

Abstract
Lipocalin 2 (Lcn2), a mammalian protein that is expressed and secreted in various pathologic states, binds siderophores, which are high-affnity iron chelators. Besides its role in limiting iron availability to pathogens in the setting of bacterial infection, Lcn2:siderophore complexes can also deliver iron to cells. In this study, we examined Lcn2 regulation in the liver of mice in situations of increased iron utilization, namely, during anemia. Anemia induced by phlebotomy, iron deprivation, or phenylhydrazine treatment was associated with upregulation of Lcn2 gene expression in the liver and elevation of serum Lcn2 protein levels. We further explored the participation of several factors known to co-occur during anemia, including hypoxia, changes in iron levels, and erythropoietic drive, in the regulation of Lcn2 by anemia. We found that hypoxia, but not iron or erythropoietin, caused an induction of Lcn2 expression. The upregulation of Lcn2 levels by anemia and hypoxia, which is not directly mediated by iron or erythropoietin, suggests a possible physiological role for Lcn2 during increased iron utilization and mobilization from stores.
Keywords: Lipocalin 2, Anemia, Hypoxia, Erythropoietin, Iron
Introduction
Iron is an essential cofactor of numerous proteins and enzymes that are critical for cell proliferation, respiration, and signal transduction. However, free iron can also be toxic because of its capacity to induce the formation of dangerous free radicals. Thus, both systemic and cellular iron balance have to be tightly maintained. Biological systems have evolved various strategies to deal with the difficulty of acquiring iron, storing it and, at the same time, avoiding its toxic potential. Each of these tasks is accomplished via the coordinated regulation of specialized molecules involved in iron absorption, transport, cellular uptake and storage [1].
In normal situations, cellular iron uptake occurs through the transferrin (Tf)-dependent pathway, involving Tf binding to transferrin receptor-1 (TfR1) on the cell membrane and its internalization by receptor-mediated endocytosis [2]. However, in conditions of altered homeostasis, iron can also be delivered to cells by alternative mechanisms, which are Tf-independent [1].
More recently, an iron-delivery pathway was identified that involves a member of the lipocalin superfamily [3]: antimicrobial protein neutrophil gelatinase-associated lipocalin (NGAL) or lipocalin 2 (Lcn2) [4].
NGAL/Lcn2 was first identified as a 25-kDa glycoprotein associated with purified human neutrophil gelatinase [5]. Homologous proteins were also identified in the mouse (Lcn2/24p3/uterocalin) and rat (α2- microglobulin-related protein/neurelated lipocalin) [6]. Lcn2 can bind catecholate-type bacterial ferric siderophores [7] (e.g. enterobactin), which are low molecular compounds that bind ferric iron and are able to acquire iron from mammalian iron-binding proteins, including Tf and lactoferrin [8]. Since Lcn2 binds enterobactin with higher affinity than the E. coli enterobactin transporter, it effectively interferes with bacterial iron uptake and, in fact, acts as a bacteriostatic agent [7].
In addition to its role as a bacteriostatic agent, Lcn2 has been shown to mediate a Tf-independent iron uptake pathway activated during kidney development [9]. The iron delivered to cells by Lcn2 is capable of regulating iron-dependent genes, such as ferritin and TfR1 [9,10], that are sensitive to cellular iron status [11], indicating that cells can utilize the iron provided by Lcn2.
While growing evidence supports the involvement of Lcn2 in a Tf-independent iron uptake pathway activated during early development, it remains unclear whether this pathway may become activated in response to disrupted iron homeostasis during adulthood.
In this study, we examined Lcn2 regulation in situations of disrupted iron homeostasis, namely, during anemia. We further explored separately the role of several factors known to co-occur during anemia, including hypoxia, changes in iron levels, and erythropoietic drive, in the regulation of Lcn2 by anemia.
Materials and methods
Animals
All procedures were performed in accordance with guidelines of the Canadian Council on Animal Care and were approved by the Institutional Animal Care Committee of the Centre Hospitalier de l’Université de Montréal (CHUM). C57BL/6 female mice aged 3 or 8 weeks were purchased from Charles River Laboratories, Inc. (Wilmington, MA). All mice were kept under strict specific pathogen-free conditions.
Animal treatments
Control mice were fed a commercial diet containing approximately 226 mg of iron per kg (Teklad Global 18% protein rodent diet TD 2018, Harlan Teklad, Madison, WI). Dietary iron overload was produced by giving 8-week-old mice the same commercial diet supplemented with 25 g carbonyl iron (Sigma-Aldrich, St. Louis, MO) per kg (TD 02030) for 4 weeks. Iron deficiency was induced by feeding 3-week-old mice the same commercial diet deficient in iron containing less than 3 mg of iron per kg (TD 80396) for 9 weeks. All mice were 12 weeks old at the time of sacrifice.
To induce anemia through phlebotomy, 0.25 mL of blood was extracted by retro-orbital puncture from anesthetized mice. The procedure was repeated 24 h later, and the animals were sacrificed 16 h after the last phlebotomy.
Hemolytic anemia was produced by i.p. administration of 40 mg/kg body weight of phenylhydrazine (PHZ, Sigma-Aldrich), once daily for 4 days. The mice were sacrificed the following day.
Cobalt chloride (CoCl2)-induced hypoxia was elicited by i.p. injection of 60 mg/kg of CoCl2 (Sigma-Aldrich) dissolved in 0.9% saline. Control mice were injected with an equivalent volume of saline. All mice were sacrificed 24 h after injection.
Normobaric hypoxia was established by diluting ambient air with nitrogen in a specially ventilated chamber, in which N2-enriched air supply was controlled with an O2 sensor-driven inlet valve. The mice were maintained in the chamber and exposed to normobaric hypoxia (10% O2) for 1, 3, and 5 days. An oxygen analyzer was used to monitor oxygen concentration in the hypoxic chamber. The control mice were kept under normoxia (room air) in the same room in which the hypoxic chamber was placed.
Erythropoiesis was induced by treating mice with 50 U of human biosynthetic erythropoietin-α (EPO) (epoetin alfa, Ortho Biotech, Bridgewater, NJ) dissolved in phosphate-buffered saline (PBS). The mice were injected i.p. daily for 4 days and were sacrificed on day 5. Control mice were similarly injected with an equivalent volume of PBS.
As a positive control for Lcn2 induction in the liver [12], mice were injected with lipopolysaccharide (LPS, Escherichia coli serotype 055: B5 – 1 mg/kg i.p., Sigma-Aldrich). Control mice were injected with an equivalent volume of saline solution. All animals were sacrificed 6 h after the injection.
Hematological measurements and transferrin saturation
EDTA-treated blood samples were obtained by orbital puncture under anesthesia. Red blood cell (RBC) count, hemoglobin (Hb), hematocrit (HCT) and mean corpuscular volume (MCV) were measured in an ABC vet counter (ABX hématologie, Montpellier, France). Serum iron, total iron-binding capacity (TIBC) and transferrin saturation were assessed by colorimetric assay [13], with the Kodak Ektachem DT60 system (Johnson & Johnson, Ortho Clinical Diagnostics, Mississauga, ON, Canada).
Measurement of liver iron concentration
Liver iron concentration was assessed by acid digestion of tissue samples, followed by iron quantification with atomic absorption spectroscopy [13].
Quantitative reverse transcription-polymerase chain reaction (RT-PCR)
Liver total RNA was isolated with Trizol reagent (Invitrogen, Burlington, ON, Canada), and RT was performed with the Thermoscript RT-PCR system (Invitrogen). Lcn2, hepcidin, and β-actin mRNA levels were measured by real-time PCR in a Rotor Gene 3000™ Real Time DNA Detection System (Montreal Biotech Inc., Kirkland, QC, Canada), with the QuantiTect SYBRGreen I PCR kit (Qiagen, Mississauga, ON, Canada) [14]. The primers were 5′-CCCATCTCTGCTCACTGTCC-3′ and 5′-TTTTTCTGGACCGCATTG-3′ for Lcn2, 5′-AGAGCTGCAGCCTTTGCAC-3′ and 5′-GAAGATGCAGATGGGGAAGT-3′ for hepcidin, and 5′-TGTTACCAACTGGGACGACA-3′ and 5′-GGTGTTGAAGGTCTCAAA-3′ for β-actin. Lcn2 and hepcidin expressions levels were normalized to the housekeeping gene β-actin.
Western blot analysis
1.5 μL of serum was boiled in loading buffer containing 4% sodium dodecyl sulfate (SDS), 20% glycerol, and bromophenol blue for 5 min. Proteins were resolved on 12% SDS-polyacrylamide gel and transferred onto nitrocellulose membranes (Amersham Biosciences, Baie d’Urfé, Québec, Canada). The membranes were blocked with 6% nonfat dry milk solution and incubated with anti-sip24/Lcn2 antibody [12] (a gift from Dr. Marit Nilsen-Hamilton, Iowa State University). To detect immunocomplexes formed, peroxidase-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., Mississauga, Ontario, Canada) was used as secondary antibody. Staining intensity was developed with the chemioluminescence system from Amersham Biosciences.
Statistical analysis
Student’s t test (unpaired, 2-tailed) was used for comparison between treatments. P<0.05 was considered significant.
Results
Lcn2 expression is induced by acute anemia
To examine whether Lcn2 is modulated in situations of increased iron utilization, anemia was induced in mice through repeated phlebotomies. This treatment led to a significant reduction of Hb and HCT values by 63% and 67%, respectively (Table 1).
Table 1.
Erythroid parameters
| Treatments |
|||||
|---|---|---|---|---|---|
| Control (n=6) | Phlebotomy (n=6) | Iron deficiency (n=6) | Phenylhydrazin (n=6) | EPO (n=8) | |
| Hb, g/dL | 13.7±0.5 | 5.1±0.6** | 12.4±0.5* | 10.2±0.9** | 15.0±0.5** |
| HCT, % | 43.9±1.6 | 14.3±1.3** | 38.1±1.2** | 13.0±1.7** | 52.3±1.7** |
Data are presented as means±SD.
P<0.005 compared to control mice.
P<0.0001 compared to control mice.
Lcn2 mRNA was dramatically increased in the liver of phlebotomized mice (Fig. 1a) compared to control mice. Conversely, and in agreement with previously-reported data [15], expression of the iron regulator hepcidin, used here as a control for the treatments, was reduced by 80% in the anemic mice (Fig. 1b). In addition, Lcn2 protein was readily detected by Western blot analysis in the sera of phlebotomized mice (Fig. 1c). These results demonstrate that Lcn2 expression is strongly evoked during acute anemia.
Fig. 1.
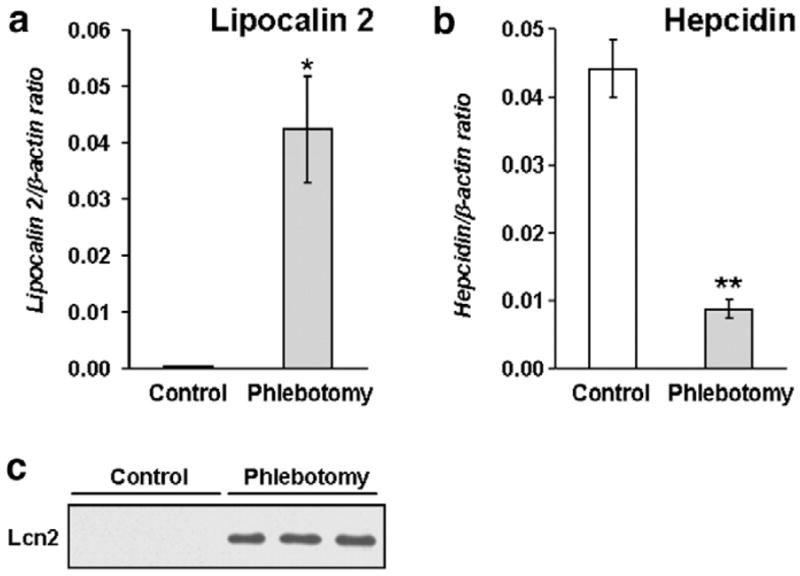
Lipocalin 2 (Lcn2) expression is induced by acute anemia. Lcn2 (a) and hepcidin (b) mRNA levels measured in the liver of control and phlebotomized mice by real-time PCR. The results are presented as means±SEM, n=6 per group. Student’s t test. *P<0.001 and **P<0.0001. (c) Lcn2 in serum as detected by Western blotting.
Lcn2 expression is induced during iron-deficiency anemia
Next, we examined whether iron-deficiency anemia would similarly affect Lcn2 expression. For this purpose, mice were fed an iron-deficient diet for 9 weeks, which led to the development of mild anemia and significant depletion of body iron stores, as revealed by modest but statistically significant decreases in Hb and HCT (Table 1), serum iron and transferrin saturation (Table 2). In addition, iron concentrations in both the liver and spleen were significantly reduced (Fig. 2a).
Table 2.
Serum iron (SI) and transferrin saturation (TS) in iron-deficient and iron-loaded mice
| Treatment | SI, μM | TS, % |
|---|---|---|
| Control (n=6) | 22±4 | 55±8 |
| Iron deficiency (n=6) | 18±2* | 35±7* |
| Carbonyl iron (n=6) | 39±13** | 96±7** |
Data are presented as means±SD.
P<0.05 compared to control mice.
P<0.0001 compared to control mice.
Fig. 2.

Lipocalin 2 (Lcn2) expression is induced during iron-deficiency anemia. (a) Iron concentrations in the liver and spleen of control mice and mice with iron-deficiency anemia (Fe-def). (b) Lcn2 and (c) hepcidin mRNA levels in the liver of control and iron-deficient anemic mice determined by real-time PCR. The results are presented as means±SEM, n=6 per group. Student’s t test: *P<0.05; **P<0.001. (d) Lcn2 in serum detected by Western blotting.
Lcn2 mRNA levels were found to be elevated by 2.6-fold in the liver (Fig. 2b), while hepcidin levels were diminished to 17% of control values (Fig. 2c). Accordingly, Lcn2 protein levels, detected by Western blotting, were increased in the serum of mice with iron-deficiency anemia (Fig. 2d). These results further demonstrate that Lcn2 production is induced by anemia.
Lcn2 expression is not affected by serum iron and liver iron levels
Both phlebotomy-induced and iron-deficiency anemia were associated with a reduction of serum iron and tissue iron levels. To determine whether reduced iron levels in serum or in tissues were responsible for the elevation of Lcn2 expression, acute anemia was induced by PHZ-treatment. In this model, unlike in phlebotomized or iron-deficient mice, serum iron, transferrin saturation and hepatic iron levels rose significantly by 2- to 3-fold over control values, as PHZ administration resulted in a large influx of iron into liver Kupffer cells following massive hemolysis (data not shown). Hb declined by 25% and HCT values decreased by 70% (Table 1).
We found that Lcn2 mRNA was dramatically increased in the liver of PHZ-treated mice compared to the controls (Fig. 3a). Conversely, and as expected, hepcidin expression was reduced by 72% in PHZ-treated mice (Fig. 3b). Lcn2 protein was readily detected by Western blot analysis in the sera of PHZ-treated mice (Fig. 3c).
Fig. 3.
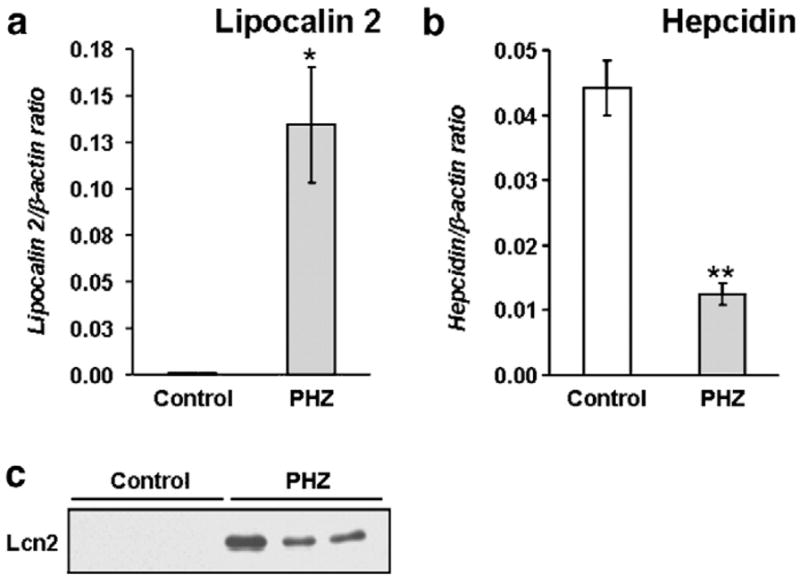
Lipocalin (Lcn2) expression is induced in mice with hemolytic anemia. (a) Lcn2 and (b) hepcidin mRNA content in the liver of control and phenylhydrazine (PHZ)-treated mice determined by real-time PCR. The results are presented as means±SEM, n=6 per group. Student’s t test: *P<0.01, **P<0.001. (c) Lcn2 in serum detected by Western blotting.
In vitro, Lcn2 expression has been shown to be regulated by intracellular iron levels [16]. To further determine whether iron itself may regulate Lcn2 expression in vivo, mice were fed an iron-enriched diet (2.5% carbonyl iron) for 4 weeks. This treatment led to an elevation of serum iron and transferrin saturation (Table 2). Iron-loading was further demonstrated by measuring liver iron concentrations, which were found to be 4 times higher in mice placed on the iron-enriched diet (Fig. 4a). Iron levels in the spleen were also significantly elevated (Fig. 4a).
Fig. 4.

Lipocalin (Lcn2) expression is not affected by serum and liver iron levels. (a) Iron concentrations in the liver and spleen of control and dietary iron-loaded mice (2.5% carbonyl iron – CI). Liver mRNA levels of (b) Lcn2 and (c) hepcidin in mice fed a standard diet (control) or CI-supplemented diet. The results are presented as means±SEM, n=6 per group. Student’s t test: *P<0.05; **P<0.0001.
Lcn2 mRNA expression remained unchanged in the liver (Fig. 4b) and spleen (data not shown) of mice fed the iron-enriched diet compared to mice kept on the standard diet. On the other hand, liver mRNA levels of hepcidin were significantly enhanced by iron-loading (Fig. 4c).
Taken together, these results show that serum and tissue iron levels are unlikely to account for the regulation of hepatic Lcn2 in situations of disrupted iron homeostasis.
Lcn2 expression is stimulated by hypoxia
Anemia results in tissue hypoxia, which is a known activator of the expression of many genes involved in iron metabolism, such as transferrin [17], TfR [18,19], ferritin [20], and ceruloplasmin [21]. To evaluate the effect of hypoxia on Lcn2 gene expression, mice were treated with CoCl2, a chemical inducer of hypoxia-like responses. As shown in Fig. 5a, an 11.6-fold increase in liver mRNA Lcn2 expression was observed 24 h after CoCl2 treatment, and an elevation of plasma Lcn2 protein levels was confirmed by Western blotting (Fig. 5b). Conversely, hepcidin gene expression was strongly down-regulated (Fig. 5c), as expected.
Fig. 5.

Lipocalin 2 (Lcn2) expression is stimulated by hypoxia. (a and d) Lcn2 and (c and f) hepcidin mRNA expression determined by real-time PCR in cobalt chloride (CoCl2)-treated mice and in mice exposed to 10% oxygen for 1, 3 or 5 days. The results are presented as means±SEM, n=6–8 per group. Student’s t test: *P<0.01, **P<0.001 and ***P<0.0001. (b and e) Lcn2 in serum detected by Western blotting.
To further test Lcn2 upregulation by hypoxia, mice were exposed to normobaric hypoxia (10% O2) for 1, 3 and 5 days before being analyzed. Hypoxia stimulated erythropoiesis, as judged by the elevated hematological indices when compared to control, normoxic mice (data not shown). In addition, hypoxia induced a decrease in circulating iron levels, as assessed by measuring serum iron and transferrin saturation, while iron concentration in the liver was increased (Table 3). As illustrated in Figs. 5d and e, Lcn2 was induced both at the mRNA and protein levels, at day 1, and returned to normal levels by day 3. Suppression of hepcidin expression was observed at days 3 and 5 (Fig. 5f).
Table 3.
Liver iron, serum iron (SI) and transferrin saturation (TS) in hypoxic mice
| Treatment | Liver iron |
||
|---|---|---|---|
| (μg iron/g dry weight) | SI, μM | TS, % | |
| Control (n=6) | 244±20 | 23±3 | 57±7 |
| Hypoxia D1 (n=6) | 298±14* | 15±4* | 36±11* |
| Hypoxia D3 (n=6) | 303±37* | 13±2* | 28±6* |
| Hypoxia D5 (n=6) | 294±31* | 16 ± 2* | 34 ± 6* |
Data are presented as means±SD.
P<0.01 compared to control mice.
These results indicate that tissue oxygenation may be a factor in Lcn2 gene regulation by anemia.
Induction of Lcn2 by anemia/hypoxia does not require increased erythropoiesis and is not directly mediated by EPO
Anemia and hypoxia induce EPO production, which, in turn, stimulates erythropoiesis [22]. To assess whether increased erythropoiesis is required for the induction of Lcn2 during anemia/hypoxia, mice were treated with EPO or, as positive controls, with LPS, a known Lcn2 inducer [23]. EPO treatment stimulated erythropoiesis, judging by the elevation of Hb and HCT (10% and 19% increase, respectively; Table 1). Moreover, a 2.4-fold increase in the spleen/body weight ratio was observed. Despite this increment of erythropoiesis activity, Lcn2 expression remained at similar levels, as seen in control mice (Fig. 6a). As expected [24], hepcidin expression was suppressed by EPO and stimulated by LPS (Fig. 6b). These results indicate that the induction of Lcn2 expression by anemia/hypoxia does not require increased erythropoiesis and is not directly mediated by EPO.
Fig. 6.
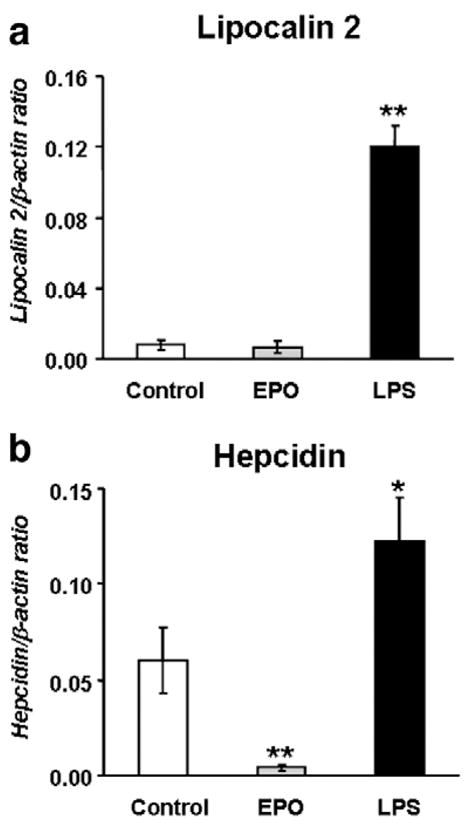
Erythropoietin is not involved in the induction of lipocalin 2 (Lcn2) by anemia/hypoxia. Lcn2 (a) and hepcidin (b) mRNA levels in the liver of controls and mice treated with erythropoietin (EPO) or lipopolysaccharide (LPS) determined by real-time PCR. The results are presented as means±SEM, n=6–8 per group. Student’s t test. *P<0.001 and **P<0.0001.
Discussion
Lcn2, an inducible protein, has been shown to bind to various iron-loaded bacterial siderophores and consequently to decrease iron availability for bacterial growth [7]. Accordingly, Lcn2 knockout mice have a profound defect in defense against E. coli challenge [23], further demonstrating the important role of Lcn2 in the innate immune response to bacterial infection by sequestering iron. Additional in vitro studies indicate that Lcn2 mediates a Tf-independent iron-delivery pathway to developing cells and, in fact, acts as an iron-trafficking protein (reviewed in [25]).
In this study, we investigated the regulation of Lcn2 expression in situations of increased iron utilization. Since the liver is the major iron storage organ from where iron can be mobilized when the erythropoietic drive is heightened, we examined the regulation of Lcn2 expression in the livers of anemic mice in vivo. We showed that Lcn2 expression in the liver was induced in mice after repeated phlebotomies as well as in iron-deficient mice. These data demonstrate that Lcn2 is stimulated by anemia.
To determine how anemia regulates Lcn2, and because phlebotomized and iron-deficient mice develop anemia with a fall in serum and tissue iron levels, tissue hypoxia, increased EPO and erythropoiesis, we further explored separately the possible role of these factors, namely, iron, hypoxia and erythropoiesis drive in the induction of Lcn2 during anemia.
Our results indicate that Lcn2 is not directly regulated by iron, because Lcn2 was upregulated in anemic mice independently of serum and liver iron levels. In fact, mice treated with PHZ had elevated serum and liver iron levels, whereas, in contrast, mice subjected to phlebotomies and mice fed an iron-deficient diet showed a fall in serum and tissue iron, yet Lcn2 was induced in all these models. Because most of the iron in PHZ-treated mice is deposited in liver Kupffer cells, and to further understand whether accumulation of excess iron predominantly in liver parenchymal cells, as opposed to Kupffer cells, affects Lcn2 levels, we examined Lcn2 expression in dietary iron-overloaded mice and found no differences compared to the controls. Interestingly, elevated Lcn2 expression has been reported in beta-thalassemia mouse models [26], which are characterized by anemia due to ineffective erythropoiesis and concomitant iron overload due to increased intestinal iron absorption [27]. Our findings in this study imply that in beta-thalassemia mouse models, Lcn2 is most likely upregulated by anemia rather than by serum and tissue iron levels.
Because anemia may lead to tissue hypoxia, we also tested whether hypoxia could regulate Lcn2 expression. Using two different mouse models, namely CoCl2 and exposure to 10% oxygen, we demonstrated that Lcn2 is also induced during hypoxia, indicating that tissue oxygenation levels may be a factor in the regulation of Lcn2 expression by anemia. Moreover, we showed that during hypoxia significant changes occur in iron distribution, as circulating iron decreased while liver iron content rose, most likely because of augmented iron uptake by hepatocytes. Interestingly, the possible involvement of Tf-independent pathways for cellular iron uptake during hypoxia, namely Lcn2, has been suggested by earlier studies disclosing that Tf-dependent iron uptake does not significantly change during hypoxia and thus does not explain the augmented iron uptake observed in cells subjected to hypoxia [20,28]. Unlike hepcidin, which is repressed at a later phase (after day 3), hypoxia seems to regulate Lcn2 at an early phase (day 1). This possibly indicates that Lcn2 may play a protective role locally related to cellular survival, for example, by favoring iron sequestration in cells to maintain enzyme function [20]. In contrast, the suppression of hepcidin necessary to match iron supply to erythropoietic demand may represent a favorable response for the adaptation of the whole organism to conditions of low oxygen availability [29].
Both anemia and hypoxia are associated with increased erythropoiesis, which could directly influence Lcn2 production through EPO production. Thus, and to further understand how anemia/hypoxia regulate Lcn2 levels, we tested whether erythropoietic drive, which we stimulated through EPO injections, was sufficient to induce Lcn2 expression. We found that while EPO treatment strongly stimulated erythropoiesis, it was insufficient to evoke the Lcn2 response, indicating that Lcn2 induction by anemia/hypoxia does not require increased erythropoiesis and is not directly mediated by EPO.
In summary, in this study, we show that Lcn2, an iron-trafficking protein and an alternative to the Tf-mediated iron-delivery pathway, is upregulated during anemia, a situation where iron utilization and mobilization from stores increases. The induction of Lcn2 may be mediated by hypoxia, but does not require heightened erythropoiesis and is independent of body iron levels.
Acknowledgments
We thank Christian Dallaire for his work with atomic absorption spectroscopy, Dongmei Wang for her technical help, and Ovid Da Silva for his editorial assistance.
This work was supported by grants from the Canadian Institutes of Health Research (Grant No. MOP44045) and the Natural Sciences and Engineering Research Council of Canada (Grant No. 298518-06). MMS is the recipient of a scholarship from the Fonds de la recherche en santé du Québec.
References
- 1.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 2.Ponka P, Lok CN. The transferrin receptor: role in health and disease. Int J Biochem Cell Biol. 1999;31:1111–1137. doi: 10.1016/s1357-2725(99)00070-9. [DOI] [PubMed] [Google Scholar]
- 3.Flower DR. The lipocalin protein family: a role in cell regulation. FEBS Lett. 1994;354:7–11. doi: 10.1016/0014-5793(94)01078-1. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan J. Mechanisms of cellular iron acquisition: another iron in the fire. Cell. 2002;111:603–606. doi: 10.1016/s0092-8674(02)01164-9. [DOI] [PubMed] [Google Scholar]
- 5.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 6.Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim Biophys Acta. 2000;1482:272–283. doi: 10.1016/s0167-4838(00)00152-7. [DOI] [PubMed] [Google Scholar]
- 7.Goetz DH, Holmes MA, Borregaard N, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 8.Winkelmann G. Microbial siderophore-mediated transport. Biochem Soc Trans. 2002;30:691–696. doi: 10.1042/bst0300691. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Goetz D, Li JY, et al. An iron delivery pathway mediated by a lipocalin. Mol Cell. 2002;10:1045–1056. doi: 10.1016/s1097-2765(02)00710-4. [DOI] [PubMed] [Google Scholar]
- 10.Li JY, Ram G, Gast K, et al. Detection of intracellular iron by its regulatory effect. Am J Physiol Cell Physiol. 2004;287:C1547–C1559. doi: 10.1152/ajpcell.00260.2004. [DOI] [PubMed] [Google Scholar]
- 11.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Nilsen-Hamilton M. Identification of a new acute phase protein. J Biol Chem. 1995;270:22565–22570. doi: 10.1074/jbc.270.38.22565. [DOI] [PubMed] [Google Scholar]
- 13.Miranda CJ, Makui H, Andrews NC, Santos MM. Contributions of beta2-microglobulin-dependent molecules and lymphocytes to iron regulation: insights from HfeRag1(−/−) and beta2mRag1(−/−) double knock-out mice. Blood. 2004;103:2847–2849. doi: 10.1182/blood-2003-09-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makui H, Soares RJ, Jiang W, et al. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood. 2005;106:2189–2195. doi: 10.1182/blood-2005-02-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler S, Röhrs S, Tickenbrock L, et al. Lipocalin 24p3 is regulated by the Wnt pathway independent of regulation by iron. Cancer Genet Cytogenet. 2007;174:16–23. doi: 10.1016/j.cancergencyto.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272:20055–20062. doi: 10.1074/jbc.272.32.20055. [DOI] [PubMed] [Google Scholar]
- 18.Lok CN, Ponka P. Identification of an erythroid active element in the transferrin receptor gene. J Biol Chem. 2000;275:24185–24190. doi: 10.1074/jbc.M000944200. [DOI] [PubMed] [Google Scholar]
- 19.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142–24146. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 20.Schneider BD, Leibold EA. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood. 2003;102:3404–3411. doi: 10.1182/blood-2003-02-0433. [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275:21048–21054. doi: 10.1074/jbc.M000636200. [DOI] [PubMed] [Google Scholar]
- 22.Jelkmann W. Erythropoietin after a century of research: younger than ever. Eur J Haematol. 2007;78:183–205. doi: 10.1111/j.1600-0609.2007.00818.x. [DOI] [PubMed] [Google Scholar]
- 23.Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 24.Nicolas G, Viatte L, Bennoun M, et al. Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis. 2002;29:327–335. doi: 10.1006/bcmd.2002.0573. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Mori K, Li JY, Barasch J. Iron, lipocalin, and kidney epithelia. Am J Physiol Renal Physiol. 2003;285:F9–F18. doi: 10.1152/ajprenal.00008.2003. [DOI] [PubMed] [Google Scholar]
- 26.Weizer-Stern O, Adamsky K, Amariglio N, et al. mRNA expression of iron regulatory genes in b-thalassemia intermedia and b-thalassemia major mouse models. Am J Hematol. 2006;81:479–483. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 27.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in {beta}-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109:5027–5035. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershko C, Link G, Pinson A. Modification of iron uptake and lipid peroxidation by hypoxia, ascorbic acid, and alpha-tocopherol in iron-loaded rat myocardial cell cultures. J Lab Clin Med. 1987;110:355–361. [PubMed] [Google Scholar]
- 29.Shih SC, Claffey KP. Hypoxia-mediated regulation of gene expression in mammalian cells. Int J Exp Pathol. 1998;79:347–357. doi: 10.1046/j.1365-2613.1998.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]