

Abstract
Huntington disease (HD) is an autosomal dominant neurodegenerative disorder characterised by chorea, cognitive impairment, dementia and personality changes, caused by the expansion of a CAG repeat in the HD gene. Often, patients with a similar clinical presentation do not carry expansions of the CAG repeat in this gene [Huntington disease-like (HDL) patients]. We report the genetic analysis of 107 Portuguese patients with an HDL phenotype. The HDL genes PRNP and JPH3, encoding the prion protein and junctophilin-3, respectively, were screened for repeat expansions in these patients. Given the partial clinical overlap of SCA17, DRPLA and neuroferritinopathy with HD, their causative genes (TBP, ATN1, and FTL, respectively) were also analysed. Finally, repeat expansions in two candidate genes, CREBBP and POU3F2, which encode the nuclear transcriptional coactivator CREB-binding protein and the CNS-specific transcription factor N-Oct-3, respectively, were also studied. Expansions of the repetitive tracts of the PRNP, JPH3, TBP, ATN1, CREBBP and POU3F2 genes were excluded in all patients, as were sequence alterations in the FTL gene. Since none of the genes already included in the differential diagnosis of HD was responsible for the disease in our sample, the genetic heterogeneity of the HDL phenotype is still open for investigation.
Keywords: Chorea, Movement disorder, Transcription factors, Triplet repeats, Neuroferritinopathy
Introduction
Huntington disease (HD) is an autosomal dominant progressive neurodegenerative disorder, usually of adult onset, associated with involuntary choreic movements, motor problems, cognitive impairment and personality alterations. HD is caused by the expansion of a CAG repeat in the coding region of the HD gene, located on chromosome 4p16.3 (The Huntington Collaborative Research Group 1993). Normal alleles contain 6–35 CAGs and pathogenic alleles carry ≥36 CAGs (The American College of Medical Genetics 1998). However, some patients with Huntington disease-like (HDL) presentations do not carry any expansion of the (CAG)n tract in the HD gene. Sequence alterations in two loci responsible for a HDL phenotype (PRNP and JPH3) have already been identified: for PRNP (HDL1), a 192-nucleotide insertion within the prion protein gene (PRNP), localised in chromosome 20p (Xiang et al. 1998), that gives rise to an expanded prion protein with eight extra octapeptide repeats (Moore et al. 2001); for JPH3 (HDL2), a CAG/CTG repeat in the junctophilin-3 gene (JPH3) (Holmes et al. 2001), localised in chromosome 16q23–24 (Kambouris et al. 2000; Nishi et al. 2000). Other possible causative genes remain unknown, although a gene associated with an autosomal recessive transmission of HDL phenotype has been mapped to chromosome 4p15.3 (Kambouris et al. 2000), and some evidence suggests that CAG repeat expansions in unidentified genes could also be responsible for a similar clinical presentation (Margolis et al. 2001).
The CREB-binding protein (CBP) is a nuclear transcriptional coactivator protein that has been found in the characteristic neuronal intranuclear inclusions of patients with several polyglutamine expansion diseases (McCampbell et al. 2000; Nucifora et al. 2001). Point mutations in the CREBBP gene have also been associated with Rubinstein–Taybi syndrome (Petrij et al. 1995). Similarly to HD, SBMA and other neurodegenerative disease genes, the CREBBP gene contains a CAG repeat tract. The POU3F2 gene, encoding the central nervous system (CNS)-specific transcription factor N-Oct-3, is another gene also containing a CAG repeat in its coding region (Atanasoski et al. 1995). This makes CREBBP and POU3F2 interesting candidate genes for autosomal dominant late-onset neurodegenerative disorders.
In movement disorders there is some clinical overlap between different diseases. This is the case for some forms of autosomal dominant cerebellar ataxia, specifically spinocerebellar ataxia type 17 (SCA17) (Koide et al. 1999; Nakamura et al. 2001), and dentatorubropallidoluysian atrophy (DRPLA) (Koide et al. 1994), which may show clinical features similar to those of HD, namely dementia, chorea, Parkinsonism and dystonia. SCA17 is caused by an expansion of a CAA/CAG repeat segment on the TATA-binding protein gene (TBP), and DRPLA is also caused by an expansion of a (CAG)n in the gene encoding athrophin-1 (ATN1). Another example of a movement disorder sharing some clinical features with HD is neuroferritinopathy. This disorder is characterised by extrapyramidal dysfunction including choreoathetosis, dystonia, spasticity and rigidity, most often without cognitive decline (Curtis et al. 2001). The dominant mutations responsible for neuroferritinopathy are either small insertions in the coding region of the ferritin light polypeptide gene (FTL), causing frame shifts and originating proteins with different C-termini (Curtis et al. 2001), or missense mutations in conserved regions (Maciel et al. 2005).
As we have previously reported (Costa et al. 2003), among our series of cases for HD diagnosis, we found a high number of HD mutation-negative patients who had both a family history and typical HD clinical features. Here, we describe a series of 107 HDL patients of Portuguese origin, who do not present (CAG)n expansions in the HD gene. In order to find the genetic cause of the HDL phenotype in these patients, we investigated the presence of repeat expansions in the two described HDL loci (PRNP and JPH3) and in the TBP, ATN1, CREBBP and POU3F2 genes. Additionally, in search of potential sequence alterations, we have sequenced the entire coding region of the FTL gene, as well as its iron responsive element (IRE) region.
Materials and methods
Subjects
From all the HD diagnostic tests performed in our laboratory between 1998 and the end of December 2003 (requested mostly by neurologists for confirmation or exclusion of a clinical diagnosis), a total of 107 samples of unrelated Portuguese patients was negative for a CAG repeat expansion in the HD gene. After the study was reviewed and approved by the research ethics committee of the Institute for Molecular and Cell Biology, and informed consent of the patients was obtained, we included these patients in the current study. For a gross clinical characterisation of the disease we have considered one group of “movement disorder” patients with the following symptoms in their clinical record: chorea, involuntary movements, tremors, Parkinsonism, dystonia, pyramidal and/or extra-pyramidal signs; another group with a mainly “psychiatric disorder” with the features: dementia, behaviour alterations, psychosis, depression and/or suicidal thoughts; and a third group with both types of manifestations. The existence of a family history of a similar disease was ascertained, and registered as present, absent or unknown. Additional laboratory investigations performed by the clinicians were requested and registered when provided (20.6% of the cases under study).
Genetic analysis
Genomic DNA was extracted from blood lymphocytes by standard methods (Sambrook et al. 1989). Repeat expansions causing HD or other disorders with a similar clinical presentation were screened in the probands: the CAG/CTG or CAA/CAG repeats in the HD, TBP, ATN1 and JPH3 genes were amplified using the pairs of primers RS1/WP2 (Andrew et al. 1994a), TBP-F/TBP-R (Nakamura et al. 2001), B37CAG repeat primers (Koide et al. 1994) and L237-1/L237-2 (Holmes et al. 2001), respectively, using the published PCR conditions for each primer pair. For screening of the HD and ATN1 genes, a positive DNA control containing 94 and 66 CAGs, respectively, was included in each set of PCR reactions. According to the guidelines for the molecular diagnosis of HD (The American College of Medical Genetics 1998), in the case of normal homoallelic individuals (both alleles with the same repeat size) at the HD gene, the presence of high repeat expansions was first excluded by a second PCR reaction including both the CAG and the CCG repeats (Andrew et al. 1994b), and finally by genomic DNA southern blotting (Guida et al. 1996). The CAA/CAG repeat tracts on the CREBBP and POU3F2 genes were also studied by PCR amplification using the primer pairs CBP1 (5′GTACCGAGAAATGTTACG3′)/CBP2 (5′GCTGCTGGAACTGGCCGTG3′) and POU3F2_1 (5′GAGACGAGCTGCACGGGCCAG3′)/POU3F2_2(5′GGCGTGGTGCACCAGATGCG3′), respectively. The PCR reaction (similar to that used for the HD gene) was performed in a final volume of 12.5 μl, 200 μM dNTPs, 1.5 mM MgCl2, primers at 200 μM and 0.75 units of Taq DNA polymerase (Fermentas, St. Leon Rot, Germany), using an initial denaturation period of 5 min at 94 °C followed by 35 cycles of 1 min at 94 °C, 1 min at 56 °C (CREBBP) or 63 °C (POU3F2), 1 min at 72 °C, and an elongation for 5 min at 72 °C. All the PCR products containing the CAG or the CAA/CAG repeats were separated in a 6% denaturing polyacrylamide gel, transferred to a nylon membrane and hybridised with a (CAG)15 oligonucleotide previously labelled with 32P. Repeat sizes were visualised by autoradiography, by which no smears (corresponding to potential high repeat expansions) were observed in the normal homoallelic individuals. The octapeptide repeat on the PRNP gene and the FTL gene were studied using methods previously described by Beck et al. (2001) and Curtis et al. (2001), respectively.
Statistical analysis
Chi-squared analysis was used to test different gender proportions for each group of patients. Student’s t-test was used to compare mean age at onset in two groups (male/female, each pair of “type of disease” and each pair of “family history”) using a 95% confidence interval of the difference. A one-way ANOVA test was used to compare mean age at onset between subgroups of type of disorder or of family history. All these tests were performed using the statistic SPSS 11.0 package. A P-value < 0.05 was considered statistically significant.
Results
The 107 Portuguese patients for whom we have molecularly excluded HD were referred to our laboratory mainly by neurologists (91.4%). Three infantile cases (age at onset 3, 5 and 7 years) and one juvenile case (age at onset 16 years) of HDL disease were included in this series. Additional clinical biochemical and molecular analyses allowed the a posteriori establishment of a different diagnosis for four of the HDL patients: one case of CNS vasculitis, one case of primary hypoparathyroidism, and two cases of recessive mitochondrial encephalopathy.
The majority of patients presented both movement and psychiatric symptoms (ntotal =58; 54.3%) (Fig. 1), and most had a positive family history of the disease (n =48; 44.8%). The largest subgroup of patients in our HDL population was that with a family history (compatible with an autosomal dominant mode of transmission) of a movement and psychiatric disorder (n =27; 25.2% of the total series and 45.8% of the movement and psychiatric group; Fig. 1).
Fig. 1.
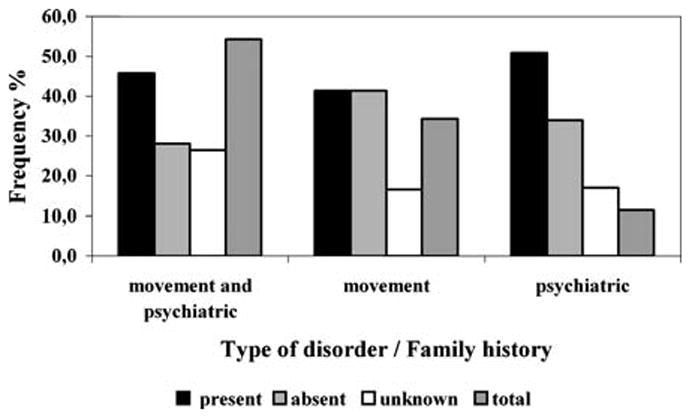
Type of disorder related with the family history of Portuguese Huntington disease-like (HDL) patients. Frequency of patients grouped by presence, absence or unknown family history for each type of disorder. Total Frequency of patients grouped by type of disorder
In relation to the type of disorder, the three groups of patients differed significantly in their mean age at onset (one-way ANOVA test, P = 0.011). This difference is due to a higher mean age at onset of patients with movement and psychiatric features (53.5±17.9 years) in relation to patients showing only a movement disorder (39.4±21.8 years)—Student’s t-test, P = 0.005—or to those presenting only psychiatric impairment (40.7±11.8 years)—Student’s t-test, P = 0.077 (Table 1). The age at onset of HDL patients did not vary depending on the existence of a present, absent or unknown family history. The gender proportions did not differ among all the groups characterised either by type of disorder, or by family history. However, the mean age at onset was significantly higher in female patients (54.1±18.1 years) than in males (38.5±19.0 years), P = 0.001 (Student’s t-test). In the subgroup of patients more represented in our HDL population (those with a family history of a movement and psychiatric disorder), the disease had a mean age at onset of 50.7±19.2 years (range: 20–78 years)—which can be considered the typical clinical presentation of HD (Table 1). In contrast, the smallest subgroup of patients (n =2) included those that showed only psychiatric alterations and an unknown family history (with a mean age at onset of 43.0 years) (Table 1).
Table 1.
Clinical features and family history of Huntington disease-like (HDL) patients
| Family history | Type of disorder |
Total (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Movement and psychiatric |
Movement |
Psychiatric |
|||||||||||
| % | Age at onset |
% | Age at onset |
% | Age at onset |
||||||||
| Range | Mean | SD | Range | Mean | SD | Range | Mean | SD | |||||
| Present | 24.8 | (20–78) | 50.7 | 19.2 | 14.3 | (5–80) | 47.3 | 21.5 | 5.7 | (25–54) | 37.8 | 12.1 | 44.8 |
| Absent | 15.2 | (25–83) | 57.7 | 18.4 | 14.3 | (3–74) | 35.8 | 22.0 | 3.8 | (33–58) | 45.5 | 17.7 | 33.3 |
| Unknown | 14.3 | (30–69) | 53.1 | 13.0 | 5.7 | (7–51) | 28.3 | 18.5 | 1.9 | (43) | 43.0 | – | 21.9 |
| Total | 54.3 | (20–83) | 53.5 | 17.9 | 34.3 | (3–80) | 39.4 | 21.8 | 11.4 | (25–58) | 40.7 | 11.8 | – |
By definition, we have excluded expansions of the CAG repeat in the HD locus for our entire HDL population; all patients presented normal or normal unstable alleles between 10 and 31 repeats. The two loci described as responsible for HDL disease—PRNP (HDL1) and JPH3 (HDL2)—were also excluded in our patients. No expansions of the octapeptide repeat were found in the PRNP gene. The CAG/CTG repeats in the JPH3 gene were all within the normal range, varying between 11 and 19 units, allele 14 being the most frequent (52.3%) followed by alleles with 16 repeats (27.6%). Homoallelism for the normal size was found in 35.5% of cases, most often (94.7%) for the commonest alleles (76.3% for allele 14, and 18.4% for allele 16); in these cases the corresponding band observed was always of higher intensity than those seen in cases of normal heteroallelism. Two other loci causing disorders with some clinical overlap with HD—TBP and ATN1—were also screened and no expansions of their repetitive tracts were found in the Portuguese HDL patients: alleles for the TBP gene varied between 29 and 41 triplets, the most frequent allele being the (CAA/CAG)38 repeat (34.1%), and 24.3% of cases were shown to be normal homoallelics (61.5% of which for allele 38); the (CAG)n at the ATN1 locus was distributed between 7 and 21 trinucleotides, with a peak at 15 CAGs (31.8%). The CAA/CAG repeat tracts in the coding regions of the CREBBP and POU3F2 genes were also screened for potential expansions. However, these repeat tracts were not polymorphic, showing 25 and 18 repeats in all patients, respectively. No smears (corresponding to potential large repeat expansions) were observed in the autoradiographs. Moreover, taking into account the similarity of clinical features between HD and neuroferritinopathy (Curtis et al. 2001), its causative gene—FTL—was entirely sequenced for all the HDL patients. No sequence alterations were observed either in the coding region, in the iron-responsive element (IRE) localised in the 5′ untranslated region, or in the acceptor or donor splice sites of the FTL gene.
Discussion
The availability of a genetic test for HD has allowed physicians not only to confirm the clinical diagnosis of patients with an established family history of HD, but also to exclude some cases presenting atypical symptoms or an unclear family history of the disease.
In this study we tried to characterise, clinically and genetically, a series of Portuguese patients with an HDL phenotype who did not carry a (CAG)n expansion in the HD locus. Our sample of HDL patients includes a large proportion of cases presenting both movement and psychiatric impairment, plus a family history of the disease (25.2%), which could be considered as true HD phenocopies.
In relation to the type of disorder, patients carrying both a movement disorder and psychiatric symptoms usually start to present the disease at a later age (53.5±17.9 years) than those presenting only one type of symptomatology (39.4±21.8 years for movement disorders, and 40.7±11.8 years for psychiatric manifestations). The cases of earliest onset of disease included in our study, three infantile and one juvenile case, all had an exclusively motor presentation, and could overlap with metabolic or mitochondrial disorders (which have been excluded), juvenile Parkinson’s disease or pantothenate kinase-associated neurodegeneration, which should be tested for these patients in the future.
Screening of the Portuguese HDL patients for octapeptide-insertions or CAG/CTG repeat expansions, respectively, for the PRNP and JPH3 loci did not reveal any expansion in these repetitive tracts. The insertion of octapeptides in the coding region of the PRNP gene has been reported in only one family (Moore et al. 2001; Xiang et al. 1998) and, as in the current report, other studies did not find this type of mutation in the PRNP gene (Bauer et al. 2004; Stevanin et al. 2003), which suggests that this must be a very rare genetic cause of disease in HDL patients. Nevertheless, the possible existence of HDL-causative point mutations in the PRNP gene in our sample remains open. Our results regarding the absence of CAG/CTG repeat expansions in the JPH3 gene are also in agreement with other reports (Bauer et al. 2002; Stevanin et al. 2002), suggesting that the mutation responsible for HDL2 is not found, so far, in Caucasian patients. The fact that this mutation was found only in patients of African ancestry could be explained by a common origin of the mutation or by a genetic predisposition for expansion in this specific population (Holmes et al. 2001; Stevanin et al. 2003). Analysis of the JPH3 locus should be of particular interest in patients of African origin with an HDL phenotype who do not present a CAG repeat expansion in the HD gene.
In contrast to other recent reports, showing that CAA/CAG repeat expansions at the TBP gene are responsible for HDL phenotype (Bauer et al. 2004; Cellini et al. 2004; Stevanin et al. 2003; Toyoshima et al. 2004), we did not find any repeat larger than 41 triplets, and until now only one intermediate allele carrying 43 repeats has been found in a Portuguese patient presenting ataxia and mental retardation (Silveira et al. 2002). This suggests that such mutations will be a rare cause of disease in our population.
Given the partial clinical overlap between DRPLA and HD and the finding of four Portuguese DRPLA families, all sharing the same haplotype with the Japanese patients (Martins et al. 2003), we determined the (CAG)n tract size on the ATN1 gene for all our HDL patients, but no expansions of this tract were found. This result is in accordance with other studies reporting no (CAG)n expansions in the ATN1 gene in HDL patients (Stevanin et al. 2002, 2003), showing that this mutation is rare in patients of non-Asian origin.
In order to exclude further mutations that could be responsible for movement disorders sharing some clinical presentation with HD, we sequenced the entire FTL gene, the gene responsible for neuroferritinopathy (Curtis et al. 2001). The adenine insertion at nucleotides 460–461, as well as other possible sequence alterations in the entire coding region of the gene and in the 5′ IRE-comprising region, were excluded in our sample. This is, to our knowledge, the first report of a large series of HDL patients in whom mutation of the FTL gene has been excluded. The results suggest that neuroferritinopathy will rarely overlap with HD, or that neuroferritinopathy is rare in the Portuguese population; we have previously identified one neuroferritinopathy family of gypsy origin, carrying a missense mutation in the FTL gene (Maciel et al. 2005).
Our study also demonstrated the first exclusion of possible expansions of the CAG repeat tracts in two candidate genes—CREBBP and the POU3F2—in patients with an HDL phenotype; these repeat tracts revealed to be stable in our series of Portuguese patients, although mutations in these genes other than expanded repetitive tracts may potentially still be responsible for the HDL presentation. Additional improvement of the detection of repeat expansions in the HDL genes and in other candidate genes is still needed.
In conclusion, in our series of HDL patients, we have excluded mutations in: (1) the described HDL loci (PRNP and JPH3); (2) genes responsible for movement disorders with clinical features similar to HD, namely the TBP, ATN1 and the FTL genes; and (3) novel candidate genes containing CAG repeat tracts—CREBBP and POU3F2. None of the genes already included in the differential diagnosis of HD was responsible for the disease in our sample, nor were any of the other possible causative genes we tested. Given previous similar reports and the high clinical heterogeneity of these patients, we suggest that the JPH3 gene should be analysed in patients of African origin, the ATN1 gene in Asian patients, and the TBP gene in patients presenting some cerebellar symptoms. Overall, the genetic cause of disease in patients showing typical HD symptoms and a family history of the disease (representing 24.5% of our population) remains unknown, suggesting that HDL disorders are clinically and genetically heterogeneous.
Acknowledgments
We would like to thank all the patients and neurologists who collaborated in this study. This project was supported by Fundação para a Ciência e a Tecnologia (FCT) and FEDER (grant CBO/33485/99). Maria do Carmo Costa, Andreia Teixeira-Castro and Marco Constante are the recipients of scholarships from FCT: SFRH/BD/9759/2003, SFRH/BI/11844/2003, and BIC included in grant CBO/33485/99, respectively.
Contributor Information
Maria do Carmo Costa, Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal.
Andreia Teixeira-Castro, Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal.
Marco Constante, Department of Medicine, CHUM, Université de Montreal Hôpital Notre-Dame, Montreal, Canada.
Marina Magalhães, Hospital Geral de Sto. António, Porto, Portugal.
Paula Magalhães, UnIGENe, Institute for Molecular and Cell Biology, Porto, Portugal.
Joana Cerqueira, UnIGENe, Institute for Molecular and Cell Biology, Porto, Portugal.
José Vale, Hospital Egas Moniz, Lisboa, Portugal.
Vitorina Passão, Hospital Júlio de Matos, Lisboa, Portugal.
Célia Barbosa, Hospital Pedro Hispano, Matosinhos, Portugal.
Conceição Robalo, Hospital Pediátrico de Coimbra, Coimbra, Portugal.
Paula Coutinho, Hospital de São Sebastião, Santa Maria da Feira, Portugal.
José Barros, Hospital Geral de Sto. António, Porto, Portugal.
Manuela M. Santos, Department of Medicine, CHUM, Universitéde Montreal Hôpital Notre-Dame, Montreal, Canada, UnIGENe, Institute for Molecular and Cell Biology, Porto, Portugal
Jorge Sequeiros, UnIGENe, Institute for Molecular and Cell Biology, Porto, Portugal, Department of Population Studies, ICBAS, University of Porto, Porto, Portugal.
Patrícia Maciel, Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal.
References
- Andrew SE, Goldberg YP, Kremer B, Squitieri S, Theilmann J, Zeisler J, Telenius H, Adam S, Almqvist EW, Anvret M, Lucotte G, Stoessl AJ, Campanella G, Hayden MR. Huntington disease without CAG expansion: phenocopies or errors in assignment? Am J Hum Genet. 1994a;54:852–863. [PMC free article] [PubMed] [Google Scholar]
- Andrew SE, Goldberg YP, Theilmann J, Zeisler J, Hayden MR. A CCG repeat polymorphism adjacent to the CAG repeat in the Huntington disease gene: implications for diagnostic accuracy and predictive testing. Hum Mol Genet. 1994b;3:65–67. doi: 10.1093/hmg/3.1.65. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Toldo SS, Malipiero U, Schreiber E, Fries R, Fontana A. Isolation of the human genomic brain-2/N-Oct 3 gene (POUF3) and assignment to chromosome 6q16. Genomics. 1995;26:272–280. doi: 10.1016/0888-7543(95)80211-4. [DOI] [PubMed] [Google Scholar]
- Bauer I, Gencik M, Laccone F, Hartmut P, Weber BHF, Holinski-Feder E, Weirich H, Moris-Rosendahl DJ, Rolfs A, Gencikova A, Bauer P, Wenning GK, Epplen JT, Holmes SE, Margolis RL, Ross CA. Trinucleotide repeat expansions in the junctophilin-3 gene are not found in Caucasian patients with Huntington’s disease-like phenotype. Ann Neurol. 2002;51:662. doi: 10.1002/ana.10184. [DOI] [PubMed] [Google Scholar]
- Bauer P, Laccone F, Rolfs A, Wullner U, Bosch S, Peters H, Liebscher S, Scheible M, Epplen JT, Weber BHF, Holinski-Feder E, Weirich-Schwaiger H, Moris-Rosendahl DJ, Andrich J, Riess O. Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington’s disease-like phenotype. J Med Genet. 2004;41:230–232. doi: 10.1136/jmg.2003.015602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JA, Mead S, Campbell TA, Dickinson A, Wientjens DPMW, Croes EA, Van Duijn CM, Collinge J. Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology. 2001;57:354–356. doi: 10.1212/wnl.57.2.354. [DOI] [PubMed] [Google Scholar]
- Cellini E, Forleo P, Nacmias B, Tedde A, Bagnoli S, Piacentini S, Sorbi S. Spinocerebellar ataxia type 17 repeat in patients with Huntington’s disease-like and ataxia. Ann Neurol. 2004;56:163. doi: 10.1002/ana.20146. [DOI] [PubMed] [Google Scholar]
- Costa MC, Magalhães P, Ferreirinha F, Guimarães L, Januário C, Gaspar I, Loureiro L, Vale J, Garrett C, Regateiro F, Magalhães M, Sousa A, Maciel P, Sequeiros J. Molecular diagnosis of Huntington disease in Portugal: implications for genetic counselling and clinical practice. Eur J Hum Genet. 2003;11:872–878. doi: 10.1038/sj.ejhg.5201055. [DOI] [PubMed] [Google Scholar]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- Guida M, Fenwick RG, Papp AC, Snyder PJ, Sedra M, Prior TW. Southern transfer protocol for confirmation of Huntington disease. Clin Chem. 1996;42:1711–1712. [PubMed] [Google Scholar]
- Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;4:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- Kambouris M, Bohlega S, Al-Tahan A, Meyer BF. Localization of the gene for a novel autosomal recessive neurodegenerative Huntington-like disorder to 4p15.3. Am J Hum Genet. 2000;66:445–452. doi: 10.1086/302744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, Saito M, Tomoda A, Miike T, Naito H, Ikuta F, Tsuji S. Unstable expansion of CAG repeat in hereditary dentatorubropallidoluysian atrophy (DRPLA) Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047–2053. doi: 10.1093/hmg/8.11.2047. [DOI] [PubMed] [Google Scholar]
- Maciel P, Cruz V, Constante-Pereira M, Iniesta I, Costa MC, Gallati S, Sousa N, Sequeiros J, Coutinho P, Santos MM. Neuroferritinopathy: missense mutation in FTL associated with early-onset bilateral pallidal involvement. Neurology. 2005;65:603–605. doi: 10.1212/01.wnl.0000178224.81169.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol. 2001;50:373–380. doi: 10.1002/ana.1312. [DOI] [PubMed] [Google Scholar]
- Martins S, Matamá T, Guimarães L, Vale J, Guimarães J, Ramos L, Coutinho P, Sequeiros J, Silveira I. Portuguese families with dentatorubropallidoluysian atrophy (DRPLA) share a common haplotype of Asian origin. Eur J Hum Genet. 2003;11:808–811. doi: 10.1038/sj.ejhg.5201054. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson HL, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, Anvret M, Prusiner SB. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385–1388. doi: 10.1086/324414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Jeong S-Y, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein–Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsh EF, Maniatis T. Molecular cloning—a laboratory manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Silveira I, Miranda C, Guimarães L, Moreira MC, Alonso I, Endonça P, Ferro A, Pinto-Basto J, Coelho J, Ferreirinha F, Poirier J, Parreira E, Vale J, Januário C, Barbot C, Tuna A, Barros J, Koide R, Tsuji S, Holmes SE, Margolis RL, Jardim L, Pandolfo M, Coutinho P, Sequeiros J. Trinucleotide repeats in 202 families with ataxia: a small expanded (CAG)n allele at the SCA17 locus. Arch Neurol. 2002;59:623–629. doi: 10.1001/archneur.59.4.623. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Camuzat A, Holmes SE, Julien C, Sahoul R, Dodé C, Hahn-Barma V, Ross CA, Margolis RL, Durr A, Brice A. CAG/CTG repeat expansions at the Huntington’s disease-like 2 locus are rare in Huntington’s disease patients. Neurology. 2002;58:965–967. doi: 10.1212/wnl.58.6.965. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dodé C, Takahashi J, Sân C, Bellance R, Brice A, Durr A. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599–1603. doi: 10.1093/brain/awg155. [DOI] [PubMed] [Google Scholar]
- The American College of Medical Genetics, American Society of Human Genetics, Huntington Disease Genetic Testing Working Group. Laboratory guidelines for Huntington Disease genetic testing. Am J Hum Genet. 1998;62:1243–1247. [PMC free article] [PubMed] [Google Scholar]
- The Huntington Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Toyoshima Y, Yamada M, Onodera O, Shimohata M, Inenaga C, Fujita N, Morita M, Tsuji S, Takahashi H. SCA17 homozygote showing Huntington’s disease-like phenotype. Ann Neurol. 2004;55:281–286. doi: 10.1002/ana.10824. [DOI] [PubMed] [Google Scholar]
- Xiang F, Almqvist EW, Huq M, Lundin A, Hayden MR, Edström L, Anvret M, Zhang Z. A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet. 1998;63:1431–1438. doi: 10.1086/302093. [DOI] [PMC free article] [PubMed] [Google Scholar]