analysis of a low-sulfated chondroitin sulfate chain in human urinary trypsin inhibitor. Biol Pharm Bull. 1993;16:945–7. doi: 10.1248/bpb.16.945. [DOI] [PubMed] [Google Scholar]
- 70.Capon C, Mizon C, Lemoine J, Rodie-Talbere P, Mizon J. In acute inflammation, the chondroitin-4 sulphate carried by bikunin is not only longer, it is also undersulphated. Biochimie. 2003;85:101–7. doi: 10.1016/s0300-9084(03)00066-x. [DOI] [PubMed] [Google Scholar]
- 71.Zhang L, Esko JD. Accumulation of a pentasaccharide terminating in alpha-N-acetylglucosamine in an animal cell mutant defective in heparan sulfate biosynthesis. J Biol Chem. 1995;270:12557–62. doi: 10.1074/jbc.270.21.12557. [DOI] [PubMed] [Google Scholar]
- 72.Ueno M, Yamada S, Zako M, Bernfield M, Sugahara K. Structural characterization of heparan sulfate and chondroitin sulfate of syndecan- 1 purified from normal murine mammary gland epithelial cells. Common phosphorylation of xylose and differential sulfation of galactose in the protein linkage region tetrasaccharide sequence. J Biol Chem. 2001;276:29134–40. doi: 10.1074/jbc.M102089200. [DOI] [PubMed] [Google Scholar]
- 73.Stern R. Hyaluronan metabolism: a major paradox in cancer biology. Pathologie Biologie. 2005;53:372–82. doi: 10.1016/j.patbio.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 74.Sugahara K, Tanaka Y, Yamada S. Preparation of a series of sulfated tetrasaccharides from shark cartilage chondroitin sulfate D using testicular hyaluronidase and structure determination by 500 MHz 1H NMR spectroscopy. Glycoconj J. 1996;13:609–19. doi: 10.1007/BF00731449. [DOI] [PubMed] [Google Scholar]
- 75.Kaneiwa T, Mizumoto S, Sugahara K, Yamada S. Identification of human hyaluronidase-4 as a novel chondroitin sulfate hydrolase that preferentially cleaves the galactosaminidic linkage in the trisulfated tetrasaccharide sequence. Glycobiology. 2009 doi: 10.1093/glycob/cwp174. [DOI] [PubMed] [Google Scholar]
- 76.Zhuo L, Hascall VC, Kimata K. Inter-alpha-trypsin inhibitor, a covalent protein-glycosaminoglycan-protein complex. J Biol Chem. 2004;279:38079–82. doi: 10.1074/jbc.R300039200. [DOI] [PubMed] [Google Scholar]
- 77.Sarrazin S, Adam E, Lyon M, et al. Endocan or endothelial cell specific molecule-1 (ESM-1): A potential novel endothelial cell marker and a new target for cancer therapy. Biochimica et Biophysica Acta (BBA)—Reviews on Cancer. 2006;1765:25–37. doi: 10.1016/j.bbcan.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 78.Tefferi A, Owen BA, Nichols WL, Witzig TE, Owen WG. Isolation of a heparin-like anticoagulant from the plasma of a patient with metastatic bladder carcinoma. Blood. 1989;74:252–4. [PubMed] [Google Scholar]
- 79.Dündar M, Müller T, Zhang Q, et al. Loss of dermatan-4-sulfotransferase-1 function results in ‘adducted thumb-clubfoot syndrome’. The American Journal of Human Genetics. 2009;85:873–82. doi: 10.1016/j.ajhg.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]