

Abstract
In this study, we show robust nicotinic excitation of pyramidal neurons in layer VI of prefrontal cortex. This layer contains the corticothalamic neurons, which gate thalamic activity and play a critical role in attention. Our experiments tested nicotinic excitation across postnatal development, using whole-cell recordings in prefrontal brain slices from rats. These experiments showed that layer VI neurons have peak nicotinic currents during the first postnatal month, a time period of intensive cortical development in rodents. We demonstrate that these currents are mediated directly by postsynaptic nicotinic receptors and can be suppressed by a competitive antagonist of α4β2* nicotinic receptors. To record from identified corticothalamic neurons, we performed stereotaxic surgery to label the neurons projecting to medial dorsal thalamus. As hypothesized, recordings from these retrogradely labeled neurons in layer VI showed prominent nicotinic currents. Finally, we examined the effects of the drug nicotine on layer VI neurons and probed for the potential involvement of the accessory subunit, α5, in their receptors. A level of nicotine similar to that found in the blood of smokers elicits a stable inward current in layer VI neurons, yet this exposure desensitizes ∼50% of the subsequent current elicited by acetylcholine. An allosteric modulator of α4β2α5 receptors resulted in a 2.5-fold potentiation of submaximal nicotinic currents. This result is consistent with the expression of the relatively rare α5 nicotinic subunit in layer VI. In summary, we show that layer VI corticothalamic neurons can be strongly excited during development by an unusual subtype of nicotinic receptor.
Keywords: corticothalamic neurons, postnatal development, nicotine, nicotinic receptors, prefrontal cortex, rat, slice
Introduction
Cortical nicotinic acetylcholine receptors are important for attention and effortful concentration (Rezvani and Levin, 2001; Granon et al., 2003; Lambe et al., 2005), and have been suggested to have long-lasting effects on the development of attention circuitry (Langley et al., 2005; Rodriguez and Bohlin, 2005; Schmitz et al., 2006). Abnormalities in cortical nicotinic binding have been demonstrated in several neurodevelopmental conditions, including autism (Perry et al., 2001; Martin-Ruiz et al., 2004), epilepsy (Picard et al., 2006), and schizophrenia (Breese et al., 2000; Marutle et al., 2001). In the human brain, nicotinic binding is highest toward the end of gestation (Court et al., 2000). The drug nicotine is a partial agonist of these nicotinic acetylcholine receptors and may disrupt the trophic effects of acetylcholine during a critical stage of development (Navarro et al., 1989; Slotkin, 2004; Liang et al., 2006). Substantial literature suggests that in humans, prenatal exposure to nicotine through maternal cigarette smoking harms the development of prefrontal attention circuitry (Langley et al., 2005; Rodriguez and Bohlin, 2005).
Rats and mice are born at a more premature stage of brain development than humans, and the first few postnatal weeks in rodents are thought to be a time period approximately equivalent to the last trimester in utero and the perinatal period in human development (Romijn et al., 1991; Watson et al., 2006). During this time, there is a marked band of high-affinity nicotinic binding in layer VI in the prefrontal cortex (Tribollet et al., 2004), the primary location of corticothalamic neurons. Recent transgenic work shows that nicotinic receptors are expressed by corticothalamic neurons and are present in corticothalamic terminals in mice (King et al., 2003). Nicotinic acetylcholine receptors have been suggested to play a trophic role in the development of cortical neurons and brain circuitry (Belluardo et al., 1999; Brown and Kolb, 2001; Liang et al., 2006).
Nicotinic receptors are excitatory, ligand-gated channels that allow the movement of Na+, K+, and Ca2+ ions across the cell membrane. At the resting membrane potential of a typical cortical neuron, the nicotinic current provides an excitatory or depolarizing influence, which is measured electrophysiologically in a voltage clamp as an inward current. Activation and desensitization parameters of nicotinic receptors depend largely on the subunit composition in these pentameric receptors (Moroni et al., 2006; Tapia et al., 2007). Layer VI nicotinic currents may have unusual properties because of the potential incorporation of an accessory α5 nicotinic receptor subunit. This subunit is expressed in layer VI (Marks et al., 1992; Winzer-Serhan and Leslie, 2005) and can substantially alter the properties of α4β2* receptors (Ramirez-Latorre et al., 1996; Tapia et al., 2007; Kuryatov et al., 2008). In mice, genetic deletion of the α5 subunit confers resistance to nicotine-elicited seizures (Salas et al., 2003). In humans, the α5 subunit has recently been identified in a cluster of genes found to predispose individuals toward heavy cigarette smoking (Saccone et al., 2007; Berrettini et al., 2008; Bierut et al., 2008) and early experimentation with tobacco (Schlaepfer et al., 2008).
Understanding the properties of nicotinic currents in layer VI neurons is critical to appreciate the potential trophic effects of acetylcholine on these neurons and how they may be altered by exposure to the drug nicotine during development. We present here the first electrophysiological study of nicotinic currents in layer VI pyramidal cells and identified corticothalamic neurons during postnatal development.
Materials and Methods
Brain slice preparation.
Coronal slices (400 μm thick) of the medial prefrontal cortex were prepared from male Charles River Sprague–Dawley rats from postnatal day (P) 7 to P67, in accordance with protocols approved by the University of Toronto Animal Care and Use Committee. These protocols conformed to international guidelines on the ethical use of animals. The brain was cooled as rapidly as possible with 4°C oxygenated sucrose artificial cerebrospinal fluid (ACSF) (254 mm sucrose was substituted for NaCl). Prefrontal slices were cut from anterior to posterior using the appearance of white matter and the corpus callosum as anterior and posterior guides to target recording to the Cg1, Cg2, and PrL regions (Paxinos and Watson, 2007). The slices were cut on a Dosaka Linear Slicer (SciMedia), and were transferred to 30°C oxygenated ACSF (containing the following, in mm: 128 NaCl, 10 d-glucose, 24 NaHCO3, 2 CaCl2, 2 MgSO4, 3 KCl, 1.25 NaH2PO4, pH 7.33) in a prechamber (Automate Scientific) and allowed to recover for at least 1 h before the beginning of an experiment. For whole-cell recordings, slices were placed in a modified chamber (Warner Instruments) mounted on the stage of an Olympus BX50WI microscope. Regular ACSF was bubbled with 95% oxygen and 5% carbon dioxide and flowed over the slice at 30°C with a rate of 3–4 ml/min.
Electrophysiology.
Whole-cell patch electrodes (2–3 MΩ) contained the following (in mm): 120 K-gluconate, 5 KCl, 2 MgCl, 4 K2-ATP, 0.4 Na2-GTP, 10 Na2-phosphocreatine, and 10 HEPES buffer (adjusted to pH 7.33 with KOH). Layer VI pyramidal neurons were patched under visual control using infrared differential interference contrast microscopy. The mean membrane properties of layer VI pyramidal neurons recorded (n = 325) were 92 ± 1 mV spike amplitude, 240 ± 11 MΩ input resistance, and −78 ± 1 mV resting potential. Because corticothalamic neurons are suggested to send a collateral axon to local circuit interneurons (West et al., 2006), we also collected data from layer VI fast-spiking interneurons. These cells were patched based on their small, circular morphology and were confirmed electrophysiologically by their characteristic, fast spiking pattern on injection of depolarizing current pulses. The properties of the fast-spiking interneurons recorded (n = 14) were 83 ± 3 mV spike amplitude, 511 ± 65 MΩ input resistance, and −70 ± 2 mV resting potential. In current clamp, neurons were recorded at their resting potentials. In voltage clamp, neurons were held at −75 mV, near the calculated equilibrium potential for chloride under these conditions, and currents were recorded using continuous single-electrode voltage-clamp mode with a Multiclamp 700b (Molecular Devices) or an EPC10 (HEKA Elektroniks), acquired and low-pass filtered at 3 kHz with either pClamp10.2/Digidata1440 (Molecular Devices) or Patchmaster 2.20 (HEKA Elektroniks).
Acetylcholine (10 μm to 1 mm, 30 s) was applied in the bath in the presence of atropine (200 nm, continuous application) to block muscarinic receptors. Brief local application of acetylcholine (1 mm, 1 s) was applied with a rapid solution exchange system (ALA Scientific Instruments) in a subset of neurons (n = 16; age range from P15 to P67) to confirm that a fast-desensitizing component was not missed. For most experiments, nicotinic currents were probed by adding 1 mm acetylcholine to the bath perfusion for a 30 s interval after a 1 min baseline and followed by a 5 min washout period. The nicotinic current was measured in Clampfit by subtracting the mean inward current at the peak (1 s) of the acetylcholine response from the mean holding current at baseline. Other drugs were also added to the bath in specific experiments: 2 μm tetrodotoxin (TTX), 10–20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 50 μm d(−)-2-amino-5-phosophonopentanoic acid (APV), 10 nm methyllycaconitine (MLA), 3 μm dihydro-β-erythroidine hydrobromide (DHβE), 100 μm 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (CPCCOEt), 30 μm 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), 300 nm nicotine hydrogen tartrate, and 100 nm or 1 μm galanthamine. All compounds were obtained from Sigma or Tocris and stored in stock solutions at −20°C, before being diluted and applied to the slice in oxygenated ACSF.
Stereotaxic surgery.
Young rats (P14–P17) were anesthetized with 2–3% isoflurane and positioned on a stereotaxic apparatus (Stoelting Co.). An incision was made to expose the midline suture and bregma. We drilled (Micro Drill; FST) a hole at empirically determined coordinates from bregma (1.5 mm lateral and 2.0 mm posterior) to target the medial dorsal thalamus with an angled injection. A 1 μl syringe (Hamilton) was lowered at a 10° angle (to avoid blood vessels in the sagittal sinus) to 6.0 mm ventral, and 0.3 μl of rhodamine microsphere solution (Lumafluor) was slowly infused. After an additional 5–10 min diffusion period, the syringe was raised and the incision closed with Vetbond tissue adhesive (3M). After recovery from anesthesia, the rat pup was returned to its home cage with its mother and siblings. Three to 12 d after the surgery, medial prefrontal brain slices were obtained as described above. The thalamus was sliced to verify infusion location in the medial dorsal thalamus. Retrogradely labeled layer VI neurons were identified under fluorescent light and patched under infared-differential interference contrast microscopy.
Statistical analysis.
The developmental changes in nicotinic currents were assessed with the Kruskal–Wallis nonparametric ANOVA and post hoc with Mann–Whitney tests, at a significance level of 0.05. Differences in responses to different pharmacological agents were determined using Student's two-tailed paired t tests, at a significance level of 0.05. Changes in the frequency and amplitude of spontaneous EPSCs (sEPSCs) were assessed with Kolmogorov–Smirnov tests of cumulative distribution, at a significance level of 0.01.
Results
Nicotinic currents in layer VI pyramidal neurons change across development
Recent work suggests that there is a band of nicotinic binding in layer VI of rat prefrontal cortex at postnatal day 15 (Tribollet et al., 2004). To examine the electrophysiological properties of potential nicotinic currents in layer VI, we blocked muscarinic receptors (the only other subtype of acetylcholine receptors) by applying atropine (200 nm) in the bath and performed whole-cell recordings from layer VI neurons to examine the effects of application of acetylcholine (10 μm to 1 mm). Recording in current clamp, we found that bath application of acetylcholine (30 s) depolarized layer VI neurons in a concentration-dependent manner, as illustrated in Figure 1A, causing the cell to fire action potentials. To quantify this nicotinic excitation, we measured nicotinic inward currents in voltage clamp. Because some types of nicotinic currents desensitize rapidly (McGehee and Role, 1995), we compared the effects of rapid local application (1 s) to bath application (30 s) of acetylcholine (1 mm). As expected, the duration of the currents elicited were different; however, the peak currents were very similar, as illustrated by the example in Figure 1B. The robustness of the peak response with bath application makes it an ideal measure to compare across different pharmacological conditions, to assess the properties of the layer VI nicotinic currents.
Figure 1.
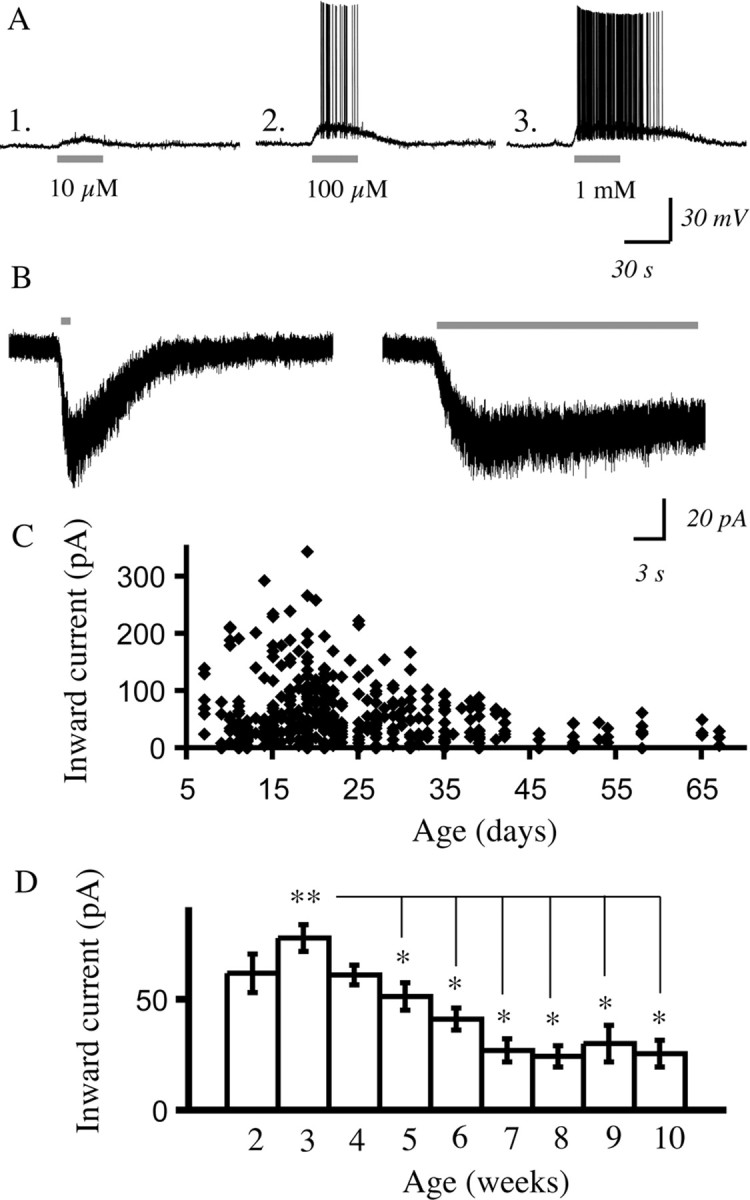
Nicotinic receptor stimulation with acetylcholine excites pyramidal neurons in layer VI in the presence of atropine to block muscarinic receptors. A2–A3, The concentration–response relationship for nicotinic depolarization of a layer VI pyramidal neuron from a P19 rat in current clamp (acetylcholine: 30 s, 5 min washout between sweeps; atropine: 200 nm present in all experiments). B, Voltage-clamp traces show that similar peak inward currents result from rapid local application of acetylcholine (1 mm, 1 s) and bath application (1 mm, 30 s). C, The scattergram shows peak inward currents from early postnatal development to adulthood elicited by acetylcholine (1 mm, 30 s). D, Summary data from the scattergram in C showing mean inward current elicited by acetylcholine in layer VI pyramidal neurons by postnatal week. The Kruskal–Wallis ANOVA shows a highly significant developmental effect (**p < 0.0001) in which the mean inward current at postnatal week 3 is significantly higher than the mean inward current in postnatal weeks 5 through 10 (*p < 0.05), as shown by Mann–Whitney tests.
In a large developmental study, we examined the inward currents elicited by acetylcholine (1 mm, 30 s) in layer VI pyramidal neurons in prefrontal cortical slices from P7 to P67. The large majority of the neurons showed nicotinic inward currents, and there was a strong developmental dependence in the magnitude of the peak current as shown in Figure 1C. Averaging by postnatal week demonstrates (Fig. 1D) that the currents are highest in the first postnatal month and then decline significantly and progressively to lower adolescent (∼P30–P50 in rat) and adult (>P50) levels. To confirm that we were not missing a rapidly desensitizing component, we examined the peak current elicited by rapid application of acetylcholine compared with bath application as in Figure 1B and found that they did not differ significantly (rapid application, 37 ± 7 pA; bath application, 40 ± 8 pA; n = 16; paired t test, p = NS; age range examined, P15–P67). This finding of a stable peak current for a given concentration of acetylcholine, regardless of the manner of application, was consistent across the age range examined (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
However, not all layer VI pyramidal neurons showed inward currents in response to acetylcholine. Even in the first postnatal month when nicotinic excitation was greatest, there were neurons that showed minimal to no response. Our preliminary analysis suggested that the firing properties of the layer VI pyramidal neurons in rat could be a useful criterion to predict which neurons were likely to have nicotinic currents, as shown in Figure 2. Recent work has suggested that corticothalamic layer VI neurons are regular spiking neurons (West et al., 2006), whereas layer VI corticocortical neurons tend to be burst-spiking neurons (Mercer et al., 2005). Therefore, we compared nicotinic inward currents between regular spiking and bursting neurons from rats from ages P15 to P42. Because the burst-firing neurons were more rare, we compared the nicotinic current in a burst-firing neuron to that of the next regular spiking neuron recorded from the same rat, providing an age-matched comparison. There were significant differences: the regular spiking neurons had a mean inward current of 60 ± 11 pA, whereas the bursting neurons had a mean current of 20 ± 7 pA (n = 7; paired t test, p < 0.01).
Figure 2.
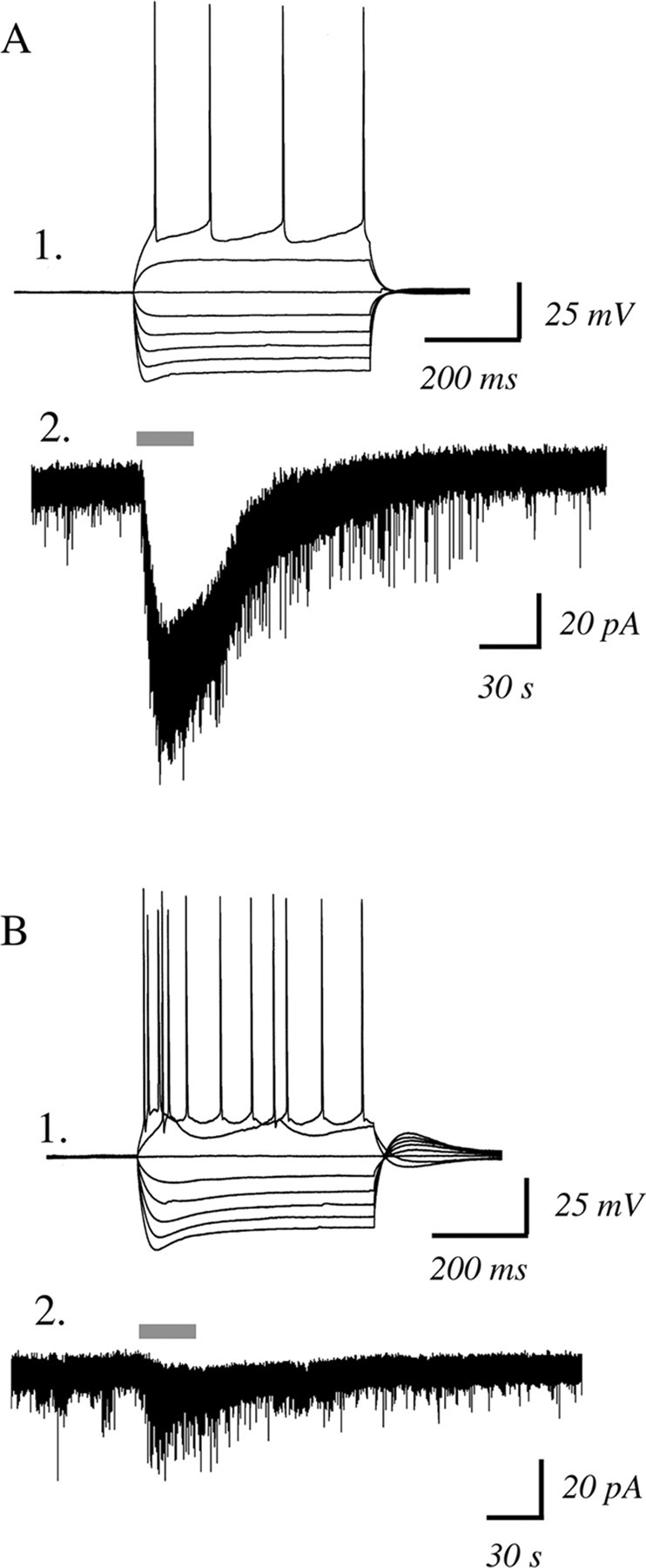
Regular-spiking, rather than bursting, layer VI neurons are more likely to have large nicotinic inward currents. A1, Current-clamp recording showing membrane potential changes in response to injecting depolarizing and hyperpolarizing current steps in a regular firing neuron from a P33 rat. A2, A voltage-clamp trace from this neuron showing a robust inward current with acetylcholine (1 mm, 30 s) in the presence of atropine (200 nm). B1, This current-clamp recording is of a burst firing neuron from the same P33 rat. B2, A voltage-clamp trace from this neuron showing minimal inward current with acetylcholine (1 mm, 30 s) in the presence of atropine (200 nm).
It can be noted in the examples in Figure 2 that both types of neurons showed a nicotinic-elicited increase in sEPSCs. In all the bursting neurons recorded, there was a significant increase in sEPSC frequency (Kolmogorov–Smirnov test, p < 0.01). In regular spiking neurons, more than half the cells also appeared to have increased sEPSCs with application of acetylcholine. However, the increase in current noise associated with the nicotinic inward current in these neurons complicates quantification of the indirect effects, and so we focused on examining the nicotinic inward currents.
Layer VI nicotinic currents are directly mediated by somatodendritic nicotinic receptors
The nicotinic inward currents in layer VI neurons appeared to be directly mediated by postsynaptic nicotinic receptors. The nicotinic currents were not suppressed by blocking action potentials with TTX (2 μm, 10 min): 100 ± 25 pA versus 102 ± 38 pA with TTX (n = 5; paired t test, p = NS; age range examined, P21–P41). Furthermore, these inward currents were not suppressed by blocking ionotropic glutamate receptors with CNQX (10–20 μm) and APV (50 μm, 10 min): 105 ± 18 pA versus 99 ± 20 pA with the blockers (n = 11; paired t test; p = NS; age range examined, P15–P50). Nor were the nicotinic currents suppressed by blocking metabotropic glutamate receptors with antagonists of mGluR1 (100 μm CPCCOEt, 10 min) and mGluR5 (30 μm MPEP, 10 min): 92 ± 7 pA versus 96 ± 10 pA with the blockers (n = 8; paired t test; p = NS; age range examined, P15-P35). These results do not appear to vary over the age range examined, as illustrated by analysis of younger and older groups shown in supplemental Figure 2, available at www.jneurosci.org as supplemental material. These pharmacological results are consistent with direct mediation of the currents by somatodendritic nicotinic receptors on the layer VI pyramidal neuron.
Consistent with a direct postsynaptic effect, large nicotinic currents decreased the average input resistance of layer VI neurons by 35 ± 5% (n = 18; paired t test, p < 0.01; age range examined, P19–P35). We used a ramp protocol to determine the reversal potential of the nicotinic currents, as illustrated in Figure 3. The protocol raised the holding potential from −115 mV to −35 mV over a 2 s period. TTX (2 μm) was present to prevent the patched cell from spiking in response to the depolarizing segment of the voltage ramp. Current-voltage plots were obtained by subtracting baseline current from the peak nicotinic current, whereas the reversal potential was determined by extrapolating the resulting line of best fit. The nicotinic currents in layer VI pyramidal neurons reverse at −16 ± 5 mV (range, −28 mV to +5 mV, n = 7; age range examined, P19–P35), consistent with a mixed cationic conductance.
Figure 3.

Nicotinic currents elicited by acetylcholine are mediated by somatodendritic nicotinic receptors on layer VI neurons. A, The current–voltage curves in TTX (2 μm) for a layer VI neuron at baseline (upper curve) and in the presence of acetylcholine (1 mm, 30 s; lower curve, which appears thicker because of increased current noise). B, The current–voltage relationship is shown, and the reversal potential was found for the nicotinic current in this neuron from a P21 rat. This reversal potential is consistent with the equilibrium potential for a nonselective cationic channel under our recording conditions.
Layer VI nicotinic currents are mediated by α4β2* nicotinic receptors
We found that the competitive α4β2* antagonist DHβE (3 μm, 10 min) significantly suppressed these nicotinic currents in layer VI neurons (n = 20; paired t test; p < 0.00001; age range examined, P7–P65), as illustrated in Figure 4, A and B. The nicotinic inward current returned completely after a 60 min washout of DHβE, as shown in Figure 4C. DHβE suppressed the nicotinic currents in brain slices from both younger and older rats, as illustrated in supplemental Figure 2, available at www.jneurosci.org as supplemental material. The nicotinic currents in layer VI pyramidal neurons were not sensitive to the α7 antagonist MLA (10 nm, 10 min; n = 11; paired t test, p = NS; age range examined, P7–P50), as illustrated in Figure 4A.
Figure 4.

Layer VI pyramidal neurons are excited by α4β2* nicotinic acetylcholine receptors. A1, Bar chart showing that the competitive α4β2* antagonist DHβE (3 μm, 10 min) suppressed the nicotinic inward current [acetylcholine (ACh) (n = 20; *p < 0.00001, paired t test; age range examined: P7–P65). A2, Bar chart showing that the α7 receptor antagonist MLA (10 nm, 10 min) does not significantly suppress this current (n = 11; p = NS, paired t test; age range examined: P7–P50). B1, B2, Voltage-clamp traces showing the effect of acetylcholine (1 mm, 30 s) before and after DHβE (3 μm, 10 min) in a neuron from a P19 rat. C1–C4, Voltage-clamp traces in a neuron from a P21 rat showing the baseline effect of acetylcholine (C1), suppression with DHβE (3 μm, 10 min) (C2), partial recovery after 30 min washout (C3), and full recovery after 60 min washout of DHβE (C4) (note that the inward current is the same as in C1, but there is a flurry of sEPSCs near the peak that cannot be resolved on this scale).
Nicotinic currents are found in retrogradely labeled corticothalamic neurons
To record from identified corticothalamic neurons, we infused retrograde tracer rhodamine microspheres (0.3 μl; Lumafluor) into the medial dorsal thalamus using stereotaxic surgery in vivo. We obtained brain slices of medial prefrontal cortex from these rats 3–12 d postoperatively and verified the infusion location in the thalamus. Low- and high-magnification images of corticothalamic neurons in prefrontal brain slice are shown in Figure 5, together with a schematic of the injection site of the retrograde tracer in medial dorsal thalamus and of the prefrontal slice, where we recorded from identified corticothalamic neurons. Whole-cell recordings from identified corticothalamic neurons (mean spike amplitude, 98 ± 2 mV; mean input resistance, 244 ± 34 MΩ; mean resting potential, −79 ± 2 mV; n = 15 neurons from eight rats) found that they were regular spiking and depolarized by nicotinic acetylcholine receptors. The properties of these neurons did not differ significantly from those of unlabeled layer VI pyramidal neurons (values given in Materials and Methods). Nicotinic currents were examined in labeled corticothalamic neurons from postnatal day 20 to 29. The nicotinic currents in these corticothalamic neurons were almost completely suppressed by DHβE (3 μm, 10 min; n = 12; paired t test, p = 0.000001), as shown in Figure 6, but were unaltered by MLA (10 nm, 10 min; n = 7; paired t test, p = NS). The time course and pharmacology of these nicotinic currents (illustrated in Fig. 6) are similar to those of the nicotinic currents in responsive neurons in layer VI in brain slices from non-operated animals.
Figure 5.

Corticothalamic neurons are labeled by stereotaxic surgery to infuse the retrograde tracer rhodamine microspheres in medial dorsal thalamus. A1, A2, Schematics show the infusion location in the thalamus (A1) as well as the coronal prefrontal slice (A2), where layer VI medial prefrontal neurons are recorded 3–12 d postoperatively. The black rectangle on the coronal slice shows the location of the low-magnification image in C. B, A high-magnification image of a labeled corticothalamic neuron during recording (the patch pipette is just above the asterisk). Scale bar, 20 μm. C, A low-magnification image of this same neuron and pipette (above asterisk) showing the laminar location of the recorded neuron within medial prefrontal cortex. Note the strong retrograde labeling in layer VI [the layer adjacent to white matter (WM)] as well as the sparse labeling of the small population of corticothalamic neurons in layer V. Scale bar, 240 μm.
Figure 6.

Identified corticothalamic neurons show robust nicotinic currents that are sensitive to the α4β2* antagonist, DHβE. A1, A2, Current-clamp trace showing the effects of acetylcholine (1 mm, 30 s) before and after DHβE (3 μm, 10 min) in a labeled corticothalamic neuron from a P21 rat. B1, B2, Voltage-clamp trace of another labeled neuron showing the effects of acetylcholine (1 mm, 30 s) before and after DHβE (3 μm, 10 min) in a P29 rat. C, Bar chart summarizing nicotinic acetylcholine (ACh) currents in labeled layer VI corticothalamic neurons before and after DHβE (n = 12; paired t test, *p < 0.000001).
Indirect excitation of interneurons in prefrontal layer VI
Because recent work suggests that corticothalamic neurons send a local collateral axon to fast-spiking interneurons in cortical layer VI (West et al., 2006), we recorded from a sample of these neurons (age range examined, P15–P39). We found that in 13 of 14 fast-spiking interneurons in layer VI, acetylcholine in the presence of atropine significantly increased the frequency of sEPSCs (Kolmogorov–Smirnov test; p < 0.01), as illustrated in Figure 7. The significant nicotinic-elicited increase in sEPSCs was 99 ± 1% suppressed by the AMPA receptor antagonist CNQX (10 μm, 10 min; n = 5), suggestive of an indirect nicotinic effect. The time course of these effects was similar to that of the nicotinic excitation of corticothalamic neurons.
Figure 7.

As hypothesized, fast-spiking interneurons in layer VI show a prominent indirect effect of nicotinic acetylcholine stimulation. A, Current-clamp trace showing membrane potential changes in response to depolarizing and hyperpolarizing current steps, with action potentials that are characteristic of a fast-spiking interneuron. B1, B2, Voltage-clamp trace showing the effects of a 30 s application of acetylcholine (1 mm). The inset magnifies the EPSCs under baseline conditions (B1) and in the presence of acetylcholine (B2). C, Bar chart summarizing the mean change in spontaneous EPSC frequency with nicotinic stimulation (n = 14; *p < 0.05, paired t test). D, Bar chart summarizing the mean change in spontaneous EPSC amplitude with nicotinic stimulation (n = 14; p = NS, paired t test).
A low level of the drug nicotine triggers a persistent inward current in layer VI neurons
Prolonged application of the drug nicotine (300 nm, 5–10 min), at a concentration similar to the peak blood level found in smokers (Henningfield et al., 1993), resulted in a small yet significant and stable mean inward current in regular spiking layer VI pyramidal neurons, as shown in Figure 8A (age range examined, P9–P41). Similar currents were observed with longer applications, as shown in Figure 8B. Even after a 45 min application of nicotine (300 nm), the nicotinic current disappeared quickly (<5 min) after termination of the nicotine application, as illustrated in supplemental Figure 3, available at www.jneurosci.org as supplemental material. Although the current elicited by nicotine itself did not appear to desensitize over 10–45 min of application, the exposure to nicotine suppressed the current evoked by acetylcholine, as illustrated in Figure 8C. A comparison of a test pulse of acetylcholine (1 mm, 30 s) before application of nicotine and after a 5 min washout of nicotine (by which time the nicotinic current had returned to baseline) showed 48 ± 4% suppression of the current elicited by acetylcholine (n = 10; paired t test, p < 0.01, age range examined, P9–P38). This desensitization of the acetylcholine response did not recover completely after 30 min of washout, as illustrated in Figure 8D.
Figure 8.
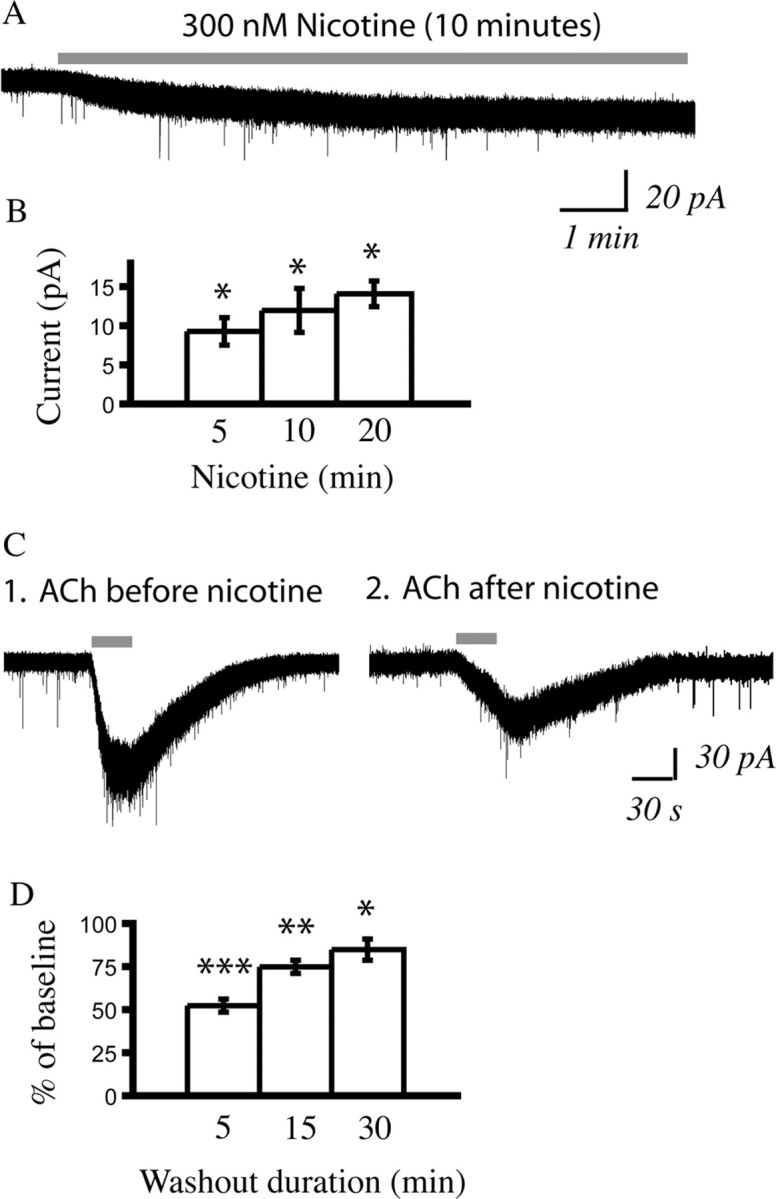
A persistent inward current is elicited by nicotine, which washes out rapidly but leaves a lingering suppression of the maximal effects of acetylcholine. A, A voltage-clamp trace showing a small but persistent inward current elicited in a neuron from a P19 rat by application of the drug nicotine (300 nm, 10 min). This concentration of nicotine is consistent with the peak blood level of nicotine seen in smokers (Henningfield et al., 1993). B, The bar chart showing the mean currents elicited by three different durations of nicotine application: 5 min (n = 31), 10 min (n = 25), and 20 min (n = 7) applications. All of the inward currents are significantly different when compared with baseline: *p < 0.001, paired t tests. The age range examined is P9-P41. C1, A voltage-clamp trace from a P21 rat showing a robust depolarization with acetylcholine (1 mm, 30 s) preceding the application of nicotine. C2, A voltage-clamp trace from the same neuron taken 5 min after the end of a 40 min nicotine application shows that the depolarization elicited by acetylcholine is decreased. D, Bar chart showing the highly significant suppression of the inward current elicited by acetylcholine (1 mm, 30 s; n = 31; ***p < 0.00001, paired t test) examined 5 min after nicotine application (300 nm, 5–10 min). This bar chart also shows the continued suppression of the responses of the neurons to acetylcholine at 15 min of washout (n = 11; **p < 0.01, paired t test) and 30 min of washout (n = 10; *p < 0.05, paired t test).
Examination of the potential involvement of the nicotinic accessory subunit, α5
We tested pharmacologically for the involvement of the α5 accessory subunit because recent work suggests that the α5 nicotinic accessory subunit is developmentally expressed in layer VI (Winzer-Serhan and Leslie, 2005) and can alter the properties of nicotinic receptors (Ramirez-Latorre et al., 1996; Tapia et al., 2007; Kuryatov et al., 2008). A recent paper by Kuryatov et al. (2008) gave us a key pharmacological tool to probe for the inclusion of α5 nicotinic subunits in the nicotinic receptors of layer VI. This group used a low concentration of galanthamine (which is an inhibitor of acetylcholinesterase at higher concentrations) and observed a doubling of the current through α4β2α5 receptors (Kuryatov et al., 2008), an order of magnitude greater than the potentiation of receptors lacking α5. We hypothesized that we would see a large increase in the layer VI nicotinic currents because the relatively rare α5 subunit is expressed in this layer (Wada et al., 1990; Marks et al., 1992; Salas et al., 2003) during postnatal development (Winzer-Serhan and Leslie, 2005). Consistent with this hypothesis, we found a 2.5-fold potentiation of nicotinic currents in layer VI neurons by galanthamine across the age range examined (P10–P41). Figure 9 shows examples and summary data for the potentiation of submaximal nicotinic currents by galanthamine (1 μm, 10 min). Because galanthamine has a micromolar IC50 for inhibiting acetylcholinesterase in brain slices, we examined whether similar potentiation could also be seen with 100 nm galanthamine. We found no statistically significant difference in the ability of 100 nm and 1 μm galanthamine to potentiate submaximal nicotinic currents [235 ± 49% (n = 5) and 258 ± 34% (n = 13) potentiation, respectively, for 10 μm acetylcholine; unpaired t test, p = NS), suggesting that allosteric modulation is responsible for the potentiation shown in Figure 9.
Figure 9.

The nicotinic currents in layer VI can be dramatically potentiated by galanthamine, suggesting the involvement of α5-containing α4β2 nicotinic receptors. A, Voltage-clamp traces showing that the inward current elicited by acetylcholine (ACh) (10 μm, 30 s) is more than doubled after application of galanthamine (1 μm, 10 min) in a recording from a P10 rat. B, The bar chart shows how much different concentrations of acetylcholine are potentiated above baseline after application of galanthamine (0.1–1 μm). The left bar shows the potentiation for 10 μm acetylcholine (n = 18; **p < 0.001, paired t test), the middle bar shows potentiation for 100 μm acetylcholine (n = 23; **p < 0.001, paired t test), and the right bar shows potentiation for 1 mm acetylcholine (n = 19; *p < 0.01, paired t test). C, The scattergram shows the percent that a submaximal concentration of acetylcholine is potentiated by galanthamine (0.1–1 μm) over the age range examined: P10–P41. The gray line indicates the level at which the initial current was not altered by galanthamine.
Discussion
The mechanisms underlying the ability of nicotine to perturb attention circuitry during development and yet enhance it in adulthood are not well understood. Here, we find that a large subset of layer VI pyramidal neurons, including identified corticothalamic neurons, are depolarized by acetylcholine through nicotinic receptors, which are sensitive to the α4β2* nicotinic antagonist DHβE. These nicotinic currents in layer VI appear to be strongly developmentally regulated and are at peak levels in the first postnatal month. The currents decrease significantly during adolescence and remain at lower levels in adulthood. Throughout postnatal development, the nicotinic inward currents are mediated directly by somatodendritic nicotinic receptors and can be elicited by concentrations of nicotine similar to those seen in the blood of smokers. Prolonged exposure to nicotine continues to elicit a small yet significant current that disappears quickly on washout. However, this exposure leaves a lasting suppression of the maximal current that can be elicited by acetylcholine. Finally, we find that submaximal nicotinic currents in layer VI neurons are dramatically potentiated by low concentrations of galanthamine. These experiments suggest that α5-containing α4β2 receptors mediate nicotinic excitation of layer VI corticothalamic neurons during postnatal development.
Developmental changes in layer VI nicotinic receptors
The presence of developmentally regulated nicotinic receptors in layer VI is supported by a high-affinity nicotinic binding study in rats (Tribollet et al., 2004). High levels of binding in layer VI are observed at P15 and much lower levels at P60. Nicotinic currents have also been reported in low-threshold spiking interneurons in layers II/III and V of visual cortex in young rats (Xiang et al., 1998), and of prefrontal cortex in young mice (Couey et al., 2007). It has been shown that knock-out mice deleted for all β2-containing nicotinic receptors have deficits in cortically mediated arousal processes (Granon et al., 2003; King et al., 2003; Cohen et al., 2005; Granon and Changeux, 2006). Interestingly, it has been found that the performance of these mice on a behavioral task of arousal can be normalized if β2 subunits are restored in corticothalamic neurons during the first 3 weeks of postnatal development (King et al., 2003).
Diversity among α4β2* nicotinic receptors
There appears to be variability in the desensitization parameters of α4β2* nicotinic receptors (Quick and Lester, 2002). Recently, emphasis has been placed on the ease with which the DHβE-sensitive nicotinic currents in interneurons of the ventral tegmental area become completely desensitized (Mansvelder et al., 2002, 2006; Wooltorton et al., 2003). However, we observe that nicotinic currents in layer VI neurons are relatively resistant to desensitization. This observation more closely parallels the nicotinic currents in the thalamus and thalamocortical axons and terminals (Gioanni et al., 1999; Lambe et al., 2005; Kawai et al., 2007). The variability in the desensitization of α4β2* nicotinic receptors may arise from differences in their subunit composition (McGehee and Role, 1995). For example, the ratio of α4 to β2 subunits has recently been shown to have profound effects on the activation and desensitization parameters of DHβE-sensitive nicotinic receptors (Moroni et al., 2006; Tapia et al., 2007). Inclusion of the α5 accessory subunit is also thought to alter several parameters of α4β2* nicotinic receptors (Brown et al., 2007; Tapia et al., 2007; Kuryatov et al., 2008). Layer VI is one of a small number of brain regions that express the α5 nicotinic subunit (Wada et al., 1990; Marks et al., 1992; Salas et al., 2003), and our data suggest that these subunits are present in the receptors mediating the layer VI nicotinic currents over the age range examined (P10–P41). Expression of the α5 subunit in layer VI also appears to be developmentally modulated (Winzer-Serhan and Leslie, 2005), showing peak expression that overlaps with the peak nicotinic currents in layer VI neurons.
Potential significance of corticothalamic nicotinic receptors for seizures
Corticothalamic neurons have been implicated in certain types of seizures (Steriade and Amzica, 2003). Intriguingly, mice that are constitutively deleted for the α5 nicotinic subunit are resistant to nicotine-elicited seizures (Salas et al., 2003). It is not known whether this protection from nicotinic seizures results from the lack of α5 in adulthood or whether the brain develops differently in the absence of α5. In contrast, genetic mutations that render α4 and β2 subunit-containing receptors hypersensitive to acetylcholine are associated with the sleep-related epilepsy syndrome: autosomal dominant nocturnal frontal lobe epilepsy (Steinlein et al., 1995; De Fusco et al., 2000; Díaz-Otero et al., 2007; Marini and Guerrini, 2007). These seizures typically begin in mid-childhood, occur in the earliest period of sleep, and, like many types of seizures, are thought to result from the synchronization of prefrontal output (Marini and Guerrini, 2007). Because corticothalamic neurons are well known to fire synchronously during sleep (Steriade, 2006) and to contribute to a variety of seizures (Steriade and Amzica, 2003), they may be particularly vulnerable to abnormal activation patterns resulting from hypersensitive nicotinic receptors.
Prenatal nicotine exposure and attention deficits
Recent human imaging data suggest that there is prolonged occupancy of cortical nicotinic receptors after one cigarette (Brody et al., 2006). Nicotine from maternal cigarette smoking tends to accumulate in the fetal brain because of its higher lipid content and the lower clearance of nicotine from the fetal compartment (Lambers and Clark, 1996). Prenatal exposure to nicotine is strongly associated with an increased incidence of attention deficit disorder (Langley et al., 2005; Schmitz et al., 2006). It has been suggested that there are many similarities between brain development in young postnatal rats and in the third-trimester human fetus. Our results suggest that prolonged exposure to nicotine during this developmental period would result in a small but persistent net activation of corticothalamic nicotinic currents but would lead to a lasting decrease in their activation by endogenous acetylcholine. In fact, our slice experiments likely underestimate the time required for recovery because complete washout of nicotine is rapid under our conditions. The effect of intermittent nicotine on the maturation of corticothalamic neurons is a critical target for future study to understand how nicotine exposure during development disrupts the prefrontal attention circuitry.
Footnotes
This work was supported by the Canadian Institutes of Health Research (Grant IHD-82660, to E.K.L.), the Canadian Foundation for Innovation (E.K.L.), the Canada Research Chairs program (E.K.L.), and the Kevin Hines Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression (E.K.L.). N.M.G. was supported by a Natural Sciences and Engineering Research Council of Canada Undergraduate Summer Research Award. We gratefully acknowledge the statistical advice of Dr. George Arhonditsis of the Department of Physical and Environmental Sciences, University of Toronto at Scarborough.
References
- Belluardo N, Mudò G, Blum M, Cheng Q, Caniglia G, Dell'Albani P, Fuxe K. The nicotinic acetylcholine receptor agonist (+/−)-epibatidine increases FGF-2 mRNA and protein levels in the rat brain. Brain Res Mol Brain Res. 1999;74:98–110. doi: 10.1016/s0169-328x(99)00266-1. [DOI] [PubMed] [Google Scholar]
- Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J, Jr, Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP, Goate AM. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008 doi: 10.1176/appi.ajp.2008.07111711. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, Marks MJ, Collins AC, Leonard S. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–364. doi: 10.1016/S0893-133X(00)00121-4. [DOI] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–915. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RW, Kolb B. Nicotine sensitization increases dendritic length and spine density in the nucleus accumbens and cingulate cortex. Brain Res. 2001;899:94–100. doi: 10.1016/s0006-8993(01)02201-6. [DOI] [PubMed] [Google Scholar]
- Brown RW, Collins AC, Lindstrom JM, Whiteaker P. Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J Neurochem. 2007;103:204–215. doi: 10.1111/j.1471-4159.2007.04700.x. [DOI] [PubMed] [Google Scholar]
- Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102:3817–3821. doi: 10.1073/pnas.0409782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Court JA, Martin-Ruiz C, Graham A, Perry E. Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat. 2000;20:281–298. doi: 10.1016/s0891-0618(00)00110-1. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, Ballabio A, Wanke E, Casari G. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275–276. doi: 10.1038/81566. [DOI] [PubMed] [Google Scholar]
- Díaz-Otero F, Quesada M, Morales-Corraliza J, Martínez-Parra C, Gómez-Garre P, Serratosa JM. Autosomal dominant nocturnal frontal lobe epilepsy with a mutation in the CHRNB2 gene. Epilepsia. 2007;49:516–520. doi: 10.1111/j.1528-1167.2007.01328.x. [DOI] [PubMed] [Google Scholar]
- Gioanni Y, Rougeot C, Clarke PB, Lepousé C, Thierry AM, Vidal C. Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur J Neurosci. 1999;11:18–30. doi: 10.1046/j.1460-9568.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- Granon S, Changeux JP. Attention-deficit/hyperactivity disorder: a plausible mouse model? Acta Paediatr. 2006;95:645–649. doi: 10.1080/08035250600719747. [DOI] [PubMed] [Google Scholar]
- Granon S, Faure P, Changeux JP. Executive and social behaviors under nicotinic receptor regulation. Proc Natl Acad Sci U S A. 2003;100:9596–9601. doi: 10.1073/pnas.1533498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- Kawai H, Lazar R, Metherate R. Nicotinic control of axon excitability regulates thalamocortical transmission. Nat Neurosci. 2007;10:1168–1175. doi: 10.1038/nn1956. [DOI] [PubMed] [Google Scholar]
- King SL, Marks MJ, Grady SR, Caldarone BJ, Koren AO, Mukhin AG, Collins AC, Picciotto MR. Conditional expression in corticothalamic efferents reveals a developmental role for nicotinic acetylcholine receptors in modulation of passive avoidance behavior. J Neurosci. 2003;23:3837–3843. doi: 10.1523/JNEUROSCI.23-09-03837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J. Roles of accessory subunits in {alpha}4{beta}2* nicotinic receptors. Mol Pharmacol. 2008;74:132–143. doi: 10.1124/mol.108.046789. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J Neurosci. 2005;25:5225–5229. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20:115–126. doi: 10.1016/s0146-0005(96)80079-6. [DOI] [PubMed] [Google Scholar]
- Langley K, Rice F, van den Bree MB, Thapar A. Maternal smoking during pregnancy as an environmental risk factor for attention deficit hyperactivity disorder behaviour. A review. Minerva Pediatr. 2005;57:359–371. [PubMed] [Google Scholar]
- Liang K, Poytress BS, Chen Y, Leslie FM, Weinberger NM, Metherate R. Neonatal nicotine exposure impairs nicotinic enhancement of central auditory processing and auditory learning in adult rats. Eur J Neurosci. 2006;24:857–866. doi: 10.1111/j.1460-9568.2006.04945.x. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB. Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology (Berl) 2006;184:292–305. doi: 10.1007/s00213-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Marini C, Guerrini R. The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol. 2007;74:1308–1314. doi: 10.1016/j.bcp.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Ruiz CM, Lee M, Perry RH, Baumann M, Court JA, Perry EK. Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res. 2004;123:81–90. doi: 10.1016/j.molbrainres.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, Perry E, Nordberg A. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat. 2001;22:115–126. doi: 10.1016/s0891-0618(01)00117-x. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- Mercer A, West DC, Morris OT, Kirchhecker S, Kerkhoff JE, Thomson AM. Excitatory connections made by presynaptic cortico-cortical pyramidal cells in layer 6 of the neocortex. Cereb Cortex. 2005;15:1485–1496. doi: 10.1093/cercor/bhi027. [DOI] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Seidler FJ, Eylers JP, Baker FE, Dobbins SS, Lappi SE, Slotkin TA. Effects of prenatal nicotine exposure on development of central and peripheral cholinergic neurotransmitter systems. Evidence for cholinergic trophic influences in developing brain. J Pharmacol Exp Ther. 1989;251:894–900. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. New York: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Perry EK, Lee ML, Martin-Ruiz CM, Court JA, Volsen SG, Merrit J, Folly E, Iversen PE, Bauman ML, Perry RH, Wenk GL. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry. 2001;158:1058–1066. doi: 10.1176/appi.ajp.158.7.1058. [DOI] [PubMed] [Google Scholar]
- Picard F, Bruel D, Servent D, Saba W, Fruchart-Gaillard C, Schöllhorn-Peyronneau MA, Roumenov D, Brodtkorb E, Zuberi S, Gambardella A, Steinborn B, Hufnagel A, Valette H, Bottlaender M. Alteration of the in vivo nicotinic receptor density in ADNFLE patients: a PET study. Brain. 2006;129:2047–2060. doi: 10.1093/brain/awl156. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380:347–351. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry. 2001;49:258–267. doi: 10.1016/s0006-3223(00)01094-5. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Bohlin G. Are maternal smoking and stress during pregnancy related to ADHD symptoms in children? J Child Psychol Psychiatry. 2005;46:246–254. doi: 10.1111/j.1469-7610.2004.00359.x. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Hum Dev. 1991;26:61–67. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M. The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol. 2003;63:1059–1066. doi: 10.1124/mol.63.5.1059. [DOI] [PubMed] [Google Scholar]
- Schlaepfer IR, Hoft NR, Collins AC, Corley RP, Hewitt JK, Hopfer CJ, Lessem JM, McQueen MB, Rhee SH, Ehringer MA. The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol Psychiatry. 2008;63:1039–1046. doi: 10.1016/j.biopsych.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M, Denardin D, Laufer Silva T, Pianca T, Hutz MH, Faraone S, Rohde LA. Smoking during pregnancy and attention-deficit/hyperactivity disorder, predominantly inattentive type: a case-control study. J Am Acad Child Adolesc Psychiatry. 2006;45:1338–1345. doi: 10.1097/S0890-8567(09)61916-X. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates. Toxicol Appl Pharmacol. 2004;198:132–151. doi: 10.1016/j.taap.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Amzica F. Sleep oscillations developing into seizures in corticothalamic systems. Epilepsia. 2003;44(Suppl 12):9–20. doi: 10.1111/j.0013-9580.2003.12006.x. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol. 2007;71:769–776. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Marguerat A, Raggenbass M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience. 2004;124:405–420. doi: 10.1016/j.neuroscience.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res. 1990;526:45–53. doi: 10.1016/0006-8993(90)90248-a. [DOI] [PubMed] [Google Scholar]
- Watson RE, Desesso JM, Hurtt ME, Cappon GD. Postnatal growth and morphological development of the brain: a species comparison. Birth Defects Res B Dev Reprod Toxicol. 2006;77:471–484. doi: 10.1002/bdrb.20090. [DOI] [PubMed] [Google Scholar]
- West DC, Mercer A, Kirchhecker S, Morris OT, Thomson AM. Layer 6 cortico-thalamic pyramidal cells preferentially innervate interneurons and generate facilitating EPSPs. Cereb Cortex. 2006;16:200–211. doi: 10.1093/cercor/bhi098. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol. 2005;481:19–30. doi: 10.1002/cne.20357. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]